

Background: The deviant ATP-binding site in the important drug resistance-linked Pdr subfamily is unique.
Results: Mutations in conserved residues exhibit significant ATPase activity, but reduced transport activity.
Conclusion: Conserved residues Cys-199, Glu-1013, and Asp-1042 are not directly involved in ATP hydrolysis but are actively involved in the transport cycle.
Significance: Our results indicate a new role for deviant ATP-binding sites.
Keywords: ABC Transporter, ATPases, Drug Transport, Membrane Proteins, Multidrug Transporters
Abstract
Pdr5 is the founding member of a large subfamily of evolutionarily distinct, clinically important fungal ABC transporters containing a characteristic, deviant ATP-binding site with altered Walker A, Walker B, Signature (C-loop), and Q-loop residues. In contrast to these motifs, the D-loops of the two ATP-binding sites have similar sequences, including a completely conserved aspartate residue. Alanine substitution mutants in the deviant Walker A and Signature motifs retain significant, albeit reduced, ATPase activity and drug resistance. The D-loop residue mutants D340A and D1042A showed a striking reduction in plasma membrane transporter levels. The D1042N mutation localized properly had nearly WT ATPase activity but was defective in transport and was profoundly hypersensitive to Pdr5 substrates. Therefore, there was a strong uncoupling of ATPase activity and drug efflux. Taken together, the properties of the mutants suggest an additional, critical intradomain signaling role for deviant ATP-binding sites.
Introduction
ATP-binding cassette (ABC)3 transporters comprise one of the largest families of integral membrane proteins. Overexpression of ABC multidrug transporters is a major problem in the treatment of pathogenic infection and cancer. An archtype ABC transporter such as mammalian P-glycoprotein has two nucleotide-binding domains (NBDs) and two transmembrane domains, each of which is contains six transmembrane helices connected by intra- and extracellular loops. Each ATP-binding site is a dimer of the two nucleotide-binding sites. An ATP sandwich is made between the Walker A, Walker B, and Q-loop of one NBD and the D-loop and Signature (C-loop) motifs of the other NBD. Transporters such as P-glycoprotein are symmetric: the motifs of the NBDs are equivalent in sequence and function.
However, a large number of clinically significant ABC transporters are asymmetric, including the CFTR chloride channel, the Mrp1 multidrug pump, and the Tap antigen transporter. In each of these, only one ATP-binding site is canonical in motif makeup. The other site is deviant (see Ref. 1 for a review of ABC transporter structure). In these transporters, the deviant site has little ATPase activity. These sites do not appear to participate actively in the transport cycle. Rather, they help to create the NBD dimer and play a positive, regulatory role (2–5). For instance, the deviant ATP-binding site of Mrp1 positively stimulates hydrolysis at the canonical site, thus allowing increased transport (3). In the work we describe in this paper, we show that the deviant ATP-binding site of the Pdr5 multidrug transporter plays an additional, critical, noncatalytic role.
Pdr5 is an ABC transporter that is the founding member of a subfamily of clinically significant multidrug efflux pumps with asymmetric nucleotide-binding motifs. The medical and economic importance of this large group of transporters cannot be overemphasized. For example, overexpression of Candida albicans Cdr1 and Cdr2 is a major problem in the treatment of fungal infections in immune-compromised patients and is a major cause of death (6–8). Interestingly, plants of agricultural significance appear to suffer from fungicide-resistant pathogens such as the gray mold Botrytis cinerea. At least some of this appears to be due to Pdr5 Subfamily member overexpression (9). The deviant ATP site of the Pdr subfamily is shown in Fig. 1A. The N-terminal NBD has deviant Walker A, Walker B, and Q-loop residues; the NBD in the center has an atypical signature domain. In these proteins, each ATP-binding site comprises the Walker A, Walker B, and Q-loop regions from one NBD and the Signature and D-loop regions of the other. It is therefore thought that one of the two Pdr5 ATP-binding sites is composed of canonical sequences from the two NBDs and the other of deviant portions (10).
FIGURE 1.
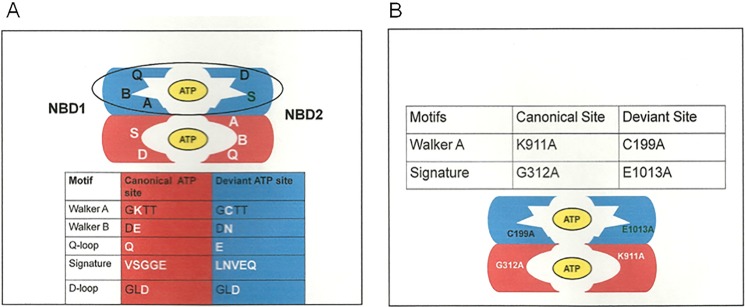
The proposed arrangement of ATP-binding sites in Pdr5. Pdr5 is an asymmetric ABC transporter. A, one ATP-binding site is deviant and comprises the Walker A and B from the N-terminal NBD and the signature and D-loop regions from the C-terminal NBD. The second site comprises canonical motifs. The major alterations in the motifs are shown in white lettering. The arrangement conforms to bioinformatic analysis described elsewhere (10). B, the initial mutations made in this study and their locations in the ATP-binding sites are illustrated.
Some controversy surrounds the functional role of the putative deviant ATP-binding site in this fungal family. Jha et al. (11) argue that it is catalytic in the closely related Cdr1 multidrug transporter in C. albicans. They found evidence that a C193A mutant protein binds but does not hydrolyze ATP and is enzymatically null. They also observed that the conserved deviant Signature residue E1004A creates a null phenotype with respect to drug resistance and has a complete loss of ATP hydrolysis, much like the canonical Signature residue G307A (12). Ernst et al. (13) provided evidence that at least in Pdr5, the comparable C199A mutant is phenotypically WT with respect to drug resistance and ATPase activity. This observation led them to posit that the deviant site, if it has any role at all, is regulatory rather than catalytic, much as is the case with the CFTR and Mrp1 transporters.
The role of the deviant Pdr5 ATP-binding site became important to us recently as we set out to map the signal interface of the Pdr5 transporter. We described a mutation S558Y in transmembrane helix 2 that uncouples ATP hydrolysis from drug transport. Thus, it hydrolyzes ATP and binds drug but exhibits a drug hypersensitivity phenotype that is almost equivalent to a pdr5 deletion mutation (14). We recovered and sequenced drug-resistant suppressor mutations that defined a signal interface pathway; its location was predicted from physical and cross-linking studies of other ABC transporters, except Pdr5 had a cis rather than a trans conformation with respect to the interaction between the Q loops and the intracellular loops. We observed suppressor mutations in intracellular loop 1 and in or near the Q-loop residues (Asp-242, Glu-244, and Asp-246) (15). Surprisingly, however, the Q-loop residues were in the deviant rather than the canonical NBD. Furthermore, we demonstrated that WT levels of drug resistance required Glu-244 and that it was nearly functionally equivalent to the canonical residue Gln-951 (the two are redundant). However, because significant ATPase activity remained in these mutants, we concluded that the Q-loop residues do not serve a catalytic function in either Pdr5 or P-glycoprotein (16). In P-glycoprotein at least, the Q-loop may be involved in nucleotide binding (17). In Pdr5, the deviant ATP-binding site was therefore certainly required to some degree, although its role remained unclear.
In the study reported here, we demonstrated that although a G312A mutation in the Signature region of the canonical ATP-binding site completely abolished drug transport and ATPase activity, the corresponding alteration in the deviant Signature (E1013A), as well as a C199A mutation in the deviant Walker A motif resulted in a protein, retained significant, albeit reduced ATPase and efflux capability. These canonical and deviant ATP-binding site residues are not functionally equivalent. We also observed that a D1042N mutant in the D-loop of the deviant ATP-binding site, although highly transport deficient, exhibited nearly WT Pdr5-specific ATPase activity. These results demonstrate that contrary to or perhaps in addition to what has been proposed for other asymmetric transporters, the deviant ATP site of the Pdr subfamily plays an active role in the transport cycle. Based on our observations, we propose a model in which ATP binding and nucleotide exchange—but not hydrolysis at the deviant site—results in a conformational change necessary for drug efflux. Hydrolysis at the canonical site returns Pdr5 to a drug-binding conformation.
EXPERIMENTAL PROCEDURES
Yeast Strains and Media
We derived strains from R-1 (14), listed in Table 1. R-1 lacks all ABC multidrug transporters but contains a PDR1–3 allele. As a result, Pdr5 is overexpressed when it is placed into R-1 by lithium acetate transformation by means of a yeast transformation kit (Sigma-Aldrich) as described in detail in recent publications (14, 15). After we constructed a mutant strain, we recovered the PDR5 coding region and sequenced the entire insert to ensure that only the desired mutation was present. Retrogen (San Diego, CA) or SeqWright (Houston, TX) carried out all routine DNA sequencing with a set of 14 primers designed to ensure at least two reads of every nucleotide. In our previous study (14), we described the construction and use of double-copy strains in the R-1 background for biochemical experiments involving plasma membrane (PM) vesicles. These strains have nearly twice the amount of Pdr5 protein and give higher signal to noise ratios. Both single- and double-copy strains are used as noted. Unless otherwise noted, we grew our strains in YEPD medium at 30 °C.
TABLE 1.
Yeast strains
All strains are isogenic to R-1 (14).
| Strain | Genotype |
|---|---|
| R-1 | MATα, PDR1–3, Δpdr5::KANMX4,ura3, yor1, snq2, pdr10, ycf1, pdr11, pdr3 |
| JG2015 | Isogenic to R-1; contains pSS607 integrated at the PDR5 gene as described (14) |
| JG2004 | Isogenic to R-1; contains two WT copies of PDR5 as described previously (14) |
| JG2052 | R-1 + C199A |
| JG2053 | R-1 + E1013A |
| JG2054 | R-1 + G312A |
| JG2064 | Double-copy G312A strain |
| JG2066 | Double-copy E1013A |
| JG2097 | D1042A (5-FOA derivative; see Ref. (9) for preparation of 5-FOA ura3 stocks) |
| JG2098 | D1042A double-copy strain |
| JG2108 | D340A 5-FOA derivative |
| JG2109 | D340A double-copy strain |
| JG2120 | D1042E 5-FOA derivative |
| JG2121 | D1042N 5-FOA derivative |
| JG2122 | D1042E,E1013A 5-FOA derivative |
| JG2126 | D1042E double-copy strain |
| JG2127 | D1042N double-copy strain |
| JG2131 | D1042E,E1013A double-copy |
Chemicals
We purchased most chemicals from Sigma-Aldrich. We dissolved cycloheximide (cyh) in sterile MilliQ water. We dissolved all other compounds in Me2SO. We added G-418 (geneticin; Research Products International, Mt. Prospect, IL) as dry powder to YEPD medium at a concentration of 200 mg/liter. We added 5-fluoorotic acid (5-FOA; Research Products International) as dry powder to supplemented SD medium at 1 mg/ml.
Site-directed Mutagenesis
We carried out site-directed mutagenesis with QuikChange II XL and Lightning kits (Agilent Technologies, Columbia, MD) as previously described (14, 15, 19), except that we designed primers with the program available on the Agilent Technologies web page. We introduced mutations into the yeast integrative plasmid pSS607, which contains a wild-type Pdr5 gene (14).
DNA Extraction and PCR Recovery of DNA
We extracted chromosomal DNA with a Qiagen Puregene Yeast/Bacteria kit B (Qiagen), and we amplified PDR5 by PCR. Consistently good amplification requires relatively pure DNA (260/280, ∼1.8–2.0; 260/230, ∼2.2) at a concentration of 60–120 ng. We performed 40 rounds of amplification as previously described (14, 15). We sent the PCR product of ∼4.5 kilobase pairs to SeqWright (Houston, TX) for purification and sequencing.
Measurements of Resistance in Liquid Culture
We determined the relative resistance of strains to cyh, clotrimazole (clo), imazalil, cyproconazole, and tebuconazole as recently described (14). We performed the plots and statistical analyses with GraphPad Prism software (GraphPad, San Diego, CA).
R6G Transport in Whole Cells
We measured R6G transport in whole cells grown at 30 °C in SD medium plus histidine and uracil medium as previously described (18). For the time course experiments, we loaded 1–2 × 106 cells suspended in 100 μl of 0.05 m Hepes buffer (pH 7.0) minus glucose for 90 min in the presence of 10 μm R6G. We did not de-energize the cells because several control experiments done with and without 20 mm 2-deoxyglucose indicated that this was unnecessary. Following loading, cells were pelleted in a microcentrifuge tube, and the supernatant was removed before resuspending in 300 μl of 0.05 m Hepes, 1 mm glucose. We incubated the tubes at 30 °C for the desired time. We terminated transport by placing the tubes in an ice-water bath. We determined cell fluorescence with a BD Biosciences FACSort with an excitation wavelength of 488 nm and an emission wavelength of 620 nm. We analyzed the data with a CellQuest program. Ten thousand cells were analyzed and used to construct the histogram plots. We expressed retained fluorescence in arbitrary units.
Preparation of Purified PM Vesicles
We prepared purified PM vesicles as recently described (18). We determined the protein concentration of PM vesicle protein with a bicinchonic acid kit (Peribo, Rockland, IL).
Gel Electrophoresis of PM Vesicle Proteins
We solubilized samples containing 20 μg of PM vesicles protein in SDS-PAGE for 30 min at 37 °C. We separated the proteins on NU-PAGE 7% Tris acetate gels (125–150 V for ∼80 min; Invitrogen).
Immunoblots of Pdr5 in PM Vesicles
We conducted Western blotting with 10 μg of PM vesicle protein as previously described (18). We performed the transfer from the gel to the nitrocellulose membrane (400 mAmp, 60 min) with an X Cell II minicell apparatus (Invitrogen). We purchased all the antibodies from Santa Cruz Biotechnology (Santa Cruz, CA). Initially (experiment in Fig. 2), we diluted the polyclonal goat anti-Pdr5 and anti-Pma1 antibodies 1:500 and 1:1,000 (yC18 and yN-20, respectively). Later (experiments in Fig. 5), we diluted the Pdr5 antibody 1:1,000 and the Pma1 antibody 1:500. We blocked the nitrocellulose membranes for 30 min with 5% nonfat milk in PBS containing 1% Tween 20. Following this, we incubated the filters in primary antibody overnight at 4 °C. We washed three times for 15 min before adding a 1:5,000 dilution of secondary antibody (donkey, anti-goat IgG horseradish peroxidase; SC2033) and incubating at room temperature for 2 h. We developed blots with a Novex ECL horseradish peroxidase chemiluminescent substrate reagent kit (Invitrogen). We determined the relative levels of Pdr5 in PM vesicles by comparison to the Pma1-loading control with Image J software as previously described (18). The ratio of Pdr5/Pma1 signal is indicated in each lane of the blots.
FIGURE 2.
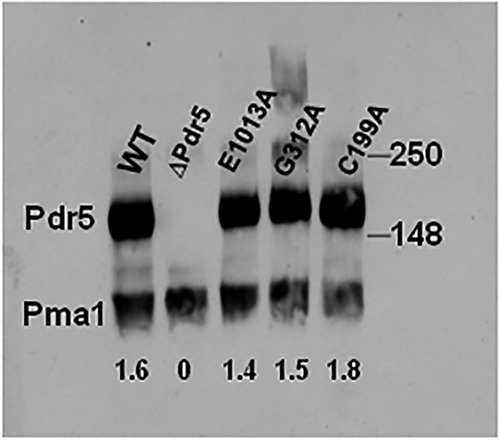
Immunoblot characterization of E1013A, C199A, and G312A (the E1013A, C199A, and G312A mutants are expressed at the plasma membrane to similar levels as WT protein). We prepared an immunoblot of WT and mutant purified PM vesicles as previously described (14, 19). We solubilized samples containing 10 μg of PM vesicle protein in SDS-PAGE for 30 min at 37 °C. We subjected the samples to gel electrophoresis and immunoblotting as described under “Experimental Procedures.” Double-copy strains are used for most of the biochemical assays in this study. The values in each lane are the ratios of Pdr5/Pma1 signal.
FIGURE 5.

The D340A and D1042A mutant proteins fail to localize to the plasma membrane. A, we determined resistance of the mutants to clo in liquid culture with isogenic WT and Δpdr5 strains, as described in Fig. 3. ■, WT; ▴ and solid line, Δpdr5; * and dashed line, D340A; × and dotted line, D1042A. B, Western blotting of PM vesicles from D340A and D1042A was performed as described under “Experimental Procedures.” We used 10 μg of PM vesicle protein made from double-copy strains in each sample and overexposed the blot. C, Western blot of the PM vesicle proteins (10 μg) made from the remaining strains used in this study. The conditions are described in B.
Assay of ATPase Activity
We measured ATPase activity as previously described for Pdr5 (19). In each experiment, we measured PM vesicles from both WT and Δpdr5 strains. Activity calculations subtracted the small amount of residual activity observed in the isogenic Δpdr5 strain (<5% WT). We verified all ATPase results by carrying out assays with at least two independent PM vesicle preparations/strain. Kinetic analyses were performed with GraphPad Prism software.
R6G Transport in Vesicles
We performed assays of R6G quenching in purified PM vesicles as described by Kolaczkowski et al. (20) with the same buffer conditions (0.05 m Hepes, pH 7.0) at 35 °C and minor modifications. We used a Varian Cary Eclipse fluorimeter (Agilent Technologies). The excitation wavelength was 529 nm, and the emission wavelength was 553 nm. Each reaction contained 30 μg of PM vesicles and 100 nm R6G. We mixed two reactions' worth (4 ml) of components in a 15-ml tube. Following this, we split the mixture and placed 2-ml portions in cuvettes. We added ATP (1.5–4 mm) to one tube and immediately placed it in the fluorimeter at 35 °C; we used a timer to keep track of elapsed time (∼30 s) between the addition of nucleotide and the start of data measurement. We monitored quenching for 15–20 min, depending on the experiment. Our kinetic analysis of quenching used Varian Cary Eclipse kinetics software.
Statistical Analyses
All statistical analyses were done with GraphPad Prism.
RESULTS
The Deviant Signature Motif Residue Glu-1013 Is Not Equivalent to Its Canonical Counterpart Gly-312
Fig. 1B shows the initial mutations used in this study. We constructed G312A and E1013A mutations in these completely conserved and analogous Signature regions of the canonical and deviant ATP-binding sites as well as the C199A mutation in the deviant Walker A motif. Ernst et al. (13) demonstrated that the canonically equivalent Walker A mutant K911A is phenotypically null. We constructed K911M-bearing yeast cells and confirmed that they are also phenotypically equivalent to our Δpdr5 strain (data not shown). We performed Western blotting (Fig. 2) to verify the presence of Pdr5 at levels comparable to WT in purified PM vesicles in all of the single-mutant strains. After introducing these mutations into the Δpdr5 strain R-1, we evaluated their resistance to two Pdr5 transport substrates: clo and cyh (Fig. 3, A and B). The G312A mutation in the canonical Signature motif resulted in cells that were as sensitive to these drugs as the Δpdr5 control strain. In contrast, the E1013A and C199A mutants retained significant drug resistance. The drug hypersensitivity of the C199A mutant was particularly modest but reproducible.
FIGURE 3.

Quantitative analysis of drug resistance and R6G transport indicates that deviant site mutants retain significant efflux capability. We evaluated drug resistance in liquid (YPD) culture as previously described (14). We seeded 2 ml of YPD broth with 0.5 × 105 cells and incubated them for 48 h at 30 °C in the presence of the drug prior to determining the cell concentration at A600. A and B, plots for clo (A) and cyh (B). We performed the plots and statistical analyses with GraphPad Prism software. The data points are the averages of at least three independent experiments. In A and B, ■, WT; ▴, Δpdr5; ▵, E1013A; ♢, C199A; ○, G312A C, R6G efflux was measured in C199A (♢) and E1013A (▵), along with a positive control (WT, ■) for 0, 5, 15, and 30 min after loading cells with 10 μm R6G for 90 min without glucose, as described under “Experimental Procedures.” We used the median of the retained fluorescence from each sample as the values in the plots, which are the averages of two independent experiments. In each sample, 10,000 cells were counted. D, comparison of WT, Δpdr5, and the G312A mutant after 30 min of efflux of R6G in 0.05 m Hepes, 1 mm glucose buffer.
Ernst et al. (13) demonstrated that the R6G transport capability of the C199A mutant is equivalent to WT. We carried out a whole cell R6G efflux experiment with E1013A and C199A mutants (Fig. 3C). Both exhibited WT efflux behavior, although we found no significant efflux in a Δpdr5 strain. Furthermore, the G312A mutant retained R6G fluorescence at levels comparable to the Δpdr5 strain even at 30 min (Fig. 3D).
E1013A and C199A Mutants Retain Significant ATPase Activity
We also measured the ATPase activity of the E1013A, C199A, and G312A mutants. Consistent with its null phenotype, the G312A mutant protein had no significant ATPase activity (Fig. 4), even though three independently produced PM vesicle preparations were tested. In contrast, E1013A and C199A mutants had reduced but significant ATPase levels. The Vmax of ∼60–80 nmol/min/mg in these mutants was approximately one-third to one-fourth that of WT.
FIGURE 4.
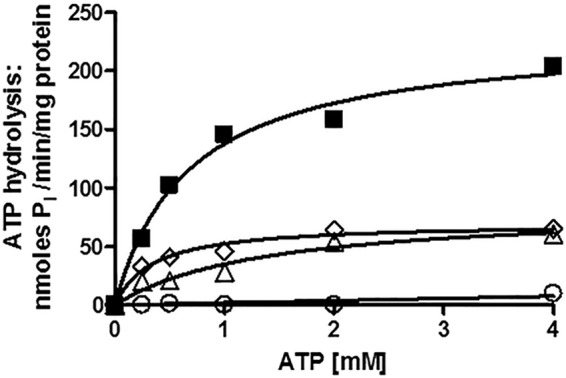
Pdr5-mediated ATPase activity in WT and mutant strains strongly suggests that the deviant ATP site is not a major source of catalytic activity. We measured Pdr5-specific ATPase activity as previously described (20) in double-copy strains with 12 μg of PM vesicle protein in each reaction, carried out for 8 min at 35 °C. Representative plots are shown (n = 3). Activity was assayed in PM vesicles prepared from double-copy strains as initially described (9). We used GraphPad Prism software to create and analyze the plots of activity versus ATP concentration. We subtracted the small (<5%) nonspecific activity observed in PM vesicles prepared from the isogenic ΔPdr5 strain before calculating the final activity. ■, WT; ▵, E1013A; ♢, C199A; ○, G312A.
The Km (ATP) of the E1013A transporter was similar to the WT; C199A was modestly lower. The Km and Vmax values for all of the experiments in this paper are found in Table 2 with their standard errors (n = 3). Assays were carried out with at least two independently prepared batches of purified PM vesicles. Over the course of this study, our PM vesicle preparations improved, and the resulting ATPase activities increased somewhat. However, the relative differences between WT and mutant preparations remained unchanged. It is now quite clear that the deviant and canonical Signature residues are not biochemically equivalent.
TABLE 2.
ATPase activity of mutant enzymes
| Substitution | Copy no. | Vmax | Km | Figure |
|---|---|---|---|---|
| nmol Pi/min/mg of protein | mm | |||
| WT | 1 | 163.4a | 0.76 | Fig. 9A |
| 1 | 197.6a | 1.25 | Fig. 9C | |
| 2 | 221.4 ± 18.81 | 0.80 ± 0.22 | Fig. 4 | |
| 2 | 287.6 ± 58.07 | 0.83 ± 0.33 | Fig. 9 (B and D) | |
| 2 | 331.0 ± 44.65 | 0.75 ± 0.27 | ||
| E1013A | 2 | 68.42 ± 10.26 | 1.00 ± 0.37 | Fig. 4 |
| 2 | 99.15 ± 10.74 | 0.29 ± 0.11 | ||
| C199A | 2 | 62.13 ± 4.836 | 0.25 ± 0.09 | Fig. 4 |
| D1042E | 1 | 67.99 + 3.083 | 0.43 ± 0.07 | Fig. 9C |
| 2 | 132.4 ± 6.176 | 0.59 ± 0.09 | Fig. 9D | |
| D1042N | 1 | 106.8 ± 19.29 | 1.16 ± 0.50 | Fig. 9A |
| 2 | 187.9 ± 18.71 | 0.60 ± 0.19 | Fig. 9B |
a Only one determination of WT activity was made in this experiment.
Analysis of D-loop Mutations
Although numerous studies have employed mutagenesis to analyze the Walker A and Signature motifs, surprisingly little is known about the role of the D-loop residues. A structural study of the Thermotoga maritima ABC transporter showed interactions between the D-loops and the Walker A motifs (21). A recent simulation derived from the structure of Sav1866 predicts that the D-loop exerts allosteric control of ATPase activity (22).
Relatively few reports phenotypically analyze site-directed mutants in this motif. In T4 Rad50, mutation of the D-loop residue Asp-512 to alanine or asparagine abolished ATPase activity by 2 orders of magnitude without changing the expression level or obvious physical properties of the protein (23). In that study, De la Rosa and Nelson provided strong support for an interaction between the D- and A-loops. A D-loop mutation in MsbA caused a reduction in ATP binding, but the nucleotides that did bind were hydrolyzed (24). These few studies therefore point to direct roles for the D-loop in ATP hydrolysis. Unlike the other motifs in the Pdr5 ATP-binding sites, both D-loop motifs of Pdr5 are canonical in sequence. To determine whether the two D-loop aspartate residues of Pdr5 are equivalent in function, we initially made alanine substitutions at each and evaluated their phenotypes. We observed clo sensitivity equivalent to the Δpdr5 control (Fig. 5A). We prepared purified PM vesicles and subjected them to Western blotting, with a Pma1 antibody as a loading control (Fig. 5B). In these preparations, the D1042A (deviant ATP-binding site) protein was almost entirely absent (lane 4), even when the film was overexposed.
In previous work, we described mutations in NBD1 that result in the loss of Pdr5 localization (25). The L183P mutation very close to Walker A of the deviant site produced a protein that was not trafficked to the PM. In that study, we obtained suppressors in a nearby residue that restored partial activity. We attempted a similar approach in the current study. We plated D1042A cells on 5 μm clo medium and recovered resistant colonies, which appeared at a relatively low frequency (∼10−7). All 10 of these independent mutations, however, were simple reversions of alanine back to aspartate.
A D1042N Mutant Is More Impaired than a D1042E Substitution
To better understand the role of Asp-1042, which is in the deviant site, we constructed D1042N and D1042E substitutions and tested their phenotypes. As with the alanine substitutions, these residues are the result of single-base pair substitutions in the codon for aspartate. The Western blot data for the Asp-1042 allelic series are found in Fig. 5C. The steady-state level of Pdr5 in PM vesicles was the same in the D1042E mutant and the WT. We observed a modest reduction of ∼25% in the D1042N mutant. This small difference was reproduced in a second set of purified PM vesicles (data not shown).
We then evaluated the relative resistance of these mutants to clo, cyh, imazalil, cyproconazole, and tebuconazole (Fig. 6). The effect of the mutations on drug sensitivity was drug-dependent. In every case, however, the D1042N allele was more sensitive than the D1042E substitution to a particular drug; D1042E actually had WT levels of resistance to imazalil. The D1042N mutant typically had one-fourth to one-sixth of the WT resistance.
FIGURE 6.

Analysis of D1042N and D1042E mutations. We determined the relative resistance of the Asp-1042 mutants to clo (A), cyh (B), imazalil (C), cyproconazole (D), and tebuconazole (E) in liquid culture as described under “Experimental Procedures.” In these experiments, n = 3. In the panels with the killing curves, ■ and green line, WT; ▴ and red line, Δpdr5; □ and violet line, D1042E; ● and blue line, D1042N.
In Fig. 7A, we present data demonstrating that, as was the case with the E1013A and C199A strains, R6G efflux in the D1042E mutant was equivalent to WT. However, yeast with the D1042N substitution showed considerably impaired transport. After 30 min of efflux in Hepes-glucose buffer, <2% of the initial fluorescence signal was retained in both D1042E and WT cells. In contrast, 54% of the initial signal remained in the D1042N mutant strain. The median fluorescence in this mutant decreased from ∼1660 arbitrary units to only ∼900. A representative histogram plot is found in Fig. 7B.
FIGURE 7.
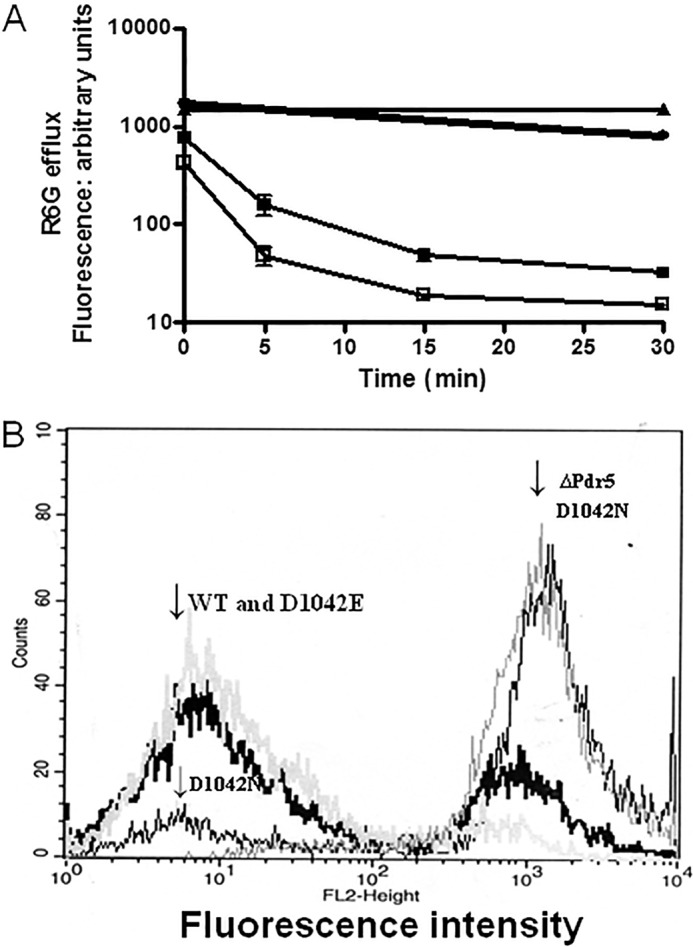
Analysis of R6G transport in whole cells demonstrates that the D1042N mutant is severely impaired. A, R6G efflux in whole cells was performed as described in the legend to Fig. 3 and under “Experimental Procedures.” ■, WT; ▴, ΔPdr5; ●, D1042N; □, D1042E. In these experiments, n = 2. B, histogram plots for cells showing the retained fluorescence after 30 min in 0.05 m Hepes, 1 mm glucose buffer. The positions of the major peaks for WT, ΔPdr5, D1042E, and D1042N are indicated.
The Behavior of Single- versus Double-Copy Strains
To explore the phenotypes of the D1042N and D1042E mutants further, we tested the clo and cyh resistance of the double-copy mutant strains and compared them with double-copy WT and single-copy mutant counterparts. The plots of growth versus increasing concentrations of clo and cyh are found in Fig. 8A and B. The resistance of the double-copy WT strain was ∼1.7 times greater than that of the single-copy WT strain for each drug. The D1042N strain showed significantly more resistance than the single one. In contrast, the D1042E double-copy strain exhibited only a very small amount of increased resistance.
FIGURE 8.

The behavior of single- versus double-copy strains. A and B, we determined the relative resistance of single- and double-copy strains in liquid culture as described under “Experimental Procedures” with clo (A) and cyh (B). The single-copy data are the same as those found in Fig. 6. For each allele, the solid line is the single-copy strain, the dashed line is the double copy. In the curves, ■ and green line, WT; □ and violet line, D1042E; ● and blue line, D1042N. C, we determined R6G efflux with the single- and double-copy strains, as described under “Experimental Procedures.” The symbols and lines are the same as those used in A and B for WT and D1042N. In these experiments, n = 4.
We also observed a clear effect of D1042N copy number on R6G efflux. We compared R6G transport in WT and D1042N strains containing one or two copies of PDR5 (Fig. 8C). The single-copy WT kinetics were similar to those shown in Figs. 3 and 6. Approximately 33% of the fluorescence remained at 5 min, and >90% was eliminated by 10 min. The double-copy WT strain, as expected, showed even stronger efflux. Only 2% of the initial fluorescence was retained at 5 min. From these curves, we estimated that the half-lives of the fluorescence in the single- and double-copy WT experiments were ∼4 and 2 min, respectively. The effect of doubling the copy number of the D1042N mutant is quite striking. In these experiments, the single-copy D1042N cells behaved much as they did in the experiment described in Fig. 6. The efflux was slow, and 57% of the time 0 signal remained at 30 min. Thus, transport in the single-copy WT was ∼8–10 times faster than in the D1042N mutant. The double-copy mutant strain, however, exhibited significant efflux, and the half-life of its fluorescence was only two to three times slower than the WT. We confirmed the difference between single- and double-copy D1042N strains with several, independent transformants (data not given).
The D1042N Mutant, although Severely Impaired, Has a Very High Level of ATPase Activity
We measured the ATPase activities of the D1042E and D1042N mutants and compared these values to the WT control. We made these measurements with at least two independent PM vesicle preparations. Because the single-copy D1042N strain is profoundly hypersensitive, we also prepared and tested mutant vesicles from single copy strains. These plots are found in Fig. 9 (A and B).
FIGURE 9.

ATPase activity of D1042N and D1042E: D1042N has nearly WT catalytic capability. ATPase activity of PM vesicles purified from single-copy (A) and double-copy (B) D1042N strains and single (C) and double-copy (D) D1042E strains was determined as previously described (20), with the reaction conditions described in the legend to Fig. 4. Representative plots are shown for each strain (see Table 2). ■, WT; □, D1042E; ●, D1042N. We monitored the effect of adding a WT Pdr5 gene on plasmid pSS607 to the D1042N mutant on resistance to clo (E) in liquid culture following transformation and strain verification. WT (■), D1042N (●), and D1042N + pSS607 (♦) were tested as described in the legend to Fig. 3.
Surprisingly, although the D1042N mutant showed the greatest drug hypersensitivity, its ATPase activity was similar to WT in single- and double-copy PM preparations. In fact, when the small (25%) reduction of D1042N protein in PM vesicles (Fig. 5C) is factored into the calculation, the activities of this substitution and WT enzyme are nearly equivalent. Furthermore, R6G transport in the other substitutions was indistinguishable from WT. The D1042N mutant, however, was noticeably deficient. This observation suggested that D1042N clearly uncoupled ATPase activity from transport. The D1042E preparations yielded activities that were lower than D1042N (Fig. 9, C and D).
The high ATPase activity and profound drug hypersensitivity of the single-copy D1042N mutant were surprising in light of the phenotypic similarity shared by the other mutants in this study. To be sure that the drug hypersensitivity we observed was attributable to a change in Pdr5 and not a chance mutation in another gene, we transformed the D1042N single-copy strain with a plasmid containing a WT PDR5 gene (pSS607). The clo resistance of this construct was similar to the WT (Fig. 9E). We obtained the same result for cyh (data not shown). Therefore, we can ascribe the drug hypersensitivity in the D1042N mutant exclusively to the altered Pdr5 protein.
Analysis of R6G Quenching in PM Vesicles with D1042N Mutant Protein Demonstrates an Uncoupling of ATPase Activity and Transport
The phenotype of the D1042N mutation strongly indicated that it largely uncoupled ATP hydrolysis from drug transport. To evaluate this quantitatively, we carried out R6G vesicle transport assays pioneered by Kolaczkowski et al. (20) and used successfully by Ernst et al. (13). The assay works on the principle that transport into the interior of inside-out vesicles results in fluorescence quenching caused by aggregate formation of the highly concentrated R6G in the vesicle lumen. Our transport studies used double-copy mutant and WT strains.
Fig. 10A depicts plots constructed from a data point for each minute of the experiment. Quenching of the signal is clearly observed. This experiment also confirms the findings of other groups (13, 20) that omitting either Pdr5 or ATP results in no quenching of the signal over a 15-min period. An actual trace (one data point every second) for the WT vesicles is found in Fig. 10B. The fit to first order kinetics is excellent, with an observed rate of 0.115/min. The mean (n = 3) rate was 0.102 ± 0.013
FIGURE 10.

Reduced R6G transport in D1042N PM vesicles demonstrates uncoupling of ATPase activity and transport. We measured quenching of R6G fluorescence in vesicles as described under “Experimental Procedures” using 100 nm R6G and 30 μg of vesicle protein. We show representative plots for each experiment. We performed these with an n value of at least 3, with three independent preparations of purified PM vesicles for each strain. A, dependence of quenching on Pdr5 and ATP in WT vesicles. ▴ = −Pdr5, +3 mm ATP; □, +Pdr5, −ATP; ■, +Pdr5, +3 mm ATP. B, a representative scan of retained fluorescence from WT vesicles in the presence of 3 mm ATP fit to first order kinetics. C, the effect of ATP concentration on the efflux of R6G from WT purified PM vesicles. ▴, 1.5 mm ATP; ▾, 4 mm ATP; ■, 3 mm ATP. D, the effect of adding 1 μm oligomycin at 13 min on quenching. □, +Pdr5, −ATP; ■, +Pdr5, +3 mm ATP; ▾, 3 mm ATP, +Pdr5, +1 μm oligomycin. E, reduced R6G quenching in the D1042N mutant. □, WT minus ATP; ■, WT plus 3 mm ATP; ○, D1042N without ATP; ●, D1042N with 3 mm ATP.
The original R6G quenching studies (13, 20) were done with 5 mm ATP, but the data in Fig. 10C show little difference in the quenching rate with 3 or 4 mm ATP. However, quenching of fluorescence was significantly reduced in assays that used 1.5 mm ATP, a concentration closer to the Km (ATPase). We used 3 mm ATP in all subsequent experiments.
Oligomycin is a potent inhibitor of Pdr5 ATPase (26), and Kolaczkowski et al. (20) demonstrated that when it is added to vesicles that are actively transporting R6G, rapid dequenching occurs, presumably because of diffusion. The data in Fig. 10D demonstrate this phenomenon in our preparations when we added 1 μm oligomycin at 13 min. Previously, we observed that 3,9-diacetylcarbazole behaves as a competitive inhibitor of R6G efflux in whole cell assays (27). We tested the effect of this Pdr5 transport substrate on R6G quenching by adding 10 μm of the compound 13 min after starting the assay with 3 mm ATP. These results were very similar to those with oligomycin (data not shown).
Fig. 10E shows representative plots (n = 3) of fluorescence quenching for WT and D1042N mutant vesicles in the presence and absence of ATP. Repeat assays with different vesicle preparations were remarkably similar. Although the mutant exhibited some quenching, the rate was obviously much slower. To quantitatively compare the two strains, we computed the relative quenching efficiency from the ATPase activity. This must be considered a minimum value. The fluorimetry measures the result of R6G aggregation, causing loss of signal rather than directly measuring transport. If aggregation is rate-limiting, the differences between WT and mutant transport capability could be larger. In the particular vesicles shown in Fig. 9D, the ATPase activity in vesicle transport buffer at 3 mm ATP was ∼275 nmol/min/mg for the WT and ∼160 nmol/min/mg for the D1042N mutant. In the 15-min period used to monitor quenching, the WT vesicles lost ∼5.0 arbitrary units/min, and the double-copy D1042N mutant lost only ∼1.6 arbitrary units/min. We used 30 μg of protein/reaction. We calculated the efficiency of ATP utilization as 0.61 arbitrary units quenched/nmol Pi released in the WT, but only 0.33 in the D1042N mutant. Results were similar in the other experiments. The differences (∼2×) between these two strains in R6G quenching and transport (2–3×) in whole cells with two copies of WT or D1042N were therefore similar (Fig. 7C). Thus, it appears that the quenching attributable to aggregation of transported R6G in vesicles is a reasonable measure of transport. When compared with its WT counterpart, even the double-copy mutant strain demonstrated significant uncoupling of ATPase activity and transport.
Double-mutant Analysis Indicates That Glu-1013 and Asp-1042 Function in the Same Biochemical Step
The partial-function mutant D1042E phenotypically resembles our Walker A and Signature mutants. The high ATPase activity and poor transport capability of the D1042N mutation indicate that Asp-1042 is required for proper signal transmission.
The use of double mutants is a powerful approach for determining whether two residues are coupled in function or are independent of each other (5, 28–30). In the latter case, the single-mutant phenotypes are strictly additive. In the former case, the phenotype of the double mutant is nonadditive relative to the single ones. To investigate whether Asp-1042 and the Signature residue Glu-1013 share the same biochemical function, we constructed an E1013A,D1042E double mutant to compare its drug resistance and ATPase activity to the single mutants. A Western blot demonstrated that the Pdr5 protein made by the double mutant was present in purified PM vesicles at steady-state levels that were ∼70% of the isogenic WT control (Fig. 5C). We compared the relative clo resistance of the single and double mutants, with the isogenic WT strain as a control (Fig. 11). The E1013A mutant was slightly more sensitive than D1042E, a result consistent with the data in previous experiments. The WT had an IC50 (7.5 μm) that was 6.3 times higher than E1013A (1.2 μm) and 4.4 times higher than D1042E (1.7 μm). The double-mutant had an IC50 of 1.3 μm, and the killing curve nearly superimposed that of E1013A. Had the effect been additive, an IC50 of 0.7 μm would have easily been observed. Instead, the double mutant showed only 5–10% inhibition at 1.0 μm. These observations demonstrated a nonadditive interaction and strongly suggested that these residues contribute to the same biochemical pathway. Using a paired t test, we determined that the E1013A and double-mutant curves were not significantly different (p < 0.05 with 99% confidence) and certainly not additive.
FIGURE 11.
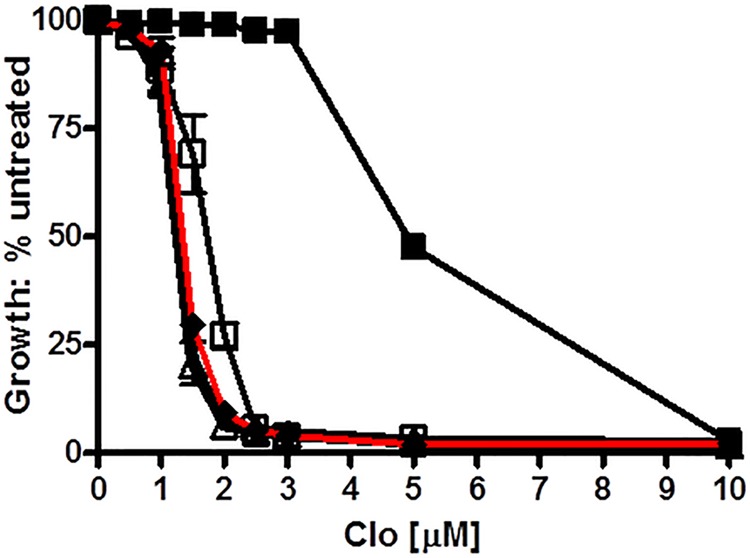
Double-mutant analysis demonstrates that Glu-1013 and Asp-1042 function in the same biochemical step. ■, WT; ▵, E1013A; □, D1042E; ♦, E1013A,D1042E. We determined the relative resistance to clo in liquid culture as previously described (10), with conditions equivalent to those in Fig. 3. The double mutant curve is indicated with a red line.
Nonadditivity was also observed with the ATPase activity. In these particular experiments, the Vmax of the WT was 331.0 ± 44.65 nmol/mg/min, which was 2.7 times that of D1042E (121.2 ± 9.569 nmol/mg/min) and 3.3 times that of E1013A (99.15 ± 10.74). Using these ratios and the standard errors, we determined that the expected activity of the D1042E,E1013A mutant ATPase would be 55.16 ± 7.612 nmol/mg/min. However, when two preparations of the double mutant vesicles were assayed, we observed a value of 79.19 nmol/mg/min ± 7.903 (n = 3). This value is significantly different from the one expected on the assumption of additivity.
The double mutant analysis therefore allows us to conclude that a signature residue that in the canonical site is required for catalysis (Gly-312) is used in the deviant site (Glu-244) to convey signal. We also constructed a C199A,E1013A double mutant for analysis. Unfortunately, the resulting double-mutant protein failed to localize to the PM (data not shown).
DISCUSSION
The members of the clinically important Pdr subfamily of fungal ABC transporters exhibit a characteristic and extreme pattern of NBD asymmetry that is distinct from any other ABC transporter. The deviant portions of NBD1 and NBD2 are thought to form a dimer and were modeled as such by Rutledge et al. (10), but the role played by these residues remained uncertain.
When mutations are introduced into highly conserved residues of the deviant Pdr5 ATP-binding site, similar phenotypes are observed in most cases. The ATPase activity and drug transport capability are usually reduced but not eliminated, as is the case with canonical ATP-binding site mutants such as the Signature allele G312A. C199A exhibits reduced but significant drug resistance and significant ATPase activity, but its canonical counterparts K911A or K911M exhibit a null phenotype. The first conclusion that we draw, therefore, is that ATP hydrolysis does not take place physiologically at the deviant site. This is supported by the lack of an obvious catalytic residue in the deviant Walker B motif. An alternative, catalytic mechanism proposed for Cys-193 in Walker A of Cdr1 (31) is not operating in vivo to any major extent in Pdr5, although it is possible that the deviant site catalyzes a very small amount of nucleotide hydrolysis as is observed with CFTR.
The second conclusion reached from analysis of a D1042N mutant is that at least some of the conserved deviant site residues communicate information from the NBDs to the drug transport sites in the transmembrane domains. This mutant therefore uncouples ATPase activity from transport, suggesting that in the deviant ATP-binding site, additional residues in addition to those of the Q-loop actually make up part of the transmission interface. The nonadditive phenotype of the D1042E,E1013A mutant implicates the Signature region in the same biochemical process. Kolaczkowski et al. (32) recently presented evidence that a residue in the deviant H-loop of the Pdr5 homologue Cdr1 is part of a signal interface.
If we had not explored the properties of the D1042N mutation, we would have assumed that the deviant site of Pdr5 functions in a manner that is similar to the mechanisms proposed for other asymmetric ABC transporters. For instance, a mutant such as E1013A showed significantly reduced ATPase activity and a similar reduction in drug resistance. We would have argued that such a mutant reduces the efficacy of dimer formation or interferes with the regulation of ATPase activity. At present, nobody has reported making and testing a mutation analogous to D1042N in the other asymmetric transporters. However, it is plausible that these mutations remain to be discovered and that the other deviant ATP-binding sites function actively in the transport cycle like Pdr5. The biochemical activities proposed for various deviant ATP-binding sites are not mutually exclusive.
Fig. 12 shows our proposed model of how the two ATP-binding sites might work. It is a variation on a plausible proposal by Gupta et al. (33). They suggested that, analogously with CFTR, the role of the deviant Pdr5 site is to bind nucleotide tightly enough to create the dimer between NBDs. Binding of ATP via nucleotide exchange and subsequent hydrolysis at the canonical ATP-binding site provide the conformational changes for drug binding and efflux. In this model, therefore, the deviant site does not play a direct role in the drug transport cycle. Significantly, our variation implicates the deviant site directly in the transport cycle. This model hypothesizes that the deviant site binds nucleotide but does not catalyze ATP hydrolysis. Instead, nucleotide exchange at the deviant site (step III-A) is used to communicate a signal to switch from a drug-binding conformation facing inward to a drug-effluxing conformation facing the cytosol. The signal is sent via residues such as Asp-1042 and Glu-1013 (and probably Cys-199), which no longer are required for catalysis, as well as the two Q-loop residues of Pdr5, which are known to function together (11). Binding and catalysis at the canonical ATP site (step III-B) resets Pdr5 in a drug-binding conformation.
FIGURE 12.
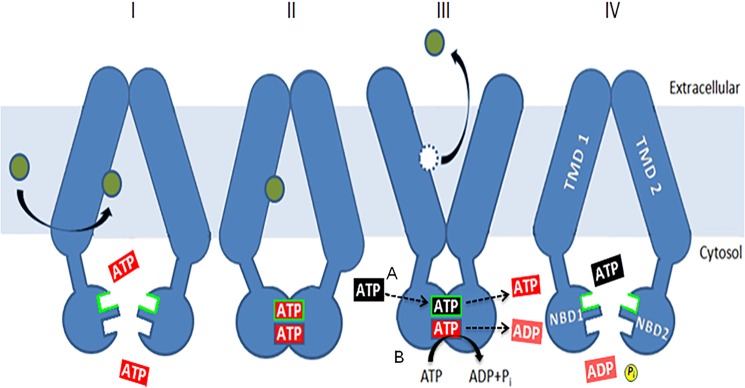
A model for the role of the deviant ATP-binding site. The model is a simple variation on one initially proposed by Gupta et al. (33). ATP (red) is bound at both sites (I), causing dimerization (II) of the NBDs, but hydrolysis is limited to the canonical one. Binding and nucleotide exchange (black for red) at the deviant site (III), indicated by the green border, allow a change from an inward-facing, drug-binding conformation to an outward-facing, drug-releasing conformation. This is mediated by residues such as Glu-1013 and Asp-1042. ATP hydrolysis at the canonical site (IV) restores the transporter to a drug-binding conformation and causes the release of unhydrolyzed ATP (blue). Allosteric inhibition of Pdr5-ATPase activity coming from the transmembrane domains via the intracellular loops works at this step. ATPase activity is not stimulated by drugs, so the cycle is thought to be constitutive in nature. Some of the mutants described here force the nucleotide exchange step to take place along with hydrolysis at the canonical site, as originally proposed by Gupta et al. (33).
Our model attempts to explain the uncoupled behavior of the D1042N and the reduced hydrolysis brought about by the other substitutions in residues of the deviant site that are clearly not involved in positioning or hydrolyzing ATP. These observations firmly establish an active role for the deviant ATP site, even if some of the details shown in Fig. 12 are incorrect. These observations are much harder to explain in the original model, in which the deviant site helps make the ATP sandwich but does not directly participate in the transport cycle. We posit that in our variation, mutant substitutions such as D1042E that reduce the ATPase activity and transport modestly because they simply slow down the cycle by preventing the deviant site from signaling the first conformational switch at a WT rate. Therefore, simply doubling the amount of mutant protein in the cell has only a small effect. Under these mutant conditions, Pdr5 behaves as described by the original Gupta et al. (33) model with both conformational changes (which may not be very different from the WT) affected by the activity at the canonical site. As a result, fewer ATP molecules are hydrolyzed per unit of time.
The behavior of the D1042N mutant, however, is distinctly different from the other substitutions we analyzed in several respects—notably, greater drug sensitivity despite very high ATPase activity. Furthermore, the strain carrying a double copy of D1042N mutant Pdr5 gene and expressing twice the amount of mutant protein demonstrates greater resistance and transport than its single counterpart. The apparent efficiency with which the energy from ATPase activity fuels R6G transport in vesicles is approximately half the WT in the double-copy strains. The D1042N mutant may be in a conformation that permits ATPase activity but transmits signal or releases drug poorly. In such circumstances, increasing the amount of mutant protein would increase signal transmission and drug transport.
Acknowledgments
We thank Sister Stephen Patrick Joly for designing Fig. 12. We appreciate Dr. Robert Ernst's critical comments on and enthusiasm for this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM07721 (to J. G.). This work was also supported by National Science Foundation Grant MCB1048838 and funds from the Intramural Research Program of the National Institutes of Health, NCI, Center for Cancer Research (to S. V. A.).
- ABC
- ATP-binding cassette
- cyh
- cycloheximide
- 5-FOA
- 5-fluoroorotic acid
- NBD
- nucleotide-binding domain
- PM
- plasma membrane
- R6G
- rhodamine 6G
- clo
- clotrimazole.
REFERENCES
- 1. Jones P. M., O'Mara M. L., George A. M. (2009) ABC transporters. A riddle wrapped in a mystery inside an enigma. Trends Biochem. Sci. 34, 520–531 [DOI] [PubMed] [Google Scholar]
- 2. Berger A. L., Ikuma M., Welsh M. J. (2005) Normal gating of CFTR requires ATP binding to both nucleotide-binding domains and hydrolysis at the second nucleotide-binding domain. Proc. Natl. Acad. Sci. U.S.A. 102, 455–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hou Y. X., Riordan J. R., Chang X. B. (2003) ATP binding to the first nucleotide-binding domain of the multidrug resistance-associated protein plays a regulatory role at low nucleotide concentration, whereas ATP hydrolysis at the second plays a dominant role in ATP-dependent leukotriene C4 transport. J. Biol. Chem. 278, 3599–35605 [DOI] [PubMed] [Google Scholar]
- 4. Linton K. J., Higgins C. F. (2007) Structure and function of ABC transporters. The ATP switch provides flexible control. Pflugers Arch. 453, 555–567 [DOI] [PubMed] [Google Scholar]
- 5. Vergani P., Lockless S. W., Nairn A. C., Gadsby D. C. (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433, 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sanglard D., Kuchler K., Ischer F., Pagani J.-L., Monod M., Bille J. (1995) mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug Transporters. Antimicrob. Agents Chemother. 39, 2378–2386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martínez M., López-Ribot J. L., Kirkpatrick W. R., Bachmann S. P., Perea S., Ruesga M. T., Patterson T. F. (2002) Heterogeneous mechanisms of azole resistance in Candida albicans clinical isolates from an HIV infected patient on continuous fluconazole therapy for oralpharyngeal candidosis. J. Antimicrob. Chemother. 49, 515–524 [DOI] [PubMed] [Google Scholar]
- 8. Loeffler J., Stevens D. A. (2003) Antifungal drug resistance. Clin. Infect. Dis. 36, S31–S41 [DOI] [PubMed] [Google Scholar]
- 9. Leroux P., Fritz R., Debieu D., Albertini C., Lanen C., Bach J., Gredt M., Chapeland F. (2002) Mechanisms of resistance to fungicides in field strains of Botrytis cinerea. Pest Manag. Sci. 58, 876–888 [DOI] [PubMed] [Google Scholar]
- 10. Rutledge R. M., Esser L., Ma J., Xia D. (2011) Toward understanding the mechanism of action of the yeast multidrug transporter Pdr5. J. Struct. Biol. 173, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jha S., Karnani N., Dhar S. K., Mukhopadhayay K., Shukla S., Saini P., Mukhopadhayay G., Prasad R. (2003) purification and characterization of the N-terminal nucleotide-binding domain of an ABC transporter of C. albicans. Uncommon cysteine-193 of Walker A is critical for ATP hydrolysis. Biochemistry 42, 10822–10832 [DOI] [PubMed] [Google Scholar]
- 12. Kumar A., Shukla S., Mandal A., Shukla S., Ambudkar S. V., Prasad R. (2010) Divergent signature motifs of nucleotide-binding domains of ABC transporter CaCdr1 of pathogenic Candida albicans are functionally asymmetric and non-interchangeable. Biochim. Biophys. Acta 1798, 1757–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ernst R., Kueppers P., Klein C. M., Schwarzmueller T., Kuchler K., Schmitt L. (2008) A mutation of the H-loop selectively affects rhodamine transport by the yeast multidrug transporter Pdr5. Proc. Natl. Acad. Sci. U.S.A. 105, 5069–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sauna Z. E., Bohn S. S., Rutledge R., Dougherty M. P., Cronin S., May L., Xia D., Ambudkar S. V., Golin J. (2008) Mutations define cross-talk between the N-terminal nucleotide-binding domain and transmembrane-helix 2 of the yeast multidrug resistance transporter Pdr5. Possible conservation of a signaling interface for coupling ATP hydrolysis to drug transport. J. Biol. Chem. 283, 35010–35022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ananthaswamy N., Rutledge R., Sauna Z. E., Ambudkar S. V., Dine E., Nelson E., Xia D., Golin J. (2010) The signaling interface of the yeast multidrug transporter Pdr5 adopts a cis configuration and there are functional overlap and equivalence of the deviant and canonical Q- loop residues. Biochemistry 49, 4440–4449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Urbatsch I. L., Gimi K., Wilke-Mounts S., Senior A. (2000) Investigation of the role of the glutamine-471 and glutamine-1114 in the two catalytic sites of P-glycoprotein. Biochemistry 39, 11921–11927 [DOI] [PubMed] [Google Scholar]
- 17. Yang R., Hou Y.-X., Campbell C. A., Palaniyandi K., Zhao Q., Bordner A. J., Chang X. B. (2011) Glutamine residues of the Q-loop of multidrug resistance-associated protein Mrp1 contribute to ATP binding via interaction with metal cofactor. Biochim. Biophys. Acta 1808, 1790–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Downes M. T., Mehla J., Ananthaswamy N., Wakschlag A., Lamonde M., Dine E., Ambudkar S. V., Golin J. (2013) The transmembrane interface of the Saccharomyces cerevisiae multidrug transporter Pdr5. Val-656 located in intracellular-loop 2 plays a major role in drug resistance. Antimicrob. Agents Chemother. 57, 1025–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Golin J., Kon Z. N., Wu C. P., Martello J., Hanson L., Supernavage S., Ambudkar S. V., Sauna Z. E. (2007) Complete inhibition of the Pdr5 multidrug efflux pump ATPase activity by its transport substrate clotrimazole suggests GTP as well as ATP maybe used as an energy source. Biochemistry 46, 13109–13119 [DOI] [PubMed] [Google Scholar]
- 20. Kolaczkowski M., van der Rest M., Cybularz-Kolaczkowska A., Soumillion J.-P., Konings W. N., Goffeau A. (1996) Anticancer drugs, ionophoric peptides and steroids as substrates of the yeast multidrug transporter Pdr5. J. Biol. Chem. 271, 31543–31548 [DOI] [PubMed] [Google Scholar]
- 21. Hohl M., Briand C., Grütter M. G., Seeger M. A. (2012) Crystal structure of a heterodimeric ABC transporter in the inward-facing conformation. Nat. Struct. Mol. Biol. 19, 395–402 [DOI] [PubMed] [Google Scholar]
- 22. Jones P. M., George A. M. (2012) Role of the D-loop in allosteric control of ATP hydrolysis in an ABC transporter. J. Phys. Chem. A. 116, 3004–3013 [DOI] [PubMed] [Google Scholar]
- 23. De la Rosa M. B., Nelson S. W. (2011) An interaction between the Walker A and D-loop motifs is critical to ATP hydrolysis and cooperativity in bacteriophage T4 Rad50. J. Biol. Chem. 286, 26258–26266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schultz K. M., Merten J. A., Klug C. S. (2011) Characterization of the E506Q and H537A dysfunctional mutants in the E. coli transporter MsbA. Biochemistry 50, 2594–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Thozée C. P., Cronin S., Goj A., Golin J., Ghislain M. (2007) Subcellular trafficking of the yeast plasma membrane transporter Pdr5 is impaired by a mutation in the N-terminal nucleotide-binding fold. Mol. Microbiol. 63, 811–825 [DOI] [PubMed] [Google Scholar]
- 26. Decottignies A., Kolaczkowski M., Balzi E., Goffeau A. (1994) Solubilization and characterization of overexpressed Pdr5 multidrug resistance nucleotide triphosphatase of yeast. J. Biol. Chem. 269, 12797–12803 [PubMed] [Google Scholar]
- 27. Hanson L., May L., Tuma P., Keeven J., Mehl P., Ferenz M., Ambudkar S. V., Golin J. (2005) The role of hydrogen bond acceptor groups in the interaction of substrates with Pdr5p, a major yeast nucleotide transporter. Biochemistry 44, 9703–9713 [DOI] [PubMed] [Google Scholar]
- 28. Horovitz A. (1996) Double-mutant cycles. A powerful tool for analyzing structure and function. Folding Des. 6, R121–R126 [DOI] [PubMed] [Google Scholar]
- 29. Quiram P. A., McIntosh J. M., Sine S. M. (2000) Pairwise interactions between neuronal α7 acetycholine receptors and α-conotoxin PnIB. J. Biol. Chem. 275, 4889–4996 [DOI] [PubMed] [Google Scholar]
- 30. Wandschneider E., Hammack B. N., Bowler B. E. (2003) Evaluation of cooperative interactions between substructures of iso-1-cytochrome C using double-mutant cycles. Biochemistry 42, 10659–10666 [DOI] [PubMed] [Google Scholar]
- 31. Prasad R., Sharma M., Rawal M. (2011) Functionally relevant residues of Cdr1p. A multidrug transporter of human pathogenic Candida albicans. J. Amino Acids 2011, 531412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kolaczkowski M., Sroda-Pomianek K., Kolaczkowska A., Michalak K. (2013) A conserved interdomain communication pathway of pseudosymmetrically distributed residues affects substrate specificity of fungal multidrug resistance transporter Cdr1. Biochim. Biophys. Acta 1828, 479–490 [DOI] [PubMed] [Google Scholar]
- 33. Gupta R. P., Kueppers P., Schmitt L., Ernst R. (2011) The multidrug transporter Pdr5. A molecular diode? Biol. Chem. 392, 53–60 [DOI] [PubMed] [Google Scholar]