

Background: The mechanism for inhibiting fork progression after DNA damage still remains.
Results: CRL4Cdt2 promotes the degradation of the p12. Cells expressing a stable form of p12 exhibit UV-resistant DNA synthesis and decreased inhibition of fork progression.
Conclusion: p12 degradation by CRL4Cdt2 is critical for inhibiting fork progression.
Significance: p12 degradation is one mechanism by which DNA damage in S-phase cells inhibits fork progression.
Keywords: Checkpoint Control, DNA Damage Response, DNA Polymerase, DNA Replication, Ubiquitination
Abstract
After acute DNA damage, the cell arrests S-phase progression by inhibiting origin initiation and fork progression to repair damaged DNA. The intra-S-phase checkpoint kinase Chk1 phosphorylates Cdc25A to target the latter for degradation by CRL1β-TrCP and so inhibit origin firing. The mechanism for inhibiting fork progression, however, has not been identified. Here, we show that degradation of p12, the fourth subunit of DNA polymerase δ, is critical for inhibiting fork progression. CRL4Cdt2 is an E3 ligase that ubiquitinates and degrades p12 after UV treatment. Cells expressing a stable form of p12 exhibit UV-resistant DNA synthesis. DNA fiber assay and alkaline-sucrose gradient assay demonstrate that the impairment of fork progression after DNA damage requires p12 degradation. These results suggest that ubiquitination of p12 through CRL4Cdt2 and subsequent degradation form one mechanism by which a cell responds to DNA damage to inhibit fork progression.
Introduction
After acute DNA damage, the intra-S-phase checkpoint arrests S-phase progression to repair damaged DNA and rescue DNA replication so that cells can later enter mitosis without any DNA sequence changes (1, 2). DNA damage during S-phase delays or arrests DNA replication by inhibiting origin initiation and fork progression. Cdc25A, a phosphatase for activating cyclin-dependent kinase 2, is targeted for degradation by CRL1β-TrCP to inhibit origin firing (3–5). The mechanism for inhibiting fork progression, however, has not been identified (6–8). p12, the smallest subunit of polymerase δ, is important for effective DNA replication and cell proliferation (9, 10). Furthermore, p12 is degraded immediately after DNA damage through polyubiquitination (11) by an unknown E3 ligase. Here, we show that p12 degradation is critical for inhibiting fork progression during the intra-S-phase checkpoint.
CRL4Cdt2 is involved in the polyubiquitination of Cdt1 (12–15), p21 (16–18), and Set8 (19–22, 31) after DNA damage. All these substrates interact with PCNA2 through a specialized PCNA-interacting motif (PIP degron) and are then recognized by CRL4Cdt2 for polyubiquitination. Here we show that p12 has a similar PIP degron and that CRL4Cdt2 ubiquitinates and degrades p12 after UV treatment. p12 degradation is critical for inhibiting fork progression.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
HCT116 and HCT116p53−/− cells have been described (23). HeLa, 293T, and WI38 cells were purchased from ATCC. p12 cDNA was purchased from Invitrogen, and mutations were induced by PCR. Stable cell lines expressing p12 mutants were generated by MSCVpuro vector (Clontech). Cells stably expressing p12 proteins were selected in 1 μg/ml puromycin (Sigma). Antibodies directed against the indicated proteins were purchased from the following sources: PCNA (PC10) and Cul4 (C-19) (Santa Cruz Biotechnologies); phospho-Chk1, Chk2, phospho-Chk2, histone-H2AX, and phospho-histone-H2AX, β-TrCP (Cell Signaling Technology); p125 (BD Biosciences); Chk1 (Novus Biologicals); and DDB1 (Invitrogen). The antibody against p12 was kindly provided by Igor Stagljar (University of Toronto) (24). siRNA transfections into cell lines were performed using Oligofectamine (Invitrogen). The following siRNA oligonucleotides were purchased from Invitrogen: β-TrCP (1+2), 5′-ACAGGAUCAUCGGAUUCCA-3′ and 5′-UCAGUGCAGUUAAGUCGUA-3′; β-TrCP (1/2), 5′-GAUGUUCCACAAAUUCCACUUTT-3′. Other siRNA oligonucleotides were described before (21).
In Vitro Ubiquitination Assays
The purification of CRL4Cdt2 complex was described previously (21). Briefly, Cul4A-Myc, DDB1-Myc, FLAG-Roc1, and FLAG-Cdt2 were transiently overexpressed in 293T cells and purified by immunoprecipitation with anti-Myc antibody. Purified complexes were mixed with GST-tagged wild type or a T8D mutant of p12 in ubiquitination reaction buffer. The ubiquitination reactions were incubated at 37 °C for 1 h and terminated by adding Laemmli sample buffer. Proteins were resolved by SDS-PAGE and immunoblotted with anti-GST antibody (Sigma).
Measurement of UV-resistant DNA Synthesis after UV Irradiation
Experiments were performed as described previously (4). Briefly, 293T cells were treated with UVC light (254 nm), as measured by a UVX digital radiometer (UVP, Inc., San Gabriel, CA) and labeled with [3H]thymidine for 15 min before harvesting (1 μCi/ml; PerkinElmer Life Sciences). After removing medium, the cells were fixed by the addition of 5% trichloroacetic acid and washed three times with 5% trichloroacetic acid. DNA was eluted in 1% SDS and 10 mm NaOH, spotted into paper for drying, and transferred to a scintillation vial. After the addition of scintillation fluid, incorporation of [3H]thymidine was measured by scintillation counting. For normalization, cells were prelabeled with [14C[thymidine for 24 h before the radiation, and DNA synthesis was measured as the 3H counts normalized to the 14C counts.
Alkaline Sucrose Gradient Centrifugation
Cells were irradiated with UV, labeled with [3H]thymidine (5 μCi/ml), and lysed in lysis buffer (0.5 m NaOH, 0.02 m EDTA, pH 12.5, 0.1% Nonidet P-40). Lysates were layered on top of a linear sucrose gradient (5–15% sucrose in 0.1 m NaOH, 0.9 m NaCl, 0.01 m EDTA, pH 12.5) and subsequently centrifuged at 26,000 rpm for 2 h at 20 °C. After collecting each fraction, DNAs were precipitated on glass microfiber filters, and radioactivity measured by scintillation counting.
DNA Fiber Assay
Cells were incubated with 50 μm CldU for 20 min. After irradiation of UV, cells were immediately incubated with 200 μm IdU for 30 min. Labeled cells were lysed (0.5% SDS, 50 mm EDTA, 200 mm Tris-HCl, pH 7.4) on glass slides and spread for DNA isolation. DNA fiber was fixed with methanol-acetic acid (3:1) and denatured with 1.5 m HCl for 30 min. For immunofluorescence, denatured DNA was stained for CldU with rat anti-BrdU monoclonal antibody (AbD Serotec, 1:200) and for IdU with mouse anti-BrdU antibody (BD Biosciences, 1:200) for 4 h at room temperature followed by an Alexa Fluor 594-conjugated anti-rat IgG (Molecular Probes) (red) and an Alexa Fluor 488-conjugated anti-mouse IgG (Molecular Probes) (green) at 1:200 dilution for 1 h at room temperature. Lengths of green tracks were determined by ImageJ software.
RESULTS AND DISCUSSION
Because p12 has a PIP box similar to the PIP degron identified in other CRL4Cdt2 substrates, we tested whether CRL4Cdt2 is responsible for p12 degradation. We depleted Cdt2 by using siRNA and measured p12 levels after UV treatment. As expected, UV treatment reduced the level of p12 in HeLa cells (Fig. 1A). Depletion of Cdt2 increased the basal level of p12 and prevented the degradation of p12 following DNA damage. CRL4Cdt2 ligase complex is also composed of DDB1 and Cul4A or Cul4B. Consistent with our hypothesis, depletion of DDB1 or Cul4A and Cul4B also abrogated p12 degradation after UV treatment (Fig. 1B). The decrease of p12 after knockdown of PCNA suggests that PCNA is required for p12 stabilization in normal cells. Although PCNA depletion decreased the basal level of p12, degradation of the residual p12 in response to DNA damage was abrogated, suggesting that the PCNA is also important for the degradation of p12 after DNA damage. Therefore, p12 degradation after UV irradiation requires components of CRL4Cdt2 and PCNA, much like the other substrates of this E3, such as Cdt1, p21, and Set8. si-Cdt2 caused accumulation of the cells in G2 as reported (15), but the depletion of Cul4A/B or DDB1 did not alter cell cycle profile (Fig. 1C), suggesting that the stabilization of p12 after depletion of CRL4 components or PCNA is not an indirect effect of cell cycle change.
FIGURE 1.
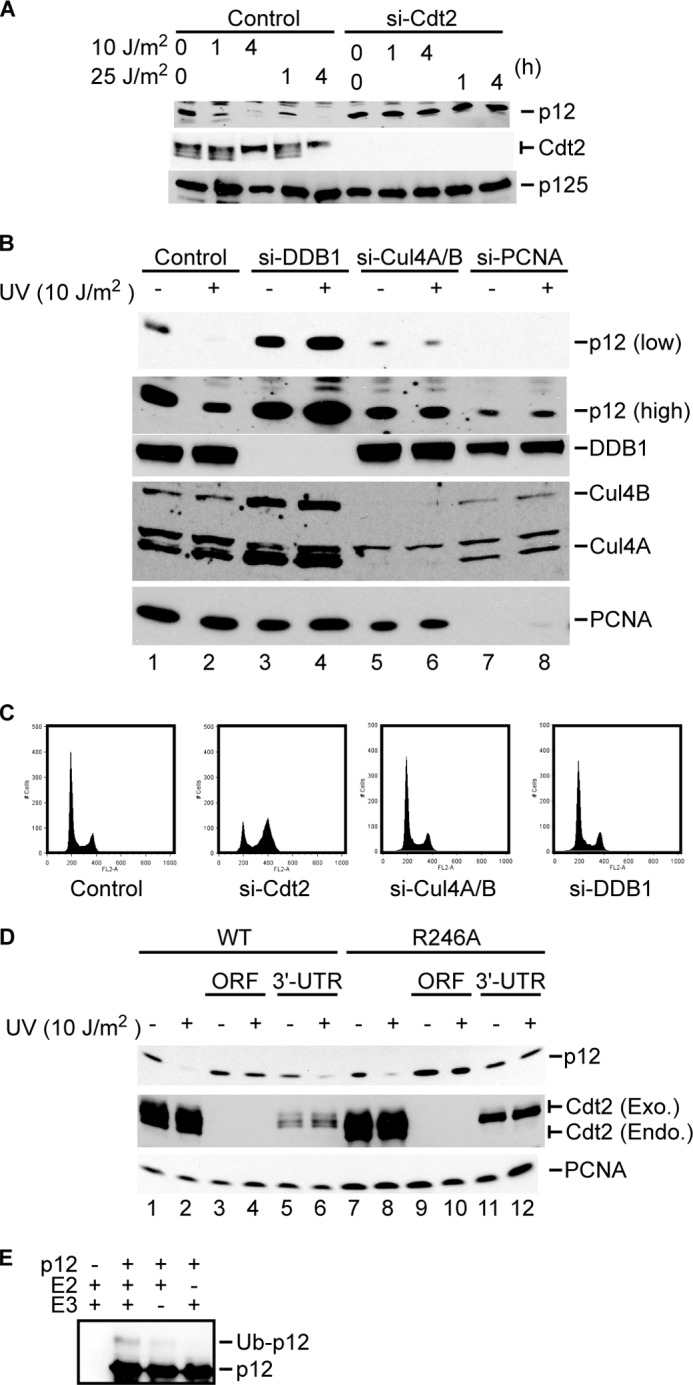
CRL4Cdt2 depletion abrogates the degradation of p12 by UV irradiation. A and B, HeLa cells were transfected with the indicated siRNA and analyzed by immunoblotting with anti-p12, anti-Cdt2, anti-p125, anti-DDB1, anti-Cul4A/B, or anti-PCNA antibody. C, HeLa cells were transfected with si-Cdt2, si-Cul4A/B, or si-DDB1. After 48 h, cell cycle was analyzed by propidium iodide staining followed by flow cytometry. D, HeLa cells stably expressing HA-tagged Cdt2 or Cdt2-R246A were transfected with si-Cdt2 targeting the ORF (ORF) or si-Cdt2 targeting the 3′-UTR (3′-UTR). Cells were irradiated with UV and lysed 1 h after irradiation. Protein lysates were separated by SDS-PAGE and analyzed by immunoblotting with anti-PCNA, anti-Cdt2, or anti-p12 antibody. Exo., exogenous; Endo., endogenous. E, immunopurified CRL4Cdt2 complexes (E3) were mixed with p12WT, E1, and UbcH5 (E2). Ubiquitinated p12 (Ub-p12) was detected by Western blotting after separating the reaction products by SDS-PAGE.
To rule out the possibility that the stabilization of p12 by si-Cdt2 was due to an off-target effect of the siRNA, we tested whether siRNA-resistant exogenous Cdt2 can restore p12 degradation in the absence of endogenous Cdt2 (Fig. 1D). Cdt2 is the substrate recognition adaptor of CRL4Cdt2 and itself interacts with the DDB1 subunit of the E3 in a manner that is dependent on Arg-246 of Cdt2 (12). HeLa cells stably expressing either wild type HA-Cdt2 or HA-Cdt2-R246A, which is deficient in binding to DDB1, were transfected with two different siRNAs, one against the open reading frame (ORF) and the other against the 3′-untranslated region (3′-UTR) of Cdt2. Because both wild type Cdt2 and Cdt2-R246A are expressed from cDNAs without the Cdt2 3′-UTR, si-Cdt2 targeting the ORF depleted both endogenous and exogenous Cdt2, whereas si-Cdt2 targeting the 3′-UTR of Cdt2 depleted only the endogenous Cdt2 (Fig. 1D). Depletion of both endogenous and exogenous Cdt2 by the ORF si-Cdt2 stabilized p12, whereas depletion of endogenous Cdt2 by 3′-UTR si-Cdt2 and sparing of exogenous wild type Cdt2 allowed p12 degradation after DNA damage (lanes 5 and 6). On the other hand, the mutant Cdt2-R246A could not restore p12 degradation in cells transfected with si-Cdt2 targeting the 3′-UTR (lanes 11 and 12). These results demonstrate that knockdown of Cdt2 specifically abrogates p12 degradation and that Cdt2 has to bind to DDB1 for p12 degradation. On the other hand, depletion of β-TrCP, the E3 ligase that targets Cdc25A, did not affect p12 degradation after UV treatment (supplemental Fig. 1).
To address whether CRL4Cdt2 ubiquitinates p12 directly, we tested its ability to ubiquitinate p12 in vitro. GST-tagged p12 was expressed in 293T cells and purified as substrate (with associated PCNA). CRL4Cdt2 E3 ligase complex was obtained as reported previously (18, 21). The ubiquitination reaction was performed in the presence of E1, UbcH5c as E2, and ATP. After incubation of p12-GST and CRL4Cdt2, only the monoubiquitinated form of p12 appeared (Fig. 1E). One possibility is that the in vitro reaction is inefficient at polyubiquitinating p12, although CRL4Cdt2 polyubiquitinates p12 in vivo, possibly because we are using the wrong E2 in the reaction as compared with what may be used in vivo. Alternatively, the monoubiquitin added by CRL4Cdt2 may be extended in vivo by an E4 type E3 ligase different from CRL4Cdt2. Finally, it has recently been shown that small proteins, less than 150 amino acids, can be degraded by monoubiquitination (25) so that it is possible that p12 is also degraded in vivo directly after monoubiquitination by CRL4Cdt2.
p12 contains an atypical PIP box (where Gln and Phe are replaced by Lys and Ser, Fig. 2A). However, GST-tagged p12WT expressed in 293T cells co-precipitated endogenous PCNA when pulled down on glutathione-Sepharose beads (Fig. 2B and supplemental Fig. 2A). Mutation of the PIP box in p12 (p12ΔPIP) totally abrogated its binding to PCNA (lane 10), confirming that this is the interaction site.
FIGURE 2.

The degradation of p12 inhibits fork progression during S-phase and is required for cell survival after DNA damage. A, alignment of PIP degrons in p12, Cdt1, and p21. Underlined residues constitute a canonical PIP box. Bold letters are highly conserved residues that predict PIP degrons, promoting ubiquitination by CRL4Cdt2. A summary of the PCNA binding and post-UV degradation of the mutants is shown. B, 293T cells were transfected with the plasmids coding GST-tagged p12 (WT or mutant), lysed, and pulled down by glutathione beads. The cell lysates (Input) and eluates (Pulldown) were analyzed by immunoblotting with anti-PCNA and anti-GST antibody. n.d., not determined. C, 293T cells stably expressing HA-tagged p12 (WT or mutant) were irradiated with UV at the indicated doses. One hour later, cells were lysed and analyzed by immunoblotting with anti-HA or anti-PCNA antibody. D, 293T cells expressing p12K4Q/S10Y or p12K4Q/S10Y/K15A were irradiated with UV (15 J/m2). 15 min before harvesting at the indicated times, cells were labeled with [3H]thymidine, and incorporated radioactivity was measured. E, 293T cells expressing the indicated p12 mutant were irradiated with UV (15 J/m2) and harvested 1 h after irradiation. 15 min before harvesting, cells were labeled with [3H]thymidine, and incorporated radioactivity was measured. Error bars indicate mean ± S.D. of three experiments. F, similar experiments were performed as in C. The size distribution of nascent DNAs was determined by alkaline-sucrose gradient. The arrow indicates a peak of λ DNA (48 Kb). G, similar experiments were performed as in F by using WI38 cells. H, a representative image of DNA fiber assay is shown. The arrows indicate ongoing forks where a green IdU track extends a red CldU track. I–K, 293T cells expressing mutant p12 were incubated with CldU for 20 min. After irradiation of UV (15 J/m2), cells were immediately incubated with IdU for 30 min. DNA was spread on a glass slide and detected by immunofluorescence. More than 100 fibers of ongoing replication fork progression (green IdU tracks that extend red CldU tracks) were measured. Histograms of green track lengths are shown in I and J. K, 293T cells without (Control) or with stable expression of p12 mutants were subjected to similar experiments as I and J. Distributions of green track lengths are shown as box plot. The p values were calculated using the Mann-Whitney U test. L, new origin firing rates were measured by counting green-only tracks divided by the total of red+green or red-only tracks. The bars and lines represent the mean ± S.E. of three independent experiments. M and N, WI38 cells, HCT116, and HCT116p53−/− cells were tested as in K. Distributions of green track lengths are shown as box plot. The p values were calculated using the Mann-Whitney U test. O, 293T cells expressing mutant p12 were irradiated with the indicated dose of UV. The number of colonies was counted 10 days after irradiation. The number of colonies relative to nonirradiated cells is shown. Error bars indicate mean ± S.D. of three experiments.
p12 binding to PCNA is reported to be essential for efficient activity of polymerase δ (9, 10). Therefore, to address the physiological role of p12 degradation, we required a mutant of p12 that still interacts with PCNA but is not degraded after radiation. Substrates of CRL4Cdt2, such as Cdt1, p21, and Set8, contain a threonine and a lysine in the context of the PIP box that are important for CRL4Cdt2-dependent degradation (15, 26) but not as important for PCNA binding, and the corresponding residues are present in p12 (Thr-8 and Lys-15) (Fig. 2A). However, mutation of Thr-8 (p12T8D) or Lys-15 (p12K15A) in p12 disrupted or decreased the interaction with PCNA (supplemental Fig. 2A), suggesting that both of these residues are important for efficient binding to PCNA in the context of the atypical PIP box of p12. We therefore substituted the Lys-4 and Ser-10 of p12 (p12K4Q/S10Y) to generate a canonical PIP box sequence (Fig. 2A). p12K4Q/S10Y still binds to PCNA at the equivalent level as p12WT (Fig. 2B). In the K4Q/S10Y context, additional mutation of Lys-15 (p12K4Q/S10Y/K15A) did not affect the affinity of p12 for PCNA. To test the stability of these mutants in response to UV, we stably expressed HA-tagged versions of these mutants in 293T cells by retroviral transduction and checked the protein levels before and after UV treatment. As with the PIP mutants of other substrates of CRL4Cdt2 ligase such as Cdt1 and p21, p12ΔPIP was not degraded after UV treatment (Fig. 2C), suggesting that binding of p12 to PCNA is required for the degradation. p12K4Q/S10Y mutant was degraded similarly as the p12WT. On the other hand, this degradation was not seen in p12K4Q/S10Y/K15A, although this protein retained its ability to bind PCNA (Fig. 2B). Therefore, this mutant would allow us to test the role of DNA damage-dependent degradation of p12 without abolishing its binding to PCNA (see below).
We next confirmed that cell cycle progression and checkpoint kinase activation were normal in cells expressing stable p12 (supplemental Fig. 2, B and C). There was no difference in cell cycle profile or phosphorylation of Chk1, Chk2, and histone H2AX after UV irradiation between cells expressing the two forms of p12. These results imply that comparison between p12K4Q/S10Y and p12K4Q/S10Y/K15A will be useful for investigating whether p12 degradation is important for the response of the cell when it is damaged in S-phase.
Because p12 is required for effective DNA synthesis and is degraded in response to DNA damage, we next tested whether p12 degradation contributes to the intra-S-phase replication arrest following DNA damage. Increasing doses of UV decreased DNA synthesis in 293T (supplemental Fig. 3A). We measured UV-resistant DNA synthesis in cells expressing UV-labile or UV-stable mutant of p12. 293T cells stably expressing p12K4Q/S10Y or p12K4Q/S10Y/K15A mutants were irradiated with UV and labeled with [3H]thymidine to label newly synthesized DNA (Fig. 2D). Cells expressing the stable p12K4Q/S10Y/K15A showed higher UV-resistant DNA synthesis after irradiation than cells expressing degradable p12K4Q/S10Y (Fig. 2, D and E), suggesting that p12 degradation is required for inhibition of DNA synthesis after DNA damage. Cells treated with ionizing radiation also showed similar results as cells treated with UV irradiation (supplemental Fig. 3B). Notably, p12ΔPIP, which is as stable as p12K4Q/S10Y/K15A, did not increase UV-resistant DNA synthesis (Fig. 2E). These results demonstrated that both the stabilization of p12 and the binding of the stable p12 to PCNA are required to rescue DNA synthesis when a cell responds to DNA damage. Similar results were obtained with WI38, a normal human fibroblast cell line (supplemental Fig. 3, D–F), and in p53 mutant H1299 lung cancer cells (supplemental Fig. 3G), suggesting that stable p12 restores DNA synthesis in damaged cells independent of cell transformation or of p53 status.
DNA damage has two immediate effects on DNA replication: decreasing the rate of fork progression and inhibiting new origin initiation. To investigate whether the degradation of p12 after radiation inhibits fork progression or initiation, we measured the size of newly synthesized DNA by separating 3H-labeled nascent single-stranded DNA using alkaline-sucrose gradient. 293T (Fig. 2F) or WI38 (Fig. 2G) cells expressing either p12K4Q/S10Y or p12K4Q/S10Y/K15A were irradiated with UV followed by 30 min of [3H]thymidine labeling. Newly labeled nascent DNA strands were fractionated by alkaline-sucrose gradient according to their size. Without UV treatment, the size distribution of newly synthesized DNA was similar between cells expressing p12K4Q/S10Y and p12K4Q/S10Y/K15A. The shorter nascent DNA near the top of the gradient arises from newly fired origins both in cells already in S-phase and in cells newly entering S-phase from G1. The longer nascent DNA nearer the bottom of the gradient is a measure of progression of already existing forks. UV irradiation of p12K4Q/S10Y-expressing cells decreases short strands due to inhibition of origin firing (including inhibition of S-phase entry) and decreases long strands due to inhibition of fork progression. On the other hand, in p12K4Q/S10Y/K15A-expressing cells irradiated with UV, the longer nascent DNA produced by fork progression was selectively spared. This result suggests that p12 degradation following radiation is responsible for the decrease in fork progression following UV damage. Similar results were obtained in 293T cells and WI38 cells (Fig. 2, F and G).
Although much is known about how DNA damage blocks firing of origins, not much is known about how fork progression is slowed by DNA damage. Because the nascent strand sizing experiments suggested the exciting possibility that p12 degradation contributes to this slowing, we decided to test this hypothesis by an independent assay. Therefore, the fork progression rate was measured by DNA fiber assay in 293T cells expressing CRL4Cdt2-insensitive p12 mutant protein. Cells were first labeled with CldU for 20 min, irradiated with UV, and then labeled with IdU for another 20 min. Genomic DNA was extracted from the cells, and single DNA molecules were stretched and denatured for immunodetection of CldU (red) and IdU (green) (Fig. 2H). Progression of existing forks was identified by a green IdU track extending a red CldU track. The rate of fork progression was measured by the lengths of these green IdU tracks (Fig. 2, I–K). The reduction of fork rate progression in response to UV treatment was smaller in cells expressing the stable p12K4Q/S10Y/K15A as compared with cells expressing the degradable p12K4Q/S10Y. Interestingly, new origin firing was still suppressed in cells expressing p12 (Fig. 2L), demonstrating the clear separation of the firing suppression and elongation suppression by DNA damage. WI38 fibroblast, HCT116 wild type, and HCT116 p53 null cells showed similar results (Fig. 2, M and N), supporting our model that p12 degradation by CRL4Cdt2 is essential for suppressing fork progression in response to DNA damage, and this is independent of cell transformation or p53 status.
It has been shown that disturbing the response of the cell to DNA damage, specifically allowing continued DNA replication immediately after DNA damage, sensitizes cells to DNA damage (1, 2). We therefore tested the sensitivity of the stable p12-expressing cells to UV irradiation by colony formation assay. Cells expressing stable p12K4Q/S10Y/K15A were significantly more sensitive than the cells expressing p12K4Q/S10Y (Fig. 2O). Collectively, these results demonstrate that p12 degradation is required for cell survival after DNA damage.
It is already known that inhibition of fork progression is dependent on Chk1 activity (7, 8, 27) and that p12 degradation is inhibited by caffeine (7), suggesting that p12 degradation is one of the pathways by which the checkpoint inhibits fork progression. However, it is entirely possible that the checkpoint kinases activate additional pathways by which to inhibit fork progression that are independent of p12. An interesting possibility is that the checkpoint pathways inactivate or degrade other components of the replication fork to inhibit fork progression.
Recently, RNF8 was reported as an E3 ubiquitin ligase for p12 (28). Under our conditions, however, siRNA-mediated knockdown of RNF8 did not stabilize p12 after UV irradiation (supplemental Fig. 4). We suspect that differences in cell lines, cellular growth conditions, extent of RNF8 knockdown, or radiation conditions account for this difference.
How does p12 degradation lead to inhibition of fork progression? In an in vitro system, the lack of p12 in the polymerase δ complex decreases polymerase activity (10, 29) so that fork progression may be retarded simply from failure of the elongating polymerase in the absence of p12. Activity of BLM, a RecQ family DNA helicase, is required for efficient replication fork restart after DNA damage (30), and depletion of p12 also affects BLM activity (24). Thus, another possibility is that degradation of p12 inhibits BLM activity at damaged forks and retards fork progression in the presence of DNA damage. The results reported in this study provide one of the long sought mechanisms by which fork progression is slowed by DNA damage to maintain genomic health in DNA-damaged cells.
Acknowledgments
We thank members of the Dutta laboratory for helpful comments.
This work was supported, in whole or in part, by National Institutes of Health Grants CA60499 and GM84465 (to A. D.) and CA140774 (to T. A.). This work was also supported by Department of Defense Breast Cancer Research Program and Uehara Memorial Foundation (to K. T.).

This article contains supplemental Figs. 1–4.
- PCNA
- proliferating cell nuclear antigen
- TrCP
- transducin repeat-containing protein
- CldU
- 5-chloro-2′-deoxyuridine
- IdU
- 5-iodo-2′-deoxyuridine
- PIP
- PCNA-interacting proteins
- BLM
- Bloom syndrome.
REFERENCES
- 1. Bartek J., Lukas C., Lukas J. (2004) Checking on DNA damage in S phase. Nat. Rev. Mol. Cell Biol. 5, 792–804 [DOI] [PubMed] [Google Scholar]
- 2. Kaufmann W. K. (2010) The human intra-S checkpoint response to UVC-induced DNA damage. Carcinogenesis 31, 751–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mailand N., Falck J., Lukas C., Syljuâsen R. G., Welcker M., Bartek J., Lukas J. (2000) Rapid destruction of human Cdc25A in response to DNA damage. Science 288, 1425–1429 [DOI] [PubMed] [Google Scholar]
- 4. Busino L., Donzelli M., Chiesa M., Guardavaccaro D., Ganoth D., Dorrello N. V., Hershko A., Pagano M., Draetta G. F. (2003) Degradation of Cdc25A by β-TrCP during S phase and in response to DNA damage. Nature 426, 87–91 [DOI] [PubMed] [Google Scholar]
- 5. Falck J., Mailand N., Syljuåsen R. G., Bartek J., Lukas J. (2001) The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410, 842–847 [DOI] [PubMed] [Google Scholar]
- 6. Miao H., Seiler J. A., Burhans W. C. (2003) Regulation of cellular and SV40 virus origins of replication by Chk1-dependent intrinsic and UVC radiation-induced checkpoints. J. Biol. Chem. 278, 4295–4304 [DOI] [PubMed] [Google Scholar]
- 7. Chastain P. D., 2nd, Heffernan T. P., Nevis K. R., Lin L., Kaufmann W. K., Kaufman D. G., Cordeiro-Stone M. (2006) Checkpoint regulation of replication dynamics in UV-irradiated human cells. Cell Cycle 5, 2160–2167 [DOI] [PubMed] [Google Scholar]
- 8. Seiler J. A., Conti C., Syed A., Aladjem M. I., Pommier Y. (2007) The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol. Cell. Biol. 27, 5806–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Meng X., Zhou Y., Lee E. Y., Lee M. Y., Frick D. N. (2010) The p12 subunit of human polymerase δ modulates the rate and fidelity of DNA synthesis. Biochemistry 49, 3545–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang Q. M., Akashi T., Masuda Y., Kamiya K., Takahashi T., Suzuki M. (2010) Roles of POLD4, smallest subunit of DNA polymerase δ, in nuclear structures and genomic stability of human cells. Biochem. Biophys. Res. Commun. 391, 542–546 [DOI] [PubMed] [Google Scholar]
- 11. Zhang S., Zhou Y., Trusa S., Meng X., Lee E. Y., Lee M. Y. (2007) A novel DNA damage response: rapid degradation of the p12 subunit of DNA polymerase δ. J. Biol. Chem. 282, 15330–15340 [DOI] [PubMed] [Google Scholar]
- 12. Jin J., Arias E. E., Chen J., Harper J. W., Walter J. C. (2006) A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol. Cell 23, 709–721 [DOI] [PubMed] [Google Scholar]
- 13. Higa L. A., Banks D., Wu M., Kobayashi R., Sun H., Zhang H. (2006) L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle 5, 1675–1680 [DOI] [PubMed] [Google Scholar]
- 14. Arias E. E., Walter J. C. (2006) PCNA functions as a molecular platform to trigger Cdt1 destruction and prevent re-replication. Nat. Cell Biol. 8, 84–90 [DOI] [PubMed] [Google Scholar]
- 15. Senga T., Sivaprasad U., Zhu W., Park J. H., Arias E. E., Walter J. C., Dutta A. (2006) PCNA is a cofactor for Cdt1 degradation by CUL4/DDB1-mediated N-terminal ubiquitination. J. Biol. Chem. 281, 6246–6252 [DOI] [PubMed] [Google Scholar]
- 16. Kim Y., Starostina N. G., Kipreos E. T. (2008) The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 22, 2507–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nishitani H., Shiomi Y., Iida H., Michishita M., Takami T., Tsurimoto T. (2008) CDK inhibitor p21 is degraded by a proliferating cell nuclear antigen-coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J. Biol. Chem. 283, 29045–29052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abbas T., Sivaprasad U., Terai K., Amador V., Pagano M., Dutta A. (2008) PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 22, 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tardat M., Brustel J., Kirsh O., Lefevbre C., Callanan M., Sardet C., Julien E. (2010) The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol. 12, 1086–1093 [DOI] [PubMed] [Google Scholar]
- 20. Oda H., Hübner M. R., Beck D. B., Vermeulen M., Hurwitz J., Spector D. L., Reinberg D. (2010) Regulation of the histone H4 monomethylase PR-Set7 by CRL4Cdt2-mediated PCNA-dependent degradation during DNA damage. Mol. Cell 40, 364–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Terai K., Abbas T., Jazaeri A. A., Dutta A. (2010) CRL4Cdt2 E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol. Cell 37, 143–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Centore R. C., Havens C. G., Manning A. L., Li J. M., Flynn R. L., Tse A., Jin J., Dyson N. J., Walter J. C., Zou L. (2010) CRL4Cdt2-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell 40, 22–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W., Vogelstein B. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 [DOI] [PubMed] [Google Scholar]
- 24. Selak N., Bachrati C. Z., Shevelev I., Dietschy T., van Loon B., Jacob A., Hübscher U., Hoheisel J. D., Hickson I. D., Stagljar I. (2008) The Bloom's syndrome helicase (BLM) interacts physically and functionally with p12, the smallest subunit of human DNA polymerase δ. Nucleic Acids Res. 36, 5166–5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shabek N., Herman-Bachinsky Y., Buchsbaum S., Lewinson O., Haj-Yahya M., Hejjaoui M., Lashuel H. A., Sommer T., Brik A., Ciechanover A. (2012) The size of the proteasomal substrate determines whether its degradation will be mediated by mono- or polyubiquitylation. Mol. Cell 48, 87–97 [DOI] [PubMed] [Google Scholar]
- 26. Havens C. G., Walter J. C. (2009) Docking of a specialized PIP Box onto chromatin-bound PCNA creates a degron for the ubiquitin ligase CRL4Cdt2. Mol. Cell 35, 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Conti C., Seiler J. A., Pommier Y. (2007) The mammalian DNA replication elongation checkpoint: implication of Chk1 and relationship with origin firing as determined by single DNA molecule and single cell analyses. Cell Cycle 6, 2760–2767 [DOI] [PubMed] [Google Scholar]
- 28. Zhang S., Zhou Y., Sarkeshik A., Yates J. R., 3rd, Thomson T. M., Zhang Z., Lee E. Y., Lee M. Y. (2013) Identification of RNF8 as a ubiquitin ligase involved in targeting the p12 subunit of DNA polymerase δ for degradation in response to DNA damage. J. Biol. Chem. 288, 2941–2950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H., Xie B., Zhou Y., Rahmeh A., Trusa S., Zhang S., Gao Y., Lee E. Y., Lee M. Y. (2006) Functional roles of p12, the fourth subunit of human DNA polymerase δ. J. Biol. Chem. 281, 14748–14755 [DOI] [PubMed] [Google Scholar]
- 30. Davies S. L., North P. S., Hickson I. D. (2007) Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat. Struct. Mol. Biol. 14, 677–679 [DOI] [PubMed] [Google Scholar]
- 31. Abbas T., Shibata E., Park J., Jha S., Karnani N., Dutta A. (2010) CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell 40, 9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]