

Background: Excessive activation of glutamate receptors is crucial to the genesis of neuropathic pain.
Results: Endogenous IL-1β in neuropathic rats enhances glutamate release by increasing presynaptic NMDA receptor activities via sphingomyelinase signaling.
Conclusion: Coupling between IL-1β receptors and presynaptic NMDA receptors contributes to the genesis of neuropathic pain.
Significance: Interruption of such coupling could be an effective approach for the treatment of neuropathic pain.
Keywords: Cell Signaling; Glia; Glutamate Receptors Ionotropic (AMPA, NMDA); Pain; Patch Clamp
Abstract
Excessive activation of glutamate receptors and overproduction of proinflammatory cytokines, including interleukin-1β (IL-1β) in the spinal dorsal horn, are key mechanisms underlying the development and maintenance of neuropathic pain. In this study, we investigated the mechanisms by which endogenous IL-1β alters glutamatergic synaptic transmission in the spinal dorsal horn in rats with neuropathic pain induced by ligation of the L5 spinal nerve. We demonstrated that endogenous IL-1β in neuropathic rats enhances glutamate release from the primary afferent terminals and non-NMDA glutamate receptor activities in postsynaptic neurons in the spinal dorsal horn. Myeloid differentiation primary response protein 88 (MyD88) is a mediator used by IL-1β to enhance non-NMDA glutamate receptor activities in postsynaptic neurons in the spinal dorsal horn. Presynaptic NMDA receptors are effector receptors used by the endogenous IL-1β to enhance glutamate release from the primary afferents in neuropathic rats. This is further supported by the fact that NMDA currents recorded from small neurons in the dorsal root ganglion of normal rats are potentiated by exogenous IL-1β. Furthermore, we provided evidence that functional coupling between IL-1β receptors and presynaptic NMDA receptors at the primary afferent terminals is mediated by the neutral sphingomyelinase/ceramide signaling pathway. Hence, functional coupling between IL-1β receptors and presynaptic NMDA receptors at the primary afferent terminals is a crucial mechanism leading to enhanced glutamate release and activation of non-NMDA receptors in the spinal dorsal horn neurons in neuropathic pain conditions. Interruption of such functional coupling could be an effective approach for the treatment of neuropathic pain.
Introduction
Treatment of neuropathic pain is hampered due to our insufficient understanding about the mechanisms by which aberrant neuronal activities in the pain pathway are induced. Excessive activation of glutamatergic receptors and release of proinflammatory cytokines, including interleukin-1β in the spinal dorsal horn, are the culprits in the genesis of neuropathic pain (1, 2). Mechanisms underlying the regulation of glutamate receptor activation by proinflammatory cytokines in the spinal dorsal horn remain obscure.
The first nociceptive sensory synapse between the primary afferent terminals and superficial dorsal horn neurons is the primary station for processing nociceptive information in the CNS. Glutamate is a major excitatory neurotransmitter released from the primary afferents in the spinal dorsal horn (3, 4). Activation of glutamate receptors is governed by the amount of glutamate release from presynaptic terminals, the rate by which glutamate is taken up by glutamate transporters, and the function of postsynaptic glutamate receptors (5). The release of glutamate from presynaptic terminals is controlled by activities of many presynaptic receptors, providing a powerful means of dynamically regulating excitatory transmission. Recently, we revealed that endogenous activation of presynaptic NMDA receptors in the central terminals of primary afferents in the spinal dorsal horn is critical to the increased glutamate release from primary afferent terminals triggered by peripheral sensory input in neuropathic rats (6). We suggested that suppression of presynaptic NMDA receptor activation is a novel approach to reduce excessive activation of glutamate receptors following peripheral nerve injury. It is not known how signaling pathways regulate the functions of presynaptic NMDA receptors in the primary afferent terminals in the spinal dorsal horn.
IL-1β is a prototypic proinflammatory cytokine critically involved in the development and maintenance of neuropathic pain (1, 2). Glial activation and subsequent release of proinflammatory cytokines, including IL-1β in the spinal dorsal horn, have been observed in neuropathic pain induced by peripheral nerve injury (7–9). Knocking out IL-1β receptors reduces behavioral hypersensitivity induced by nerve injury (10, 11). Intrathecal administration of IL-1β in normal rats enhances both the acute response and the wind-up activity of dorsal horn neurons and increases mechanical allodynia and hyperalgesia (12–14). Recent studies have shown that perfusion of exogenous IL-1β onto normal spinal slices increases the frequency and amplitude of spontaneous EPSCs and AMPA- and NMDA-induced currents in dorsal horn neurons, suggesting that IL-1β can enhance both the release of glutamate from presynaptic neurons and functions of AMPA and NMDA receptors in postsynaptic neurons (15). Yet, it remains unclear whether and how glutamate release from the primary afferent terminals is regulated by the endogenous IL-1β in neuropathic pain conditions.
Previous studies showed that levels of ceramide in the spinal dorsal horn are increased in neuropathic animals following nerve injury (16, 17) or in morphine-tolerant rats (18). Ceramide is a potent proinflammatory sphingolipid that is generated by hydrolysis of sphingomyelin on the membrane by acid or neutral sphingomyelinase (19). Inhibition of ceramide production suppresses hyperalgesia in neuropathic rats (17) and morphine-tolerant rats (18, 20). Studies of forebrain areas suggest that the neutral sphingomyelinase/ceramide pathway is an important pathway for IL-1β to enhance postsynaptic NMDA receptor functions (21). The role of neutral sphingomyelinase and its product ceramide in regulating glutamatergic synaptic activities in the spinal dorsal horn has not been investigated. Furthermore, the regulation of presynaptic NMDA receptors by proinflammatory cytokines has not been addressed in any systems in the CNS. In this study, we tested our hypothesis that endogenous IL-1β in the spinal dorsal horn of neuropathic rats, acting through the neutral sphingomyelinase/ceramide signaling pathway, enhances presynaptic NMDA receptor activities, resulting in increased glutamate release from the primary afferent terminals.
EXPERIMENTAL PROCEDURES
Animals
Young adult male Sprague-Dawley rats (weight range, 160–220 g) were used. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Georgia and were fully compliant with the National Institutes of Health Guidelines for the Use and Care of Laboratory Animals.
Ligation of the L5 Spinal Nerve and Behavioral Tests
The animals were randomly divided into spinal nerve ligation injury (SNL)2 group and sham-operated (control) group. SNL injury was performed as previously described (22). Briefly, under 2–3% isoflurane, a midline incision above the lumbar spine and deep dissection through the paraspinal muscles were made to expose the left L6 transverse process, and the process was then removed. The L5 spinal nerve was isolated and tightly ligated with a 4-0 silk suture distal to the dorsal root ganglia and proximal to the formation of the sciatic nerve. The incisions were then closed. Sham-operated rats underwent the same operation and handling as the SNL group but without nerve ligation. No drugs were used after the surgery. Behavioral tests were performed to determine the hind paw mechanical sensitivity 1 day before the operation and on days 7–14 post-surgery prior to electrophysiological and molecular experiments (6, 23). Briefly, the animals were placed on wire mesh, loosely restrained under a Plexiglas cage (12 × 20 × 15 cm), and allowed to acclimate for at least 30 min. von Frey monofilaments with bending forces ranging from 0.1 to 12.4 g were applied below the mesh onto the mid-plantar side of each hind paw to evoke paw withdrawal responses. Each hind paw was stimulated 10 times with each von Frey monofilament, and the frequency (percentage) of paw withdrawal responses to 10 stimulations was recorded. The least bending force that evoked withdrawal in more than half the trials was assigned as the 50% withdrawal threshold.
In Vitro Whole Cell Recordings and Data Analysis
Spinal Slice and Dorsal Root Ganglion Slice Preparations
Rats were deeply anesthetized via isoflurane inhalation. Surgery was made to expose and remove the spinal lumbar enlargement segment or L4 and L5 dorsal rood ganglions. To make spinal slices, the lumbar spinal cord section was placed in ice-cold sucrose artificial cerebrospinal fluid (aCSF) presaturated with 95% O2 and 5% CO2. The sucrose aCSF contained 234 mm sucrose, 3.6 mm KCl, 1.2 mm MgCl2, 2.5 mm CaCl2, 1.2 mm NaH2PO4, 12.0 mm glucose, and 25.0 mm NaHCO3. The pia-arachnoid membrane was removed from the section. The L4–5 spinal segment, identified by the large dorsal roots, was attached with cyanoacrylate glue to a cutting support, which was then glued onto the stage of a vibratome (Series 1000, Technical Products International, St. Louis, MO). To make dorsal root ganglion slices, a single dorsal root ganglion was embedded in agar (2%) that was then mounted onto the stage of the vibratome. Transverse spinal cord slices (400 μm) or dorsal root ganglion slices (450 μm) were cut in the ice-cold sucrose aCSF and then preincubated in Krebs solution oxygenated with 95% O2 and 5% CO2 at 35 °C for at least 2 h before they were transferred to the recording chamber perfused with the Krebs solution. The Krebs solution contained 117.0 mm NaCl, 3.6 mm KCl, 1.2 mm MgCl2, 2.5 mm CaCl2, 1.2 mm NaH2PO4, 11.0 mm glucose, and 25.0 mm NaHCO3.
Whole Cell Voltage Clamp Recordings
Following preincubation, a single spinal slice or dorsal root ganglion slice was placed in the recording chamber (volume, 1.5 ml), perfused with Krebs solution at 35 °C, and saturated with 95% O2 and 5% CO2. Borosilicate glass recording electrodes (resistance, 3–5 megohms) were pulled. Electrodes used for recording neurons in the spinal slices were filled with an internal solution containing 135.0 mm potassium gluconate, 5.0 mm KCl, 2.0 mm MgCl2, 0.5 mm CaCl2, 5.0 mm HEPES, 5.0 mm EGTA, 5.0 mm ATP-Mg, 0.5 mm Na-GTP, 10 mm QX-314, and 1 mm MK-801. MK-801 was used to block postsynaptic NMDA receptors (24–26). Electrodes for recording neurons in the dorsal root ganglion slices were filled with the solution containing 110.0 mm Cs2SO4, 2.0 mm MgCl2, 0.5 mm CaCl2, 5.0 mm HEPES, 5.0 mm EGTA, 5.0 mm ATP-Mg, 0.5 mm Na-GTP, and 10.0 mm lidocaine N-ethyl bromide (QX314). Live dorsal horn neurons in the spinal lamina I and outer lamina II or small dorsal root ganglion neurons (≤30 μm) were visualized using a microscope system and approached using a three-dimensional motorized manipulator (Sutter Instrument Co.), and whole cell configurations were established by applying moderate negative pressure after electrode contact. A seal resistance of at least 2 gigohms and an access resistance of 20–35 megohms were considered acceptable. The series resistance was optimally compensated by at least 70% and constantly monitored throughout the experiments (6, 27, 28). Experiments showing any evidence of loss of voltage control were discarded. Signals were amplified using an Axopatch 700B (Molecular Devices) and displayed and stored in a personal computer.
When neurons in spinal slices were recorded, EPSCs were evoked using constant-current electrical stimuli (0.2-ms duration repeated every 40 s) at a fixed stimulating intensity (0.8 mA) applied with a concentric bipolar stimulating electrode placed at the dorsal root entry zone (6, 29, 30). To specifically determine the function of presynaptic NMDA receptors in the primary afferent terminals, only neurons receiving monosynaptic input from primary afferent input were recorded. Monosynaptic input was based on a constant latency with graded intensity and high frequency repetitive stimulation (20 Hz) (6, 31, 32). Miniature EPSCs (mEPSCs) were recorded in the presence of tetrodotoxin (TTX, 1 μm) to block voltage-dependent sodium channels (action potentials). All recordings from spinal slices were made in the presence of bicuculline (10 μm) and strychnine (5 μm) in the external solution to block GABAA and glycine receptors at a membrane potential of −70 mV.
When neurons in dorsal root ganglion slices were recorded, NMDA currents were evoked by exogenous NMDA (50 μm) injected onto the recorded neuron by puff application (pressure, 3 p.s.i.; duration, 20 ms, repeated every 60 s) through a glass pipette with an opening tip size of 8–10 μm. Recordings from dorsal root ganglion slices were made in the presence of TTX (1 μm), 6,7-dinitroquinoxaline-2,3-dione (10 μm, which blocks non-NMDA glutamate receptors), bicuculline (10 μm), and strychnine (5 μm) in the external solution and at a membrane potential of 40 mV to remove the voltage-dependent Mg2+ block from NMDA receptors (23).
Data Analysis
Data were recorded using Axopatch 700B amplifiers, digitized at 10 kHz, and analyzed off-line. The means of four EPSCs evoked by electrical stimulation at base line, in the presence of tested drugs, and after washout of tested drugs were measured using the Clampfit software program (version 10.2; Molecular Devices, Sunnyvale, CA). In some neurons, we also measured the inverse squared coefficient of variation (CV−2) of the peak amplitudes of 10 evoked EPSCs, where CV represents the ratio of the standard deviation to the mean. The frequency and amplitude of mEPSCs in 3 min before and after perfusion of tested drugs were analyzed and averaged using a peak detection program (MiniAnalysis; Synaptosoft Inc., Decatur, GA). The means of four NMDA currents evoked by exogenous NMDA at base line, in the presence of tested drugs, and after washout of tested drugs were measured. The data were presented as the mean ± S.E. The Student's t test was used to determine statistical differences between data obtained in the absence and presence of tested drugs (paired t test). A p value of less than 0.05 was considered statistically significant.
Western Blot Experiments
Animals were deeply anesthetized with urethane (1.3–1.5 g/kg, intraperitoneally) 7 days after ligation of the L5 spinal nerve. The L4 to L5 spinal segment was exposed by surgery and removed from the rats. The dorsal quadrant of the spinal segment ipsilateral to the operated side was isolated and quickly frozen in liquid nitrogen and stored at −80 °C for later use. Frozen tissues were homogenized with a hand-held pellet in lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.1% SDS, 1% deoxycholic acid, 2 mm orthovanadate, 100 mm NaF, 1% Triton X-100, 0.5 mm phenylmethylsulfonyl fluoride, 20 μm leupeptin, 100 IU ml−1 aprotinin) for 0.5 h at 37 °C. The samples were then centrifuged for 20 min at 12,000 × g at 4 °C, and the supernatants containing proteins were collected. The quantification of protein contents was made by the BCA method. Protein samples (40 μg) were electrophoresed in 8% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membrane (Millipore, Bedford, MA). The membranes were blocked with 5% milk and incubated overnight at 4 °C with a polyclonal goat anti-IL-1β (1:500, Millipore, Bedford, MA) or a monoclonal mouse anti-β-actin (1:2000, Sigma) primary antibody as a loading control. Then the blots were incubated for 1 h at room temperature (RT) with a corresponding HRP-conjugated secondary antibody (1:5000; Santa Cruz Biotechnology), visualized in ECL solution (SuperSignal West Pico Chemiluminescent Substrate, Pierce) for 1 min, and exposed to the FluorChem HD2 system. The intensity of immunoreactive bands was quantified using ImageJ 1.46 software (National Institutes of Health). Results were expressed as the ratio of IL-1β to β-actin control. The Student's t test (nonpaired t test) was used to determine statistical differences between the neuropathic and sham-operated groups. A p value of less than 0.05 was considered statistically significant.
Materials
Bicuculline, strychnine, NMDA, MK-801, C2-ceramide, and TTX were obtained from Sigma. d-Aminophosphonovaleric acid (D-AP5) and d-serine were obtained from Tocris Bioscience (Minneapolis, MN). 3-O-Methylsphingomyelin (3-OMS) was obtained from Enzo Life Sciences (Paris, France). Recombinant human IL-1β and IL-1ra proteins were purchased from R&D Systems (Minneapolis, MN). The myeloid differentiation primary response protein 88 (MyD88) inhibitory peptide and the control peptide for MyD88 inhibitory peptide (Pepinh-control) were purchased from Invivogen (San Diego). All pharmacological agents were applied via perfusion into the recording chamber unless indicated.
RESULTS
All rats receiving ligation of L5 spinal nerve had mechanical allodynia prior to undergoing the electrophysiological recordings and molecular experiments that took place between days 7 and 14 post-surgery. This was evidenced by the fact that mechanical thresholds of hind paw withdrawal responses ipsilateral to the L5 ligation decreased significantly from 7.15 ± 0.36 g at base line to 1.09 ± 0.15 g (p < 0.001, 38 rats) in neuropathic rats. The mechanical threshold in 34 sham-operated rats was not significantly altered (from 7.66 ± 0.39 to 7.82 ± 0.47 g).
Endogenous IL-1β in Neuropathic Rats Enhances Glutamate Release from the Primary Afferent Terminals and Non-NMDA Glutamate Receptor Activities in Dorsal Horn Postsynaptic Neurons in the Spinal Dorsal Horn
We recently reported that an increased glutamate release from the presynaptic terminals contributes to the enhanced activation of non-NMDA glutamate receptors in the first sensory synapse in the spinal dorsal horn of neuropathic rats induced by SNL (6). To investigate endogenous mechanisms leading to the increased glutamate release in neuropathic rats, we first determined levels of IL-1β in the spinal dorsal horn ipsilateral to the surgery side in sham-operated rats and SNL rats 7 days post-operation. Protein expression of IL-1β was significantly increased by 213.02 ± 29.16% in rats (n = 4) receiving L5 ligation compared with sham-operated rats (n = 4) (Fig. 1), which is in line with previous reports that production of IL-1β is increased between days 7 and 20 in the same region in the same animal model (33, 34). Next, we determined the role of endogenous IL-1β in regulating non-NMDA glutamate receptor activities in the first sensory synapse in the spinal dorsal horn by recording mEPSCs in neurons receiving monosynaptic inputs from the primary afferents in the presence of TTX (1 μm). TTX was used to block voltage-gated sodium channels and the action potential-dependent glutamate release from the presynaptic neurons. Electrodes used for recording in this study were filled with an internal solution containing the NMDA receptor blocker MK801 (1 mm) to block NMDA receptors in the recorded postsynaptic neuron (6, 35). An increase or decrease in mEPSC frequencies indicates an increase or decrease in the presynaptic transmitter release probability, respectively, whereas an increase or decrease in mEPSC amplitudes indicates an increase or decrease in the post-synaptic receptor activities, respectively (36). After base-line mEPSCs were recorded, we applied a selective IL-1β receptor antagonist IL-1ra (100 ng/ml) (37, 38) into the recording chamber through bath perfusion. IL-1ra significantly and reversibly reduced mEPSC frequencies (n = 8, p < 0.001) and amplitudes (n = 8, p < 0.001) in spinal slices obtained from neuropathic rats (Fig. 2, A and E) but had no significant effects (n = 7) in sham-operated rats (Fig. 2, B and F). These data suggested the following: (a) endogenous IL-1β in neuropathic rats enhances glutamate release from presynaptic terminals, and (b) non-NMDA glutamate receptor activities in dorsal horn postsynaptic neurons were enhanced by endogenous IL-1β in neuropathic rats. This notion was supported by the fact that mEPSC frequencies (n = 10, p < 0.01) and amplitudes (n = 10, p < 0.01) in spinal slices of sham-operated rats were significantly and reversibly increased by exogenous bath application of IL-1β (10 ng/ml) (Fig. 2, C and G). However, exogenous bath application of IL-1β (10 ng/ml) had no effects on either mEPSC frequencies (n = 7) or amplitudes (n = 7) in spinal slices of neuropathic rats (Fig. 2, D and H), suggesting that the effects induced by exogenous IL-1β seen in sham-operated animals were occluded by endogenous IL-1β in neuropathic rats.
FIGURE 1.

Protein expression of IL-1β in the spinal dorsal horn ipsilateral to the injury site in neuropathic rats is increased. Samples of IL-1β expression in the spinal dorsal horn at the L4 to L5 segment in neuropathic (n = 4) and sham-operated (n = 4) rats are shown. The bar graph shows the mean (+S.E.) relative density to β-actin in each group. ***, p < 0.001.
FIGURE 2.

Endogenous IL-1β in neuropathic rats enhances glutamate release from presynaptic terminals and activities of non-NMDA glutamate receptors in dorsal horn postsynaptic neurons in neuropathic rats. Inhibition of IL-1β receptors with bath application of IL-1ra (100 ng/ml) significantly and reversibly reduced both the frequency and amplitudes of mEPSCs in neuropathic rats (A and E) but had no effects on mEPSCs in sham-operated rats (B and F). Bath perfusion of IL-1β enhanced both the frequency and amplitudes of mEPSCs in sham-operated rats (C and G) but had no effects on mEPSCs in neuropathic rats (D and H). Bar graphs show the mean (+S.E.) amplitude and frequency for each tested agent. The number of neurons included in each group for the analysis is shown in each bar. **, p < 0.01; ***, p < 0.001; NS, no statistical significance.
To specifically determine the role of IL-1β in the regulation of glutamate release from presynaptic terminals and the signaling pathway used by IL-1β to alter non-NMDA glutamate receptor activities in postsynaptic neurons, we included the MyD88 inhibitory peptide (1 mm) in the recording electrode internal solution. MyD88 is a proximal mediator of the IL-1β signaling pathway demonstrated in immune cells (39, 40). Under this condition, bath perfusion of IL-1ra (100 ng/ml) reversibly and significantly reduced the mEPSC frequency (n = 10, p < 0.001) but not the mEPSC amplitude (n = 10) in spinal slices of neuropathic rats (Fig. 3, A and C). Bath perfusion of IL-1β (10 ng/ml) significantly increased the mEPSC frequency (n = 7, p < 0.001) but not the mEPSC amplitude in spinal slices of sham-operated rats (Fig. 3, B and D). For control, in another set of experiments, the control peptide for MyD88 inhibitory peptide (Pepinh-control, 1 mm) was included in the recording electrode internal solution. Under such a condition, perfusion of IL-1β (10 ng/ml) significantly (p < 0.05) increased both the frequency and amplitude of mEPSCs (n = 4) in neurons obtained from sham-operated rats (data not shown). These data further confirm that glutamate release from presynaptic terminals in neuropathic rats is enhanced by endogenous IL-1β. The lack of effects induced by IL-1ra in neuropathic rats and by IL-1β in sham-operated rats on mEPSC amplitudes also suggested that MyD88 is a mediator used by IL-1β to alter non-NMDA glutamate receptor activities in the spinal dorsal horn neurons.
FIGURE 3.

MyD88 is a mediator used by IL-1β to alter non-NMDA glutamate receptor activities in the spinal dorsal horn neurons. Recordings were obtained with electrodes containing the MyD88 inhibitory peptide (1 mm) in the intracellular solution. Under this condition, bath perfusion of IL-1ra (100 ng/ml) reversibly and significantly reduced the mEPSC frequency but not the mEPSC amplitude in spinal slices of neuropathic rats (A and C). Bath perfusion of IL-1β (10 ng/ml) significantly increased the mEPSC frequency but not the mEPSC amplitude in spinal slices of sham-operated rats (B and D). Bar graphs show the mean (+S.E.) amplitude and frequency for each tested agent. The number of neurons included in each group for the analysis is shown in each bar. **, p < 0.01; ***, p < 0.001; NS, no statistical significance.
It is likely that synaptic input from the primary afferent fibers constitutes only a fraction of the total excitatory input to superficial dorsal horn neurons. The mEPSCs we recorded above might well be a reflection of overall excitatory inputs from both the primary afferents and excitatory interneurons to the recorded neuron. To specifically address the role of IL-1β in the regulation of glutamate release from the primary afferents, a pair of EPSCs was evoked by a pair of electric stimulating pulses (50 ms apart) applied to the spinal dorsal root (6). We measured the paired pulse ratio (PPR) of EPSCs (i.e. the ratio of the second peak amplitude over the first peak amplitude induced by paired pulse stimulation). Analysis of PPRs is a classic approach to determine the transmitter release probability from presynaptic terminals (27, 41–44). A decrease of PPR indicates an increased probability of neurotransmitter release from presynaptic terminals. Vice versa, an increase of PPRs indicates a decreased probability of neurotransmitter release from presynaptic terminals. Bath application of IL-1ra (100 ng/ml) significantly and reversibly reduced the first peak amplitudes by 29.08 ± 2.48% (n = 16, p < 0.001). This was accompanied by a significant increase of PPRs from 0.64 ± 0.02 to 0.75 ± 0.01 (n = 16, p < 0.001) (Fig. 4A). In contrast, amplitudes and PPRs (n = 8) recorded from sham-operated rats were not altered by the same treatment (Fig. 4B). When we bath-applied IL-1β (10 ng/ml) in spinal slices of sham-operated rats, the first peak amplitudes of evoked EPSCs were significantly increased by 41.97 ± 3.80% (n = 15, p < 0.001), and PPRs were reduced from 0.75 ± 0.01 to 0.65 ± 0.01 (n = 15, p < 0.001) (Fig. 4C). By contrast, amplitudes of evoked EPSCs and PPRs (n = 7) recorded from spinal slices of neuropathic rats were not altered by bath-applied IL-1β (10 ng/ml) (data not shown).
FIGURE 4.

Endogenous IL-1β in neuropathic rats increases glutamate release from the primary afferent terminals in neuropathic rats. Samples of EPSCs evoked by a pair of electrical pulses applied to the spinal dorsal root before (base line) and after application of IL-1ra (100 ng/ml) recorded from neuropathic rats (A) and sham-operated rats (B). C, samples of EPSCs evoked by a pair of electrical pulses applied to the spinal dorsal root recorded from sham-operated rats at base line and after bath application of IL-1β (10 ng/ml). Bar graphs show the mean percentage (+ S.E.) of base line amplitude and mean (+ S.E.) P2/P1 ratios for each tested agent. The number of neurons included in each group for the analysis is shown in each bar. *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, no statistical significance.
Furthermore, we analyzed the inverse squared coefficient of variation (CV−2) of the peak amplitudes of 10 evoked EPSCs before and during perfusion of IL-1ra (100 ng/ml). An increase of presynaptic release is expected to cause an increase in CV−2 (45, 46). The CV−2 in neuropathic rats was significantly reduced by IL-1ra from 2070.14 ± 145.50 to 494.57 ± 42.85 (n = 7, p < 0.001) (Fig. 5, A and C). Furthermore, bath perfusion of IL-1β (10 ng/ml) significantly increased the CV−2 from 561.64 ± 48.22 to 2277.26 ± 404.78 (n = 15, p < 0.001) in spinal slices of sham-operated rats (Fig. 5, B and C). Together, these results indicated the following: (a) endogenous activation of presynaptic IL-1β receptors in the spinal dorsal horn of neuropathic rats increases glutamate release from the primary afferent terminals in the first sensory synapse; (b) glutamate release from the primary afferent terminals in the spinal dorsal horn of normal rats is not regulated by endogenous IL-1β.
FIGURE 5.

Activation of IL-1β receptors increases the inverse squared coefficient of variation. A, samples of variability of 10 evoked EPSCs collected from a neuron from neuropathic rats before and after bath perfusion of IL-1ra (100 ng/ml). B, samples of variability of 10 evoked EPSCs collected from a neuron from sham-operated rats before and after bath perfusion of IL-1β (10 ng/ml). C, bar graphs show the mean (+ S.E.) CV−2 before (base line) and after perfusion of each tested agent. The number of neurons included in each group for the analysis is shown in each bar. ***, p < 0.001.
Presynaptic NMDA Receptors Are Effector Receptors Used by Endogenous IL-1β to Enhance Glutamate Release from the Primary Afferents in Neuropathic Rats
We recently reported that endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferent terminals in the spinal dorsal horn (6). Functional coupling between NMDA receptors and IL-1β receptors in postsynaptic neurons in the brainstem was previously reported (47). We next determined the possible functional coupling between IL-1β receptors and presynaptic NMDA receptors at the primary afferent terminals. In the following experiments, both the MyD88 inhibitory peptide (1 mm) and MK-801 (1 mm) were included in the electrode intracellular solution. We recorded EPSCs in sham-operated rats. After the effects of IL-1β (10 ng/ml) on the amplitude and PPR of evoked EPSCs were documented, we added a selective NMDA receptor inhibitor D-AP5 (25 μm) into the recording chamber in the presence of IL-1β (10 ng/ml). D-AP5 significantly reduced the first peak amplitudes by 23.54 ± 4.36% (n = 6, p < 0.01) but increased PPRs from 0.66 ± 0.03 to 0.73 ± 0.01 (n = 6, p < 0.05) (Fig. 6A), which basically reversed the effects induced by IL-1β on the amplitude and PPR of evoked EPSCs. In the next set of experiments in sham-operated rats, we first determined the effects of DAP5 on the PPR and amplitudes of EPSCs. Consistent with our recent study (6), bath application of D-AP5 (25 μm) had no effects on the PPR and amplitudes of EPSCs in sham-operated rats. In the presence of D-AP5 (25 μm), further addition of IL-1β (10 ng/ml) (n = 6) into the recording chamber did not induce changes in PPRs and amplitudes of EPSCs (data not shown). In other words, presynaptic NMDA receptors are effector receptors used by the endogenous IL-1β to enhance glutamate release from the primary afferents in neuropathic rats. We recently reported that exogenous application of NMDA alone or NMDA plus d-serine increases glutamate release from the primary afferent terminals in the spinal dorsal horn of neuropathic rats but not in sham-operated rats (6). Here, we further confirm this conclusion by showing that bath application of NMDA (50 μm) plus d-serine (200 μm) increased the first amplitude of EPSCs and decreased the PPR in neuropathic rats. When we further added IL-1ra (100 ng/ml) to block IL-1β receptors in the presence of NMDA (50 μm) plus d-serine (200 μm), the first amplitude of EPSCs (n = 8) was significantly decreased, and the PPR (n = 8) was significantly increased in neuropathic rats. Interestingly, the reduction of EPSC amplitudes and the increase of PPRs induced by IL-1ra reached a level that made them significantly different from their base-line counterparts (Fig. 6B). Furthermore, in another set of experiments, after the effects of IL-1ra (100 ng/ml) on the amplitude and PPR of evoked EPSCs in spinal slices of neuropathic rats were documented, we perfused NMDA (50 μm) plus d-serine (200 μm) into the recording chamber in the presence of IL-1ra. Addition of NMDA plus d-serine did not alter the amplitude and PPR under this condition (Fig. 6C). Collectively, these data indicated that endogenous IL-1β in neuropathic rats enhances glutamate release from the primary afferents in the spinal dorsal horn through coupling with presynaptic NMDA receptors. In other words, presynaptic NMDA receptors are effector receptors used by the endogenous IL-1β to enhance glutamate release from the primary afferents in neuropathic rats.
FIGURE 6.

Presynaptic NMDA receptors are effector receptors used by the endogenous IL-1β to enhance glutamate release from the primary afferents in neuropathic rats. A, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of sham-operated rats at base line, during perfusion of IL-1β (10 ng/ml), and then during addition of the NMDA receptor inhibitor D-AP5 (25 μm) in the presence of IL-1β. B, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron from neuropathic rats at base line, during perfusion of NMDA (50 μm) plus d-serine (200 μm), and then during addition of IL-1ra (100 ng/ml) in the presence of NMDA plus d-serine. C, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron from neuropathic rats at base line, during perfusion of IL-1ra (100 ng/ml), and then during addition of NMDA (50 μm) plus d-serine (200 μm) in the presence of IL-1ra. Bar graphs (right) show the mean percentage (+ S.E.) of base-line amplitude and mean (+ S.E.) P2/P1 ratios for the tested agents. The number of neurons included in each group for the analysis is shown in each bar. *, p < 0.05; **, p < 0.01; ***, p < 0.001. NS, no statistical significance.
NMDA Currents Recorded from Small Neurons in the Dorsal Root Ganglion of Normal Rats Are Enhanced by IL-1β
The enhancement of presynaptic NMDA receptor activities by IL-1β was further confirmed by the following experiments conducted in sham-operated adult rats. As no techniques were currently available to record NMDA receptor activities directly at the presynaptic terminals, we chose to record NMDA receptor activities at the soma of the primary afferent terminals (i.e. neurons in the dorsal root ganglion of L4 and L5 spinal segments), and we examined the regulation of NMDA receptor activities by IL-1β. The somas of Aδ and C fibers (small neurons with diameters ≤30 μm) (48, 49) in the dorsal root ganglion were recorded because the superficial spinal dorsal horn neurons, as we recorded, are known to receive inputs from Aδ and C fiber primary afferent terminals (50, 51). Exogenous NMDA (50 μm) was applied directly to the recorded neuron by a puffing pipette placed about 20 μm away from the recorded neuron. After recording the base-line NMDA currents, we applied IL-1β (10 ng/ml) into the recording bath for 2 min. As shown in Fig. 7, IL-1β significantly increased amplitudes of NMDA currents by 65.40 ± 10.51% (p < 0.001). These data indicate that NMDA receptor activities in small primary sensory neurons are indeed increased by IL-1β, which is consistent with and confirms our conclusion that NMDA receptor activities in the primary afferent terminals are enhanced by IL-1β.
FIGURE 7.
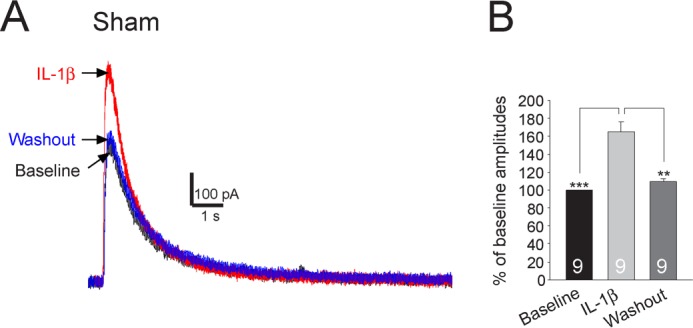
NMDA currents recorded from small neurons in the dorsal root ganglion are significantly enhanced by IL-1β. Recordings of NMDA currents evoked by NMDA (50 μm) injected onto the recorded neuron by a puff electrode at base line, during, and after washout of IL-1β are shown in A. The mean (+S.E.) amplitudes of NMDA currents at base line, during, and after washout of IL-1β are shown in bar graphs (B). The number of neurons included for the analysis is shown in each bar. **, p < 0.01; ***, p < 0.001.
Functional Coupling between IL-1β Receptors and Presynaptic NMDA Receptors in the Primary Afferent Terminals Is Mediated by the Neutral Sphingomyelinase/Ceramide Signaling Pathway
Studies of forebrain brain areas (21) and cultured neurons (52) suggested that neutral sphingomyelinase and its product ceramide are used by IL-1β to enhance NMDAR functions in postsynaptic neurons. To determine whether this is the case for IL-1β in the spinal dorsal horn to enhance presynaptic NMDA receptors at the presynaptic site, a pair of EPSCs was recorded from spinal slices of sham-operated rats before, during, and after washout of C2-ceramide (a membrane-permeable form of ceramide, 2 μg/ml) (53). C2-ceramide significantly increased the first amplitude of EPSCs by 57.93 ± 5.20% (n = 9, p < 0.001), and reduced the PPR from 0.75 ± 0.01 to 0.61 ± 0.02 (n = 9, p < 0.001) in sham-operated rats (Fig. 8A). These effects were basically reversed by bath perfusion of the NMDA inhibitor D-AP5 (25 μm) (Fig. 8A). Vice versa, in sham-operated rats, bath application of C2-ceramide (2 μg/ml) in the presence of D-AP5 (which blocks NMDA receptors) had no effects on either the amplitude or PPR of EPSCs (n = 8) (data not shown). These data indicated that C2-ceramide uses presynaptic NMDA receptors as effector receptors to increase glutamate release from the primary afferent terminals.
FIGURE 8.

Presynaptic NMDA receptors are effector receptors used by the sphingomyelinase/ceramide pathway to enhance glutamate release from the primary afferents in neuropathic rats. A, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of sham-operated rats at base line, during perfusion of C2-ceramide (2 μg/ml), and then during addition of the NMDA receptor inhibitor D-AP5 (25 μm) in the presence of C2-ceramide. B, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats at base line, during, and after washout of the neutral sphingomyelinase inhibitor (3-OMS, 30 μm). C, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of neuropathic rats at base line, during perfusion of NMDA (50 μm) plus d-serine (200 μm), and then during addition of 3-OMS (30 μm) in the presence of NMDA plus d-serine. Bar graphs (right) show the mean percentage (+S.E.) of base-line amplitude and mean (+ S.E.) P2/P1 ratios for the tested agents. The number of neurons included in each group for the analysis is shown in each bar. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine whether activities of presynaptic NMDA receptors in neuropathic rats are regulated by endogenous activation of the neutral sphingomyelinase/ceramide signaling, we determined the function of presynaptic NMDA receptors in the presence of the sphingomyelinase inhibitor (3-OMS). We found that perfusion of 3-OMS (30 μm) (52, 55) into the recording chamber significantly reduced the first amplitude of EPSCs by 29.85 ± 1.68% (n = 6, p < 0.001) and increased the PPR from 0.64 ± 0.01 to 0.75 ± 0.02 (n = 6, p < 0.01) (Fig. 8B). In the presence of 3-OMS (30 μm), bath application of NMDA (50 μm) plus d-serine (200 μm) did not significantly alter the amplitude (n = 7) and PPR of EPSCs (n = 7) in neuropathic rats (data not shown). In another set of experiments, after the effects induced by bath perfusion of NMDA (50 μm) plus d-serine (200 μm) on the amplitude and PPR of evoked EPSCs in spinal slices of neuropathic rats were documented, we added 3-OMS (30 μm) into the recording bath to suppress sphingomyelinase activities. Under this condition, addition of 3-OMS significantly reduced the first amplitude (n = 7, p < 0.001) and increased the PPR (n = 7, p < 0.001) (Fig. 8C) to such a degree that it made them significantly different from their base-line counterparts. We further conjectured that activation of neutral sphingomyelinase in neuropathic rats should result in elevation of its product ceramide levels, which would attenuate or occlude the effects induced by C2-ceramide seen in sham-operated rats (Fig. 8A). As expected, bath perfusion of C2-ceramide (2 μm) did not significantly change the amplitudes (base line, 1195.5 ± 43.05 pA; C2-ceramide, 1231.16 ± 49.35 pA) and the PPRs (base line, 0.60 ± 0.02; C2-ceramide, 0.62 ± 0.02) of EPSCs (n = 7) recorded from neuropathic rats. Together, these data indicated that presynaptic NMDA receptors are effector receptors used by the sphingomyelinase/ceramide pathway to enhance glutamate release from the primary afferents in neuropathic rats.
Finally, we determined whether the sphingomyelinase/ceramide pathway is used by IL-1β to enhance the function of presynaptic NMDA receptors and glutamate release from the primary afferent terminals in the spinal dorsal horn. In spinal slices obtained from sham-operated rats, after the effects of IL-1β (10 ng/ml) on the amplitude and PPR of evoked EPSCs were recorded, 3-OMS (30 μm) was added into the recording bath to suppress the neutral sphingomyelinase activity. Addition of 3-OMS essentially erased the effects induced by IL-1β on the first amplitude and the PPR (Fig. 9A). In another set of experiments, after recording base-line amplitudes and PPRs of EPSCs in slices of sham-operated rats, 3-OMS (30 μm) was first applied into the recording bath. Addition of 3-OMS did not alter the amplitude and PPR of evoked EPSCs (Fig. 9B). These data suggested that glutamate release from the primary afferent terminals in the spinal dorsal horn is not maintained by the tonic activities of sphingomyelinase under normal conditions. Furthermore, in the presence of 3-OMS (30 μm), addition of IL-1β did not significantly alter the amplitude and PPR of EPSCs recorded from sham-operated rats (Fig. 9B). In other words, the enhanced glutamate release by IL-1β in sham-operated rats (see Fig. 3C) was abolished when activities of sphingomyelinase were suppressed. From the data, we concluded that neutral sphingomyelinase/ceramide mediates functional coupling between IL-1β receptors and presynaptic NMDA receptors in the primary afferent terminals.
FIGURE 9.

Enhancement of glutamate release from the primary afferent terminals by IL-1β is mediated by the sphingomyelinase/ceramide pathway. A, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of sham-operated rats at base line, during perfusion of IL-1β (10 ng/ml), and then during addition of the neutral sphingomyelinase inhibitor (3-OMS, 30 μm) in the presence of IL-1β. B, samples of EPSCs evoked by a pair of electrical pulses recorded from a neuron of sham-operated rats at base line, during perfusion of 3-OMS (30 μm), and then during addition of IL-1β (10 ng/ml) in the presence of 3-OMS. Bar graphs (right) show the mean percentage (+S.E.) of base-line amplitude and mean (+S.E.) P2/P1 ratios for the tested agents. The number of neurons included in each group for the analysis is shown in each bar. *, p < 0.05; **, p < 0.01. NS, no statistical significance.
DISCUSSION
Given that excessive activation of glutamate receptors and increased production of proinflammatory cytokines in the spinal dorsal horn are the culprits in the development and maintenance of many types of pathological pain conditions, including neuropathic pain, identifying the mechanisms underlying interactions between activation of glutamate receptors and proinflammatory cytokines would provide a road map to conquer pathological pain. This study identified an endogenous mechanism used by IL-1β to enhance glutamatergic synaptic activation in the first sensory synapse in the spinal dorsal horn. We first confirmed that ligation of spinal nerve increases IL-1β levels in the spinal dorsal horn. We demonstrated that glutamate release from the primary afferent terminals and non-NMDA glutamate receptor activities in postsynaptic neurons in the spinal dorsal horn of neuropathic rats are increased by endogenous IL-1β. MyD88 is a mediator used by IL-1β to enhance non-NMDA glutamate receptor activities in postsynaptic neurons in the spinal dorsal horn. Presynaptic NMDA receptors are effector receptors used by IL-1β in neuropathic rats to increase glutamate release from the primary afferent terminals in the spinal dorsal horn. We further elucidated that the neutral sphingomyelinase/ceramide signaling pathway mediates the functional coupling between IL-1β receptors and presynaptic NMDA receptors in the primary afferent terminals. Our study for the first time identified the primary afferent central terminal as an important site for the interactions between glutamate receptors and proinflammatory cytokines and the signaling pathway underlying these interactions.
Mechanisms Used by Glial Cells to Alter Glutamatergic Synaptic Activation in Pathological Pain Conditions
Activation of glutamatergic synapses depends on three key factors as follows: the number and properties of glutamate receptors in postsynaptic neurons, the amount of glutamate released from presynaptic terminals, and the rate by which glutamate is taken up by glutamate transporters (56, 57). These three factors can be altered by glial cells under pathological pain conditions. For example, down-regulation of glial glutamate transporter protein expression in spinal dorsal horn astrocytes is associated with allodynia and hyperalgesia induced by chronic nerve injury (58, 59), chemotherapy (e.g. paclitaxel) (60), or opioids (61). At the synaptic level, we demonstrated that deficient glutamate uptake by glial glutamate transporters enhances activation of AMPA and NMDA receptors and causes glutamate to spill to the extrasynaptic space and activate extrasynaptic NMDA receptors in spinal sensory neurons (23, 27).
Besides glial glutamate transporters, bioactive substances released from activated glial cells induced by nerve injury or peripheral inflammation may alter the number and function of glutamate receptors in postsynaptic neurons. Among the many bioactive substances released from activated glial cells, proinflammatory cytokines, including IL-1β, IL-6, and TNFα, are well known to be involved in the genesis of pathological pain (1, 2). Surprisingly, only a handful of studies directly address the impacts created by these cytokines on spinal glutamatergic synaptic activities. In a rat model of inflammatory pain induced by injecting complete Freund's adjuvant, IL-1β produced by activated astrocytes increased phosphorylation of NMDA receptors in the spinal dorsal horn (62). In another model of inflammatory pain induced by carrageenan, it was demonstrated that elevated levels of TNFα in the spinal dorsal horn increase AMPA receptor activities by enhancing trafficking of AMPA receptors from the cytosol to the membrane (63). Functions of AMPA and NMDA receptors in the dorsal horn postsynaptic neurons in normal rats are increased by exogenous IL-1β, IL-6, and TNFα (15). It remains unknown whether and how AMPA and NMDA receptor activities in the spinal dorsal horn neurons are altered by endogenous IL-1β in neuropathic conditions. Our study advances this research area by directly demonstrating that endogenous IL-1β in neuropathic rats enhances non-NMDA glutamate receptor activities in spinal dorsal horn neurons. Interestingly, we found that the IL-1β-induced enhancement of non-NMDA glutamate receptor activities was inhibited when the MyD88 inhibitory peptides were microdialyzed into the recorded neuron. MyD88 is an adaptor molecule widely known to be a proximal mediator for the biological action of IL-1β in the peripheral immune system (39, 40). Little is known about the role of MyD88 in regulating the effects induced by IL-1β on non-NMDA glutamate receptor activities in the CNS. This study, for the first time, reveals that MyD88 is a key molecule mediating the action of IL-1β on non-NMDA glutamate receptor activities in spinal dorsal horn neurons.
Glutamate release from presynaptic terminals in the dorsal horn is also regulated by proinflammatory cytokines. It was demonstrated previously by others that perfusion of exogenous IL-1β, IL-6, or TNFα increases spontaneous EPSC frequencies in dorsal horn neurons in normal spinal rats (15). It remains unknown whether and how glutamate release from the primary afferent terminals are regulated by proinflammatory cytokines in pathological pain conditions. Our study is the first to show that glutamate release from the primary afferent terminals is regulated by endogenous IL-1β in neuropathic pain conditions. In keeping with this, functional expression of IL-1β receptors in the soma of the primary afferents was previously demonstrated (64) and further confirmed in this study (Fig. 7).
Primary Afferent Central Terminus Is an Important Site for the Functional Coupling between NMDA Receptors and IL-1β
Glutamate release from the primary afferent terminals in the spinal dorsal horn is regulated by activities of many presynaptic receptors. For example, glutamate release from the primary afferents in the spinal dorsal horn is reduced upon activation of μ-opioid receptors (65), α2-adrenoreceptors (66), CB1 cannabinoid receptors (67), GABAA receptors (68), GABAB receptors (69), and group II and group III metabotropic glutamate receptors (70). It is worth noting that group III metabotropic glutamate receptors can be activated by both glutamate and N-acetylaspartylglutamate (71). However, activation of group I metabotropic glutamate receptors (72) or TRPV1 receptors (73) increases glutamate release from the central terminals of primary afferents. Added to this complexity of presynaptic regulation mechanisms is the presynaptic NMDA receptor reported recently by us and others. Endogenous activation of NMDA receptors in the central terminals of primary afferents is critical to the increased glutamate release from primary afferent terminals triggered by peripheral sensory input in neuropathic rats (6) or in rats with morphine tolerance (74). These studies indicate that regulation of presynaptic receptor activities offers a powerful means of dynamically regulating excitatory transmission in the spinal dorsal horn. Understanding the regulation of presynaptic receptors by proinflammatory cytokines would reveal synaptic mechanisms by which glial cells alter spinal nociceptive processing. In this regard, it was recently reported that TNF-α increases glutamate release from presynaptic terminals via TRPV1 receptors (73). Our findings that presynaptic NMDA receptors in the primary afferent terminals are the effector receptors used by endogenous IL-1β to increase glutamate release from the primary afferent terminals are the first to reveal the functional coupling between presynaptic NMDA receptors and IL-1β. This conclusion is in line with previous reports showing functional expressions of NMDA receptors and IL-1β receptors in the dorsal root ganglion neurons (i.e. the soma of the primary afferents) (64) and the enhanced NMDA receptor activities by IL-1β in postsynaptic neurons in different nervous systems (21, 52). It is conceivable to speculate that activation of presynaptic NMDA receptors causes influx of Ca2+ through the NMDA receptors into the central terminals of primary afferents and subsequent increase of glutamate release, as shown in hippocampal slices (75). The influx of Ca2+ could come directly from the opening of NMDA receptors (76) and/or from voltage-gated calcium channels in response to depolarization due to the opening of NMDA receptors (77, 78).
It is worth noting that in addition to blocking effects induced by IL-1β, IL-1ra also blocks the action of IL-1α. The following facts support that the effects induced by IL-1ra on slices of neuropathic rats in this study are due to its inhibitory effects on IL-1β but not IL-1α. First, IL-1α expression in the spinal dorsal horn is not altered after nerve injury (79). Second, intrathecal injection of IL-1α in naive rats does not influence nociceptive transmission but reduces nociceptive behaviors in animals with neuropathic pain (79). Third, we found that treatment of IL-1β in the spinal slices of the sham-operated animals induces opposite effects on the synaptic transmission in comparison with the effects induced by IL-1ra in neuropathic rats.
Role of Ceramide in Pathological Pain
Ceramide appears to be a critical mediator in the generation of both peripheral and spinal central sensitization in pathological pain conditions. Peripheral sensitization and hyperalgesia induced by intradermal injection of TNFα is in part mediated by ceramide (80). Intradermal injection of the cell penetrating analog of ceramide (C2-ceramide) produces hyperalgesia in rats (80, 81). Exogenous application of neutral sphingomyelinase or C2-ceramide increases TTX-resistant sodium currents in the soma of primary sensory neurons (54). In the spinal dorsal horn, neutral sphingomyelinase and ceramide levels are increased in neuropathic rats induced by nerve injury (16). Suppression of ceramide production in the spinal dorsal horn ameliorates neuropathic pain, which is accompanied by decreased microglial activation in the spinal dorsal horn (17). However, it remains unknown about the mechanisms by which ceramide alters dorsal horn synaptic transmission. Our data showed that inhibition of neutral sphingomyelinase suppressed glutamate release from the primary afferent central terminals in neuropathic animals but had no effects in sham-operated animals (Fig. 8). These findings identified the primary afferent central terminal as a site of action for endogenous ceramide in neuropathic rats to enhance glutamatergic synaptic transmission in the spinal dorsal horn. Most intriguingly, we further demonstrated that ceramide mediates the functional coupling between IL-1β and presynaptic NMDA receptors in the primary afferent terminals. Hence, inhibition of ceramide synthesis blocks both the IL-1β and presynaptic NMDA receptor activities at the presynaptic site in neuropathic conditions. Our findings are in line with previous studies of forebrain brain areas and cultured neurons showing that neutral sphingomyelinase/ceramide is used by IL-1β to enhance NMDAR functions in postsynaptic neurons (21, 52).
In conclusion, this study reveals that functional coupling between IL-1β receptors and presynaptic NMDA receptors at the primary afferent terminals is a crucial mechanism leading to enhanced glutamate release and activation of non-NMDA receptors in the spinal dorsal horn neurons in neuropathic pain conditions. Interruption of such functional coupling could be an effective approach for the treatment of neuropathic pain.
This work was supported, in whole or in part, by National Institutes of Health Grant RO1 NS-064289 from NINDS (to H. R. W.).
This article was selected as a Paper of the Week.
- SNL
- spinal nerve ligation
- TTX
- tetrodotoxin
- EPSC
- excitatory postsynaptic current
- mEPSC
- miniature EPSC
- PPR
- paired pulse ratio
- CV
- coefficient of variation
- aCSF
- artificial cerebrospinal fluid
- D-AP5
- d-aminophosphonovaleric acid
- 3-OMS
- 3-O-methylsphingomyelin.
REFERENCES
- 1. Ren K., Dubner R. (2010) Interactions between the immune and nervous systems in pain. Nat. Med. 16, 1267–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Milligan E. D., Watkins L. R. (2009) Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 10, 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Biasi S., Rustioni A. (1988) Glutamate and substance P coexist in primary afferent terminals in the superficial laminae of spinal cord. Proc. Natl. Acad. Sci. U.S.A. 85, 7820–7824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Willis W. D. (2002) Long-term potentiation in spinothalamic neurons. Brain Res. Brain Res. Rev. 40, 202–214 [DOI] [PubMed] [Google Scholar]
- 5. Danbolt N. C. (2001) Glutamate uptake. Prog. Neurobiol. 65, 1–105 [DOI] [PubMed] [Google Scholar]
- 6. Yan X., Jiang E., Gao M., Weng H. R. (2013) Endogenous activation of presynaptic NMDA receptors enhances glutamate release from the primary afferents in the spinal dorsal horn in a rat model of neuropathic pain. J. Physiol. 591, 2001–2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee H. L., Lee K. M., Son S. J., Hwang S. H., Cho H. J. (2004) Temporal expression of cytokines and their receptors mRNAs in a neuropathic pain model. Neuroreport 15, 2807–2811 [PubMed] [Google Scholar]
- 8. DeLeo J. A., Colburn R. W., Rickman A. J. (1997) Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Res. 759, 50–57 [DOI] [PubMed] [Google Scholar]
- 9. Shamash S., Reichert F., Rotshenker S. (2002) The cytokine network of Wallerian degeneration: tumor necrosis factor-α, interleukin-1α, and interleukin-1β. J. Neurosci. 22, 3052–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wolf G., Gabay E., Tal M., Yirmiya R., Shavit Y. (2006) Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain 120, 315–324 [DOI] [PubMed] [Google Scholar]
- 11. Kleibeuker W., Gabay E., Kavelaars A., Zijlstra J., Wolf G., Ziv N., Yirmiya R., Shavit Y., Tal M., Heijnen C. J. (2008) IL-1β signaling is required for mechanical allodynia induced by nerve injury and for the ensuing reduction in spinal cord neuronal GRK2. Brain Behav. Immun. 22, 200–208 [DOI] [PubMed] [Google Scholar]
- 12. Reeve A. J., Patel S., Fox A., Walker K., Urban L. (2000) Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in the spinal cord neuronal responses to nociceptive stimuli in the rat. Eur. J. Pain 4, 247–257 [DOI] [PubMed] [Google Scholar]
- 13. Kwon M. S., Shim E. J., Seo Y. J., Choi S. S., Lee J. Y., Lee H. K., Suh H. W. (2005) Differential modulatory effects of cholera toxin and pertussis toxin on pain behavior induced by TNF-α, interleukin-1β, and interferon-γ injected intrathecally. Arch. Pharm. Res. 28, 582–586 [DOI] [PubMed] [Google Scholar]
- 14. Falchi M., Ferrara F., Gharib C., Dib B. (2001) Hyperalgesic effect of intrathecally administered interleukin-1 in rats. Drugs Exp. Clin. Res. 27, 97–101 [PubMed] [Google Scholar]
- 15. Kawasaki Y., Zhang L., Cheng J. K., Ji R. R. (2008) Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1β, interleukin-6, and tumor necrosis factor-α in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci. 28, 5189–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patti G. J., Yanes O., Shriver L. P., Courade J. P., Tautenhahn R., Manchester M., Siuzdak G. (2012) Metabolomics implicates altered sphingolipids in chronic pain of neuropathic origin. Nat. Chem. Biol. 8, 232–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kobayashi Y., Kiguchi N., Maeda T., Ozaki M., Kishioka S. (2012) The critical role of spinal ceramide in the development of partial sciatic nerve ligation-induced neuropathic pain in mice. Biochem. Biophys. Res. Commun. 421, 318–322 [DOI] [PubMed] [Google Scholar]
- 18. Muscoli C., Doyle T., Dagostino C., Bryant L., Chen Z., Watkins L. R., Ryerse J., Bieberich E., Neumman W., Salvemini D. (2010) Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J. Neurosci. 30, 15400–15408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 20. Ndengele M. M., Cuzzocrea S., Masini E., Vinci M. C., Esposito E., Muscoli C., Petrusca D. N., Mollace V., Mazzon E., Li D., Petrache I., Matuschak G. M., Salvemini D. (2009) Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J. Pharmacol. Exp. Ther. 329, 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balosso S., Maroso M., Sanchez-Alavez M., Ravizza T., Frasca A., Bartfai T., Vezzani A. (2008) A novel non-transcriptional pathway mediates the proconvulsive effects of interleukin-1β. Brain 131, 3256–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim S. H., Chung J. M. (1992) An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 50, 355–363 [DOI] [PubMed] [Google Scholar]
- 23. Nie H., Weng H. R. (2010) Impaired glial glutamate uptake induces extrasynaptic glutamate spillover in the spinal sensory synapses of neuropathic rats. J. Neurophysiol. 103, 2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nie H., Weng H. R. (2009) Glutamate transporters prevent excessive activation of NMDA receptors and extrasynaptic glutamate spillover in the spinal dorsal horn. J. Neurophysiol. 101, 2041–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berretta N., Jones R. S. (1996) Tonic facilitation of glutamate release by presynaptic N-methyl-d-aspartate autoreceptors in the entorhinal cortex. Neuroscience 75, 339–344 [DOI] [PubMed] [Google Scholar]
- 26. Drdla R., Gassner M., Gingl E., Sandkühler J. (2009) Induction of synaptic long-term potentiation after opioid withdrawal. Science 325, 207–210 [DOI] [PubMed] [Google Scholar]
- 27. Weng H. R., Chen J. H., Pan Z. Z., Nie H. (2007) Glial glutamate transporter 1 regulates the spatial and temporal coding of glutamatergic synaptic transmission in spinal lamina II neurons. Neuroscience 149, 898–907 [DOI] [PubMed] [Google Scholar]
- 28. Wu L. J., Toyoda H., Zhao M. G., Lee Y. S., Tang J., Ko S. W., Jia Y. H., Shum F. W., Zerbinatti C. V., Bu G., Wei F., Xu T. L., Muglia L. J., Chen Z. F., Aueberson Y. P., Kaang B. K., Zhuo M. (2005) Up-regulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J. Neurosci. 25, 11107–11116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weng H. R., Chen J. H., Cata J. P. (2006) Inhibition of glutamate uptake in the spinal cord induces hyperalgesia and increased responses of spinal dorsal horn neurons to peripheral afferent stimulation. Neuroscience 138, 1351–1360 [DOI] [PubMed] [Google Scholar]
- 30. Yoshimura M., Nishi S. (1993) Blind patch-clamp recordings from substantia gelatinosa neurons in adult rat spinal cord slices: pharmacological properties of synaptic currents. Neuroscience 53, 519–526 [DOI] [PubMed] [Google Scholar]
- 31. Yoshimura M., Jessell T. M. (1989) Primary afferent-evoked synaptic responses and slow potential generation in rat substantia gelatinosa neurons in vitro. J. Neurophysiol. 62, 96–108 [DOI] [PubMed] [Google Scholar]
- 32. Kohno T., Moore K. A., Baba H., Woolf C. J. (2003) Peripheral nerve injury alters excitatory synaptic transmission in lamina II of the rat dorsal horn. J. Physiol. 548, 131–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang W., Mei X. P., Chen L., Tang J., Li J. L., Wu S. X., Xu L. X., Li Y. Q. (2012) Triptolide prevents and attenuates neuropathic pain via inhibiting central immune response. Pain Physician 15, E995–1006 [PubMed] [Google Scholar]
- 34. Chen T., Jiang J., Huang H., Wang D., Liu Y., Hong Y. (2012) Role of bovine adrenal medulla 22 (BAM22) in the pathogenesis of neuropathic pain in rats with spinal nerve ligation. Eur. J. Pharmacol. 685, 24–29 [DOI] [PubMed] [Google Scholar]
- 35. Bindoni M., Perciavalle V., Berretta S., Belluardo N., Diamantstein T. (1988) Interleukin 2 modifies the bioelectric activity of some neurosecretory nuclei in the rat hypothalamus. Brain Res. 462, 10–14 [DOI] [PubMed] [Google Scholar]
- 36. Lissin D. V., Carroll R. C., Nicoll R. A., Malenka R. C., von Zastrow M. (1999) Rapid, activation-induced redistribution of ionotropic glutamate receptors in cultured hippocampal neurons. J. Neurosci. 19, 1263–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cunningham A. J., Murray C. A., O'Neill L. A., Lynch M. A., O'Connor J. J. (1996) Interleukin-1β (IL-1β) and tumour necrosis factor (TNF) inhibit long-term potentiation in the rat dentate gyrus in vitro. Neurosci. Lett. 203, 17–20 [DOI] [PubMed] [Google Scholar]
- 38. Rossi S., Furlan R., De Chiara V., Motta C., Studer V., Mori F., Musella A., Bergami A., Muzio L., Bernardi G., Battistini L., Martino G., Centonze D. (2012) Interleukin-1β causes synaptic hyperexcitability in multiple sclerosis. Ann. Neurol. 71, 76–83 [DOI] [PubMed] [Google Scholar]
- 39. Watters T. M., Kenny E. F., O'Neill L. A. (2007) Structure, function, and regulation of the Toll/IL-1 receptor adaptor proteins. Immunol. Cell Biol. 85, 411–419 [DOI] [PubMed] [Google Scholar]
- 40. Muzio M., Ni J., Feng P., Dixit V. M. (1997) IRAK (Pelle) family member IRAK-2 and MyD88 as proximal mediators of IL-1 signaling. Science 278, 1612–1615 [DOI] [PubMed] [Google Scholar]
- 41. Zucker R. S. (1989) Short-term synaptic plasticity. Annu. Rev. Neurosci. 12, 13–31 [DOI] [PubMed] [Google Scholar]
- 42. Manabe T., Wyllie D. J., Perkel D. J., Nicoll R. A. (1993) Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J. Neurophysiol. 70, 1451–1459 [DOI] [PubMed] [Google Scholar]
- 43. Foster T. C., McNaughton B. L. (1991) Long-term enhancement of CA1 synaptic transmission is due to increased quantal size, not quantal content. Hippocampus 1, 79–91 [DOI] [PubMed] [Google Scholar]
- 44. Xu H., Wu L. J., Wang H., Zhang X., Vadakkan K. I., Kim S. S., Steenland H. W., Zhuo M. (2008) Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J. Neurosci. 28, 7445–7453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Korn H., Faber D. S. (1991) Quantal analysis and synaptic efficacy in the CNS. Trends Neurosci. 14, 439–445 [DOI] [PubMed] [Google Scholar]
- 46. Bekkers J. M., Stevens C. F. (1990) Presynaptic mechanism for long-term potentiation in the hippocampus. Nature 346, 724–729 [DOI] [PubMed] [Google Scholar]
- 47. Guo W., Wang H., Watanabe M., Shimizu K., Zou S., LaGraize S. C., Wei F., Dubner R., Ren K. (2007) Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J. Neurosci. 27, 6006–6018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Harper A. A., Lawson S. N. (1985) Conduction velocity is related to morphological cell type in rat dorsal root ganglion neurones. J. Physiol. 359, 31–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma C., LaMotte R. H. (2005) Enhanced excitability of dissociated primary sensory neurons after chronic compression of the dorsal root ganglion in the rat. Pain 113, 106–112 [DOI] [PubMed] [Google Scholar]
- 50. Sugiura Y., Lee C. L., Perl E. R. (1986) Central projections of identified, unmyelinated (C) afferent fibers innervating mammalian skin. Science 234, 358–361 [DOI] [PubMed] [Google Scholar]
- 51. Basbaum A. I., Bautista D. M., Scherrer G., Julius D. (2009) Cellular and molecular mechanisms of pain. Cell 139, 267–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tsakiri N., Kimber I., Rothwell N. J., Pinteaux E. (2008) Differential effects of interleukin-1 α and β on interleukin-6 and chemokine synthesis in neurones. Mol. Cell Neurosci. 38, 259–265 [DOI] [PubMed] [Google Scholar]
- 53. Nakabo Y., Pabst M. J. (1997) C2-ceramide and C6-ceramide inhibited priming for enhanced release of superoxide in monocytes, but had no effect on the killing of leukaemic cells by monocytes. Immunology 90, 477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang Y. H., Vasko M. R., Nicol G. D. (2002) Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. J. Physiol. 544, 385–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen B. C., Chang H. M., Hsu M. J., Shih C. M., Chiu Y. H., Chiu W. T., Lin C. H. (2009) Peptidoglycan induces cyclooxygenase-2 expression in macrophages by activating the neutral sphingomyelinase-ceramide pathway. J. Biol. Chem. 284, 20562–20573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jonas P. (2000) The time course of signaling at central glutamatergic synapses. News Physiol. Sci. 15, 83–89 [DOI] [PubMed] [Google Scholar]
- 57. Clements J. D. (1996) Transmitter time course in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 19, 163–171 [DOI] [PubMed] [Google Scholar]
- 58. Sung B., Lim G., Mao J. (2003) Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J. Neurosci. 23, 2899–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Xin W. J., Weng H. R., Dougherty P. M. (2009) Plasticity in expression of the glutamate transporters GLT-1 and GLAST in spinal dorsal horn glial cells following partial sciatic nerve ligation. Mol. Pain 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weng H. R., Aravindan N., Cata J. P., Chen J. H., Shaw A. D., Dougherty P. M. (2005) Spinal glial glutamate transporters down-regulate in rats with taxol-induced hyperalgesia. Neurosci. Lett. 386, 18–22 [DOI] [PubMed] [Google Scholar]
- 61. Mao J., Sung B., Ji R. R., Lim G. (2002) Chronic morphine induces down-regulation of spinal glutamate transporters: implications in morphine tolerance and abnormal pain sensitivity. J. Neurosci. 22, 8312–8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang R. X., Li A., Liu B., Wang L., Ren K., Zhang H., Berman B. M., Lao L. (2008) IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain 135, 232–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Choi J. I., Svensson C. I., Koehrn F. J., Bhuskute A., Sorkin L. S. (2010) Peripheral inflammation induces tumor necrosis factor-dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain 149, 243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Binshtok A. M., Wang H., Zimmermann K., Amaya F., Vardeh D., Shi L., Brenner G. J., Ji R. R., Bean B. P., Woolf C. J., Samad T. A. (2008) Nociceptors are interleukin-1β sensors. J. Neurosci. 28, 14062–14073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Heinke B., Gingl E., Sandkühler J. (2011) Multiple targets of μ-opioid receptor-mediated presynaptic inhibition at primary afferent Aδ- and C-fibers. J. Neurosci. 31, 1313–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kawasaki Y., Kumamoto E., Furue H., Yoshimura M. (2003) α2 adrenoceptor-mediated presynaptic inhibition of primary afferent glutamatergic transmission in rat substantia gelatinosa neurons. Anesthesiology 98, 682–689 [DOI] [PubMed] [Google Scholar]
- 67. Lever I. J., Malcangio M. (2002) CB(1) receptor antagonist SR141716A increases capsaicin-evoked release of Substance P from the adult mouse spinal cord. Br. J. Pharmacol. 135, 21–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Willis W. D. (2006) John Eccles' studies of spinal cord presynaptic inhibition. Prog. Neurobiol. 78, 189–214 [DOI] [PubMed] [Google Scholar]
- 69. Ataka T., Kumamoto E., Shimoji K., Yoshimura M. (2000) Baclofen inhibits more effectively C-afferent than Aδ-afferent glutamatergic transmission in substantia gelatinosa neurons of adult rat spinal cord slices. Pain 86, 273–282 [DOI] [PubMed] [Google Scholar]
- 70. Gerber G., Zhong J., Youn D., Randic M. (2000) Group II and group III metabotropic glutamate receptor agonists depress synaptic transmission in the rat spinal cord dorsal horn. Neuroscience 100, 393–406 [DOI] [PubMed] [Google Scholar]
- 71. Neale J. H., Olszewski R. T., Gehl L. M., Wroblewska B., Bzdega T. (2005) The neurotransmitter N-acetylaspartylglutamate in models of pain, ALS, diabetic neuropathy, CNS injury, and schizophrenia. Trends Pharmacol. Sci. 26, 477–484 [DOI] [PubMed] [Google Scholar]
- 72. Song J. H., Park E. S., Han S. M., Han S. R., Ahn D. K., Youn D. H. (2009) Signal transduction mechanisms underlying group I mGluR-mediated increase in frequency and amplitude of spontaneous EPSCs in the spinal trigeminal subnucleus oralis of the rat. Mol. Pain 5, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Park C. K., Lü N., Xu Z. Z., Liu T., Serhan C. N., Ji R. R. (2011) Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J. Neurosci. 31, 15072–15085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zeng J., Thomson L. M., Aicher S. A., Terman G. W. (2006) Primary afferent NMDA receptors increase dorsal horn excitation and mediate opiate tolerance in neonatal rats. J. Neurosci. 26, 12033–12042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. McGuinness L., Taylor C., Taylor R. D., Yau C., Langenhan T., Hart M. L., Christian H., Tynan P. W., Donnelly P., Emptage N. J. (2010) Presynaptic NMDARs in the hippocampus facilitate transmitter release at theta frequency. Neuron 68, 1109–1127 [DOI] [PubMed] [Google Scholar]
- 76. Glitsch M., Marty A. (1999) Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J. Neurosci. 19, 511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Awatramani G. B., Price G. D., Trussell L. O. (2005) Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron 48, 109–121 [DOI] [PubMed] [Google Scholar]
- 78. Christie J. M., Jahr C. E. (2008) Dendritic NMDA receptors activate axonal calcium channels. Neuron 60, 298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mika J., Korostynski M., Kaminska D., Wawrzczak-Bargiela A., Osikowicz M., Makuch W., Przewlocki R., Przewlocka B. (2008) Interleukin-1α has antiallodynic and antihyperalgesic activities in a rat neuropathic pain model. Pain 138, 587–597 [DOI] [PubMed] [Google Scholar]
- 80. Joseph E. K., Levine J. D. (2004) Caspase signalling in neuropathic and inflammatory pain in the rat. Eur. J. Neurosci. 20, 2896–2902 [DOI] [PubMed] [Google Scholar]
- 81. Doyle T., Chen Z., Muscoli C., Obeid L. M., Salvemini D. (2011) Intraplantar-injected ceramide in rats induces hyperalgesia through an NF-κB- and p38 kinase-dependent cyclooxygenase 2/prostaglandin E2 pathway. Faseb J. 25, 2782–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]