

Background: MLL fusion proteins use similar strategy for leukemic transformation through DOT1L recruitment.
Results: Ten amino acids in DOT1L were identified as essential for binding and transformation by MLL-AF9.
Conclusion: Biochemical and functional results indicate that blocking DOT1L recruitment represents a promising therapeutic strategy for mixed lineage leukemia.
Significance: Identified DOT1L peptide will lay a foundation toward discovery of chemical tools able to block DOT1L recruitment.
Keywords: Fusion Protein, Histone Methylation, Peptides, Protein-Protein Interactions, Surface Plasmon Resonance (SPR), AF9, DOT1L, ENL, Mixed Lineage Leukemia
Abstract
The MLL fusion proteins, AF9 and ENL, activate target genes in part via recruitment of the histone methyltransferase DOT1L (disruptor of telomeric silencing 1-like). Here we report biochemical, biophysical, and functional characterization of the interaction between DOT1L and MLL fusion proteins, AF9/ENL. The AF9/ENL-binding site in human DOT1L was mapped, and the interaction site was identified to a 10-amino acid region (DOT1L865–874). This region is highly conserved in DOT1L from a variety of species. Alanine scanning mutagenesis analysis shows that four conserved hydrophobic residues from the identified binding motif are essential for the interactions with AF9/ENL. Binding studies demonstrate that the entire intact C-terminal domain of AF9/ENL is required for optimal interaction with DOT1L. Functional studies show that the mapped AF9/ENL interacting site is essential for immortalization by MLL-AF9, indicating that DOT1L interaction with MLL-AF9 and its recruitment are required for transformation by MLL-AF9. These results strongly suggest that disruption of interaction between DOT1L and AF9/ENL is a promising therapeutic strategy with potentially fewer adverse effects than enzymatic inhibition of DOT1L for MLL fusion protein-associated leukemia.
Introduction
Epigenetic regulation of transcription through covalent modifications of histones is a fundamental mechanism of transcriptional regulation that is important for stem cell self-renewal as well as lineage commitment during development. Two broad groups of transcriptional regulators, the trithorax (Trx) and polycomb group Pc(G) proteins are the pivotal positive and negative regulators in this mechanism. Deregulation of transcription by disruption of both of these families of epigenetic regulators is emerging as a common cause of human malignancies.
Mixed lineage leukemia (MLL)2 protein, is a Trx group protein that can be disrupted by fusion to a variety of translocation partners in human lymphoid and myeloid acute leukemia. As a result of chromosome translocation, the MLL N terminus becomes fused to one of more than 60 partner proteins forming chimeric oncogenes that up-regulate the expression of HOX genes, blocking the hematopoietic differentiation, and ultimately lead to acute leukemia (1–4). Leukemia mediated by MLL rearrangements possess unique clinical and biological features, and they are present in over 70% cases of infant leukemia (5) and in general account for ∼5% of acute lymphoblastic leukemia, 5–10% of acute myeloid leukemia, and almost all cases of mixed lineage leukemia (6). New therapeutic strategies are needed, because patients with leukemia harboring MLL translocations have highly unfavorable prognoses with current treatment (7).
Emerging findings from a number of groups suggest that the common MLL fusions, including MLL-AF4, MLL-AF9, and MLL-ENL, use a similar strategy for leukemic transformation. This involves recruitment of the histone methyltransferase, DOT1L (disruptor of telomeric silencing 1-like), an enzyme that lacks the canonical SET domain (Su(var)3–9, Enhancer of Zeste, and Trithorax) and is solely responsible for catalyzing the methylation of histone 3 at lysine 79 (H3K79) (8–11). Several multiprotein complexes involved in transcriptional activation/elongation were independently identified and studied, showing that all of them contain MLL fusion proteins together with DOT1L and/or p-TEFb (a complex of cyclinT and CDK9, which phosphorylates RNA polymerase II) (8–14). It was reported that the MLL translocation partners AF4, AF9, AF5q31, and ENL interact in a complex named ENL-associated protein complex, which in addition to the core translocation partners contains the p-TEFb together with the DOT1L (10). These findings were modified by several following reported studies describing similar complexes associated with DOT1L and p-TEFb as “effector units” but importantly not both at the same time. For example, it was demonstrated that ENL exists in two distinct complexes: one with DOT1L and one within an endogenous higher ordered complex (designated AEP) in which ENL associates with AF4, AF5q31, and p-TEFb (12). In this model, ENL has dual roles and interacts with AEP and DOT1L recruiting them sequentially to the same target chromatin. This study showed that the AEP complex is required for sustained transcription of target genes and transformation of hematopoietic progenitors, whereas the recruitment of DOT1L by MLL-ENL fusion protein plays a role in the maintenance of transcriptional memory. The C-terminal AF9 domain in MLL-AF9 was also shown to form distinct higher order complexes through direct associations with AF4/p-TEFb and with DOT1L (13). Notably, MLL-AF9-mediated target gene (HOXA 9) expression and cell transformation depend on these protein-protein interactions (PPI) and formation of the higher order complexes. These findings that DOT1L associates with MLL fusion proteins in several reported complexes suggested that aberrant H3K79 methylation might be a shared mechanism of oncogenic transcriptional activation in MLL rearrangement leukemia. It is known that the recruitment of DOT1L results in hypermethylation of H3K79 on the prominent MLL fusion downstream target loci Hoxa9 and Meis1 (15). Genome-wide analysis revealed a distinct pattern of H3K79 methylation in human MLL-rearranged primary leukemia samples compared with normal proB cells and leukemia with other abnormalities (16). Transient knockdown or conditional knock-out mice models have demonstrated that DOT1L is required for MLL fusion-mediated leukemic transformation and in vivo leukemia development and maintenance, indicating that there is a strong functional interconnection between complexes formed by MLL fusion proteins and DOT1L (9, 17–20). These findings illustrate the central role of the DOT1L recruitment and H3K79 methylation in leukemogenesis by controlling transcription of hematopoietic genes and implicate PPI between DOT1L and MLL oncogenic fusion proteins as a potential therapeutic target.
To this end, we characterized the AF9/ENL-DOT1L interaction at the biochemical, biophysical, and functional levels. Binding studies demonstrate that only 10 amino acids in DOT1L are essential for the interaction and recruitment of DOT1L by MLL fusion proteins, AF9 and ENL. Importantly, the functional studies show that this interaction is required for transformation by MLL-AF9. These results strongly suggest that disruption of this PPI represents a promising therapeutic strategy for MLL fusion protein-associated leukemia.
EXPERIMENTAL PROCEDURES
Plasmids and Cloning
The full-length hDOT1L was generously provided by Dr. Yi Zhang (University of North Carolina at Chapel Hill). Different DOT1L plasmids, tested in this study (see Table 1), were constructed using full-length hDOT1L as a template. The plasmids for AF9, ENL, and AF4 were made using MLL-AF9, MLL-ENL, and MLL-AF4 fusion proteins as templates. The obtained constructs for protein expression were cloned by ligation-independent cloning (LIC) methods as described before (21). Different DOT1L constructs (see Table 1) and AF4 protein (amino acids 749–775) were cloned into pMocr-LIC vector. ENL (amino acids 489–559) was cloned into pMSCG9-LIC vector, and AF9 (amino acids 497–568) was cloned into pGB1-LIC vector. MSCV-based HA-tagged wild type mouse DOT1L, methyltransferase inactive full-length mouse DOT1L with following mutations, G163R, S164C, and G165R, designated as mDOT1L (RCR), and neomycin vector constructs have been reported before (18). In the MSCV-HA-mDOT1L 10aaΔ construct, 10 residues identified as the AF9/ENL interaction site (amino acids 863–872) were deleted by using the QuikChange site-directed mutagenesis kit (Agilent Technologies). These residues in mouse are conserved and correspond to the identified human DOT1L interaction site, amino acids 865–874 (see Fig. 2B). To construct the pCMV-Myc CXXC-AF9 plasmid, a fragment of an MLL-AF9 fusion protein was cut from an MLL-AF9 fusion protein expression vector using MfeI and XhoI and inserted into the pCMV-Myc vector (Clontech) following digestion with EcoR1 and XhoI. A dual nuclear localization signal was inserted into the SfiI site downstream of the Myc tag. The sequence of MLL includes amino acids 1116–1422 and is followed by AF9 sequence including amino acids 479–568. The positive clones for all desired constructs were confirmed by DNA sequencing (University of Michigan DNA sequencing core).
TABLE 1.
Binding affinities of AF9 and ENL proteins to full-length DOT1L and different constructs of DOT1L immobilized on a CM5 sensor chip and determined by SPR
| Recombinant immobilized proteins (residues) | AF9 (497–568) KD ± S.D. | ENL (489–559) KD ± S.D. |
|---|---|---|
| nm | nm | |
| DOT1L (1–1537)a | 33 ± 4 | 206 ± 80 |
| DOT1L (826–1095) | 111 ± 10 | 238 ± 72 |
| DOT1L (826–958) | 92 ± 8 | 122 ± 16 |
| DOT1L (826–937) | 78 ± 15 | 81 ± 6 |
| DOT1L (854–1095) | 135 ± 4 | 148 ± 14 |
| DOT1L (925–1095) | 1,825 ± 276 | 6,220 ± 113 |
| DOT1L (854–925) | 42 ± 2 | 90 ± 12 |
a FLAG-tagged full-length human DOT1L protein.
FIGURE 2.

Mapping the AF9/ENL-binding site in DOT1L protein. A, alignment of human DOT1L (865–880) with human AF4 (761–774). The conserved residues are marked with asterisks, and the similar amino acids are marked with dots. B, alignment of the identified AF9/ENL-binding site in DOT1L protein from different species. C, SPR competitive binding curves of DOT1L 16-, 10-, and 7-mer and AF4 14-mer peptides against AF9 (497–568) and ENL (489–559) proteins running different tested concentrations over CM5 chip with immobilized DOT1L (826–1095). D, binding affinity of fluorescent-labeled DOT1L 10-mer peptide against AF9 and ENL. E, ITC of MBP-ENL (489–559) (80 μm) with a solution of DOT1L 16-mer (400 μm) and 10-mer (400 μm). For DOT1L 7-mer peptide (500 μm), 110 μm MBP-ENL was used. For all titrations, the raw data are shown in the upper panels, and the integrated heat data are shown in the lower panels. No binding was observed when the DOT1L peptides were tested against the MBP tag only. F, FP competitive binding curves of AF4 (749–775) recombinant protein and AF4 14-mer peptide using fluorescein-labeled DOT1L 10-mer peptide. mP, milli-polarization units. RU, response units.
Protein Expression and Purification
All recombinant proteins were expressed in Escherichia coli strain BL21 (DE3) (Invitrogen). The proteins were induced with 200 μm isopropyl β-d-1-thiogalactopyranoside at 20 °C except GB1-AF9 (497–568), which was induced at 16 °C. The cells were harvested after 20 h and resuspended in cold lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.01% β-mercaptoethanol) and purified by affinity chromatography employing nickel-agarose (Qiagen). Mocr-DOT1L and -AF4 proteins were further purified by ion exchange chromatography in 25 mm Tris-HCl, pH 7.5, with NaCl gradient ranging from 25 mm to 1 m, and 3 mm DTT, whereas MBP-ENL (489–559) and GB1-AF9 (497–568) proteins were purified with size exclusion chromatography in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 3 mm DTT. The MBP and GB1 tags from ENL (489–559) and AF9 (497–568), respectively, were removed by proteolysis using tobacco etch virus protease, followed by nickel column and size exclusion chromatography using the same buffer as above to obtain purified cleaved proteins. Purified recombinant proteins were stored at −80 °C for further experiments.
Peptide Synthesis
Peptides were synthesized using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) solid phase peptide synthesis techniques on an ABI 433A automated peptide synthesizer. NovaPEG Rink amide resin (EMD) was used to prepare all C-terminal amide-capped peptides. Standard side chain protecting groups were used for all amino acids. All the peptides were acetylated on the N terminus. Peptides containing an N-terminal biotin group or fluorescein are coupled to lysine and two β-alanine residues as a spacer. All crude peptides were purified by semipreparative reverse phase HPLC, and their sequence and purity were verified by electrospray ionization mass spectrometry and analytical reverse phase HPLC.
Surface Plasmon Resonance (SPR) Binding Studies
All SPR-based experiments were performed on a BIAcore 2000 (GE Healthcare) instrument. Different tested DOT1L recombinant proteins were immobilized on a CM5 sensor chip by standard 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide coupling chemistry followed by ethanolamine deactivation of the surfaces. For immobilization of full-length FLAG-DOT1L protein, anti-FLAG antibody (anti-FLAG M2; Sigma) was immobilized on the CM5 chip by the amine coupling chemistry. HEK293 whole cell lysate with overexpressed FLAG-DOT1L at 2.4 μg/μl total protein concentration was injected over the FLAG antibody surface giving ∼4000 response units immobilization level. For determination of AF9 and ENL binding affinity, the proteins were tested at different concentrations (0.01–3 μm) using HBS-P as a running buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 0.005% (v/v) P20; GE Healthcare) and 20 mm NaOH as the regeneration buffer. The Fc1 surface was used as a control surface and was treated in the same manner as the active surfaces but in the absence of immobilized protein. Binding parameters kon, koff, and KD were calculated by simultaneous nonlinear regression using BIAEvaluation software.
Solution competitive SPR-based assay was performed to determine the IC50 values of DOT1L peptides. The tested DOT1L wild type and alanine-mutated peptides were preincubated with AF9 or ENL proteins (500 nm) for at least 30 min, and then the reaction mixture was injected over the surface of Mocr-DOT1L (826–1095) immobilized CM5 chip. Response units were measured at 15 s in the dissociation phase, and the specific binding was calculated by subtracting the control surface (Fc1) signal from the surfaces with immobilized Mocr-DOT1L. IC50 values were determined by nonlinear least squares analysis using Graph Pad Prism 5.0 software.
Fluorescence Polarization (FP) Assay
FP experiments were performed in 96-well, black, round-bottomed plates (Corning) and read using a Biotek H1 hybrid plate reader. Fluorescein-tagged DOT1L 10-mer peptide (Flu-DOT1L) was used as a fluorescent probe in the FP-based binding assays. The KD values of Flu-DOT1L tracer with GB1-AF9 and MBP-ENL proteins were determined using a fixed concentration of the probe (10 nm Flu-DOT1L) and serial dilutions of tested proteins (0.01–5 μm), in assay buffer (100 mm NaH2PO4, pH 7.4, 150 mm NaCl, 0.01% Triton X-100) with 4% Me2SO to produce a total reaction volume of 125 μl. The plate was mixed on a shaker and incubated at room temperature to reach equilibrium. The polarization values in milli-polarization units were measured at an excitation wavelength at 485 nm and an emission wavelength at 530 nm. Polarization data were analyzed using GraphPad Prism 5.0 by nonlinear fitting with a one-site binding model.
Competitive FP binding experiments were carried out in 96-well plates with serial dilutions of tested protein or peptides, and fixed concentration of GB1-AF9 protein (500 nm) and Flu-DOT1L (10 nm) in the same assay buffer and final volume of 125 μl. The polarization values were measured after 3 h of incubation. Negative controls containing GB1-AF9 protein and probe (equivalent to 0% inhibition) and positive controls containing only free Flu-DOT1L probe (equivalent to 100% inhibition) were included on each assay plate. IC50 values were determined by nonlinear regression fitting of the competition curves (GraphPad Prism 5.0).
Isothermal Titration Calorimetry
Isothermal titration calorimetry (ITC) was carried out using a Nano-ITC Micro Calorimeter (TA Instruments) at 20 °C. MBP-ENL (489–559) was dialyzed against 50 mm Na2HPO4 (pH 7.5) and 100 mm NaCl. 80 μm of MBP-ENL was used for titration studies of DOT1L 16-mer and 10-mer peptides, and 110 μm was used for DOT1L 7-mer titration. DOT1L 16-mer and 10-mer peptides (400 μm) and DOT1L 7-mer (500 μm) dissolved in the same buffer were tested by injecting 2-μl aliquots into the protein sample, at time intervals of 30 s, to ensure that the titration peak returned to the base line. The ITC data were analyzed by NanoAnalyze software package using a one-site binding model.
Circular Dichroism
DOT1L peptides were dissolved in phosphate buffer (50 mm Na2HPO4, pH 7.4, 100 mm NaCl). CD measurements were performed at room temperature using a Jasco J-715 and a quartz flow cell with a 1-mm path length. The spectra were averaged from 10 scans, and the base line (buffer scan) was subtracted from each spectrum.
Homology Modeling
The solution NMR structure of the C-terminal hydrophobic domain of AF9 in complex with the elongation factor AF4 (Protein Data Bank code 2LM0) (22) was employed for the homology modeling of the complex between ENL and DOT1L. The reported NMR structure contains 10 conformers, and the fourth was randomly selected as the template because the alignment of these conformers showed that they are virtually identical except the very flexible loops, which have not been employed for the building of the homology model of ENL-DOT1L complex. For the modeling purpose, the residues 497–568 from AF9 and 761–767 from AF4 were used, and the remaining residues in the flexible loops were deleted. This simplified structure was minimized using the program of LigX of MOE 2010.10 (Chemical Computing Group Inc.). The corresponding sequence of ENL (489–559) was aligned with this minimized AF9-AF4 complex using the MOE-align. The homology model was built using the homology model module of MOE 2010.10. During this process, the AF4 peptide, LMVKITL, was kept as environment for induced fit. In the top-scored model produce by MOE, the LMVKITL peptide was mutated to DOT1L 7-mer peptide, LPISIPL, and the obtained homology model was minimized using the program of LigX of MOE 2010.10.
Cell Culture, Transfections, and Immunoprecipitation
HEK293 cells were plated in 100-mm culture dishes and cultured in DMEM supplemented with 10% FBS and antibiotics. Co-transfection with FLAG-DOT1L and Myc-CXXC-AF9 was performed using Lipofectamine 2000 (Invitrogen). Forty-eight hours post-transfection, cells were collected and lysed using BC-300 lysis buffer (20 mm Tris-HCl, pH 8.0, 300 mm KCl, 1 mm EDTA, 10% glycerol, 0.1% Nonidet P-40, and protease inhibitor mixture; Roche Applied Science). The lysates was precleared for 2 h in mouse IgG-agarose (Sigma-Aldrich). The precleared lysates were incubated with different concentrations of DOT1L 10-mer peptide at 4 °C overnight. The next day, the cell lysates were immunoprecipitated with anti-FLAG M2 magnetic beads (Sigma-Aldrich) at 4 °C for 2 h. After incubation, the beads were washed extensively, boiled in SDS loading buffer, and analyzed by Western blotting using mouse monoclonal anti-FLAG M2 (Sigma-Aldrich) and goat monoclonal anti-Myc tag (Abcam) antibody.
Pulldown Assay
HEK293 cells transfected with Myc-CXXC-AF9 were lysed in BC-300 lysis buffer in the same way as the immunoprecipitation experiment. The supernatant was precleared for 2 h in streptavidin-agarose beads (Thermal Scientific). The precleared lysates were incubated with different concentrations of biotin-labeled DOT1L 10-mer peptide at 4 °C overnight. The next day, the cell lysate were incubated with streptavidin-agarose at 4 °C for 2 h. After extensive washing the beads with the BC-300 lysis buffer without Nonidet P-40, the pulldown samples were applied to SDS-PAGE, and pulldown Myc-CXXC-AF9 protein was probed with goat monoclonal anti-Myc tag antibody (Abcam).
Retroviral Transductions and Colony-forming Unit (CFU) Assay
Retroviral production and transduction of bone marrow progenitor cells were carried out as described (18). Briefly, retroviruses were generated by transfecting MSCV-HA wild type mouse DOT1L, methyltransferase mutant mDOT1L (RCR), mDOT1L deletion (10aaΔ; deletion of the residues 863–872 amino acids) constructs, and neomycin vector control into Plat-E cell line with FuGENE 6 (Roche Applied Science). Fresh viral supernatants were used for transducing MLL-AF9 transformed cells described (18). 150,000 cells were used for transduction per viral supernatant from a 10-cm dish. The cells were then plated on Methocult medium (M3234; Stem Cell Technologies) with 1% penicillin/streptomycin (Invitrogen), 10 ng/ml IL3 (R&D Systems), 5 nm 4-hydroxytamoxifen (Sigma) or 100% ethanol, and 1 mg/ml G418 (Invitrogen). Colonies were scored 5–7 days after plating for two rounds. In the final round, colonies were stained with 0.1% p-iodonitrotetrazolium violet (Sigma) for visualization.
RNA Extraction, cDNA Generation, and Protein Extraction
RNA was extracted from cells using TRIzol reagent (Invitrogen) and converted to cDNA using SuperScript II (Invitrogen) according to the manufacturer's instructions. Whole cell lysate samples were prepared by directly resuspending cells in Tris-glycine SDS sample buffer (Novex) and sonicating for 15 min (Bioruptor, Diagenode). Primers used for the quantitative PCR: 5 S rRNA, TCTACGGCCATACCACCCTGA and GCCTACAGCACCCGGTATTCC; and HA-Dot1l, GCCACCATGTACCCCTACGACGTG and GATTTCCTCGCAGACCCACCGGAT.
RESULTS
Mapping the AF9/ENL-Binding Site in DOT1L
AF9 and ENL are two of the most common MLL fusion proteins (2) and belong to the YEATS domain protein family (23). Both of these two proteins consist of N-terminal YEATS domain and a C-terminal hydrophobic domain (24). AF9 and ENL shared high homology in the C-terminal domain. Previously it has been shown that this domain of AF9/ENL is involved in the PPI that is crucial for MLL leukemic transformation (10, 12, 25, 26). The interaction of AF9 or ENL with DOT1L has been reported previously by co-immunoprecipitation and yeast two-hybrid studies (10, 12, 13, 24, 27). However, there is a lack of detailed biochemical and biophysical characterization of this interaction. To understand the molecular basis of DOT1L interaction with MLL fusion proteins, we analyzed these PPI using surface plasmon resonance. We developed SPR assay to quantify the binding affinity between full-length DOT1L protein and MLL fusion proteins. For that purpose, the FLAG-tagged full-length DOT1L protein was transiently transfected in HEK293 cells, and after 48 h of transfection, the DOT1L protein was captured from the HEK293 whole cell lysate on an anti-FLAG antibody-coated biosensor chip. The recombinant C-terminal domain from human AF9 (residues 497–568) and the corresponding segment from ENL protein (residues 489–559) were expressed and purified for the biochemical binding studies (Fig. 1A). Using this system, we determined that AF9 and ENL bind to the immobilized full-length DOT1L with a dissociation constant (KD) of 33 and 206 nm, respectively, agreeing well with a 1:1 interaction model (Fig. 1B). These studies confirm that DOT1L directly interacts with AF9 and ENL and for the first time the binding affinity of DOT1L to AF9 and ENL was quantitatively determined.
FIGURE 1.

Interaction between DOT1L and MLL fusion proteins, AF9 and ENL, measured by surface plasmon resonance. A, schematic presentation of AF9/ENL and DOT1L proteins used for the binding studies. B and C, sensorgrams representing the concentration-dependent binding of the C-terminal domain of AF9 and ENL tested in concentration range from 0.01 to 3 μm, with full-length FLAG-DOT1L (B) and Mocr-DOT1L (826–1095) (C), both immobilized on a CM5 sensor chip. The kon, koff, and KD were calculated by simultaneous nonlinear regression using 1:1 binding model and BIAevaluation 3.1 software. The experimental data are shown in black, whereas the global fit analyses are shown in red. RU, response units.
The C-terminal unstructured region of DOT1L has been reported to be involved in the interaction with AF9 and ENL (10, 12). Therefore we cloned and expressed the DOT1L 826–1095 fragment and studied the interaction of this recombinant protein with AF9/ENL. SPR analysis showed that this segment of the DOT1L protein has KD values of 111 and 238 nm to AF9 and ENL protein, respectively (Fig. 1C), confirming that this region in DOT1L protein is essential for interactions with AF9/ENL proteins.
To further map the region of DOT1L required for the AF9/ENL interaction, a series of truncated constructs of DOT1L, devised according to predicted stability, were tested for their binding to AF9 and ENL (Fig. 1A and Table 1). The binding studies show that truncations of the DOT1L protein on the N-terminal site up to 854 residues and C-terminal deletions up to 925 residues did not affect the interactions with AF9 and ENL. The corresponding DOT1L fragment (amino acids 854–925) binds to AF9 and ENL with KD values of 42 and 90 nm, respectively, similar to the full-length DOT1L protein (Table 1). These results confirm that this segment is crucial for interactions with MLL fusion proteins AF9 and ENL.
Identification of a Conserved Peptide Motif in DOT1L Essential for Binding AF9/ENL
It is known that the C-terminal hydrophobic AF9/ENL domains in MLL fusions retain the ability to form independently higher order complexes with AF4/p-TEFb and with DOT1L, demonstrating that the associations of AF9/ENL with AF4 and DOT1L are mutually exclusive (12, 13). The AF9-binding domain of AF4 is known and is conserved among the AF4 homologs (28). Based on these findings, we predicted that the AF9/ENL interacting site in DOT1L might share certain similarity to AF4 protein. By aligning the shortest fragment of DOT1L (854–925) involved in AF9/ENL interactions, with the AF9-binding domain of AF4 (761–774), a small 16-residue region in DOT1L (865–880) was identified. This region shares sequence homology with AF4, consisting of four conserved and four similar residues (Fig. 2A). Importantly, alignment of DOT1L from different species demonstrate that this fragment is highly conserved (Fig. 2B). Two peptides, DOT1L-10-mer (865–874) and DOT1L-16-mer (865–880), were synthesized and tested for their binding to AF9 and ENL. The competitive SPR assay demonstrates that both peptides can block the PPI between DOT1L/AF9 and DOT1L/ENL with IC50 values of 490 and 320 nm, respectively, against AF9, and 1,340 nm and 1,560 nm, respectively, against ENL (Fig. 2C). The fluorescent labeled DOT1L 10-mer peptide binds AF9 and ENL with KD values of 65 and 495 nm, respectively (Fig. 2D). The binding affinity to ENL was further confirmed using ITC assay (Fig. 2E). Both peptides, DOT1L 16- and 10-mer, bind to the ENL protein with KD of 900 and 1,100 nm, respectively, in 1:1 stoichiometry, consistent with FP-based results. The obtained binding results for DOT1L 16- and 10-mer peptides provide strong evidence that the identified peptide motif in DOT1L is both required and sufficient for interaction with AF9 and ENL.
Furthermore, we expressed and purified AF4 recombinant protein (residues 749–775), synthesized the AF4 14-mer peptide (residues 761–774) (28), and tested whether AF4 and DOT1L compete for the same conserved C-terminal domain of AF9 and ENL. As was expected, the recombinant AF4 protein and AF4 14-mer peptide efficiently competed away the binding of DOT1L 10-mer fluorescein-labeled peptide to the C-terminal domain of AF9 protein with IC50 values of 560 and 280 nm (Fig. 2F). The SPR solution competitive binding assay confirmed these results and showed that the AF4 14-mer peptide is inhibiting the PPI between AF9-DOT1L and ENL-DOT1L, with similar potency as DOT1L 16- and 10-mer peptides, showing IC50 values of 200 and 430 nm against AF9 and ENL, respectively (Fig. 2C). These findings clearly demonstrate that DOT1L and AF4 bind with similar binding affinity and compete for the same AF9/ENL interaction site, the C-terminal hydrophobic domain.
Alanine-scanning Mutagenesis Studies of the Interaction between DOT1L 10-mer Peptide and MLL Fusion Proteins
Systematic alanine mutagenesis studies of the identified AF9/ENL-binding site in DOT1L were performed to test the contribution of each residue for the interactions with MLL fusion proteins. DOT1L residues from Leu865 to Val874 were mutated to alanine, thus creating 10 different DOT1L mutants, and determined their potency to inhibit the PPI in SPR-based competitive assay (Table 2 and Fig. 3A). The three conserved residues in DOT1L, Leu865, Ile869, and Leu871, as well as the similar residue, Ile867, failed to tolerate alanine substitution, with a significant decrease in binding affinity to both AF9 and ENL proteins. These results demonstrate the importance of the hydrophobic interactions, which is consistent with the notion of a conserved C-terminal hydrophobic domain in both AF9 and ENL. The unconserved residues, Pro866, Ser868, and Pro870 had from 10- to 40-fold reductions in their ability to inhibit the PPI. Interestingly, mutation of the last three residues in the DOT1L 10-mer peptide, Ser872, Thr873, and Val874 was well tolerated and showed only a 2–6-fold decrease in the binding affinity compared with the wild type peptide. We then synthesized wild type 7-mer DOT1L peptide (865–871) and tested its binding to AF9 and ENL. Consistent with the mutation data, this peptide showed 8-fold reduction in the binding to AF9 and 5-fold reduction in binding affinity to ENL protein in comparison with the 10-mer peptide (Figs. 2C and 3A). The binding affinity of DOT1L 7-mer peptide to ENL protein was confirmed in ITC assay showing KD of 3,400 nm, 3-fold lower in comparison with the 16- and 10-mer DOT1L peptides (Fig. 2E). Therefore, the alanine scanning mutagenesis studies indicate that the heptapeptide binding motif of DOT1L is essential for interactions with MLL fusion proteins and is the minimum required fragment. In addition, the obtained results show a similar pattern in the binding of all tested peptides against AF9 and ENL proteins, suggesting that they share similar structural requirements for this interaction, and a single amino acid mutation of the hydrophobic conserved residues is sufficient to disrupt the PPI. We also investigated the role of the secondary structure of DOT1L peptides to further understand the interactions between DOT1L peptide and MLL fusion proteins. For this purpose, CD experiments were performed, and DOT1L 16- and 10-mer peptides were tested. The obtained results showed that both peptides have unordered (random-coil) secondary structure (Fig. 3B). Overall, these findings provide a rationale toward future efforts in identifying chemical probes that can disrupt the interaction between DOT1L and the MLL fusion proteins.
TABLE 2.
Peptide sequence and IC50 values of wild type and alanine-mutated DOT1L peptides against AF9 and ENL proteins obtained by competitive SPR-based assay using CM5 chip with immobilized DOT1L protein
| Peptide | Sequence | IC50 ± S.D. |
|
|---|---|---|---|
| AF9 | ENL | ||
| μm | |||
| DOT1L 10-mer | Ac-LPISIPLSTV-NH2 (amino acids 865–874) | 0.49 ± 0.22 | 1.34 ± 0.34 |
| L865A | Ac-APISIPLSTV-NH2 | 98.5 ± 15.1 | 27.5 ± 6.8 |
| P866A | Ac-LAISIPLSTV-NH2 | 3.3 ± 0.6 | 6.5 ± 1.2 |
| I867A | Ac-LPASIPLSTV-NH2 | 57.2 ± 17.3 | >200 |
| S868A | Ac-LPIAIPLSTV-NH2 | 5.3 ± 1.7 | 29.1 ± 0.8 |
| I869A | Ac-LPISAPLSTV-NH2 | >200 | >200 |
| P870A | Ac-LPISIALSTV-NH2 | 2.8 ± 0.1 | 11.6 ± 1.7 |
| L871A | Ac-LPISIPASTV-NH2 | 30.7 ± 10.3 | 146.8 ± 11.5 |
| S872A | Ac-LPISIPLATV-NH2 | 0.55 ± 0.21 | 2.8 ± 0.6 |
| T873A | Ac-LPISIPLSAV-NH2 | 0.058 ± 0.017 | 0.36 ± 0.02 |
| V874A | Ac-LPISIPLSTA-NH2 | 0.7 ± 0.02 | 4.2 ± 0.6 |
| DOT1L 7-mer | Ac-LPISIPL-NH2 | 3.9 ± 0.1 | 7.3 ± 0.3 |
| DOT1L 16-mer | Ac-LPISIPLSTVQPNKLP-NH2 (amino acids 865–880) | 0.32 ± 0.01 | 1.56 ± 0.09 |
| AF4 14-mer | Ac- LMVKITLDLLSRIP-NH2 (amino acids 760–773) | 0.20 ± 0.06 | 0.43 ± 0.15 |
FIGURE 3.

Characterizing the DOT1L 10-mer peptide and its interactions with MLL fusion proteins. A, binding affinities of alanine mutated DOT1L 10-mer peptides to AF9 and ENL in comparison to the wild type DOT1L 10-mer peptide. B, circular dichroism spectra of DOT1L 16- and 10-mer peptides.
Interactions of DOT1L with the C-terminal Domain of AF9 and ENL
Solution NMR structure of AF9-AF4 complex determined that the C-terminal hydrophobic domain in AF9 has three conserved helical segments (Fig. 4A) (22). Deleting these helical regions completely abrogated the transforming activity of MLL-AF9 and MLL-ENL fusion proteins (26). To determine whether the helical regions are important for the binding of these two MLL fusion proteins to DOT1L, we prepared two recombinant fragments of ENL protein, ENL (489–544) consisting of helices 1 and 2, and ENL (523–559), which has only helix 3. It was determined that ENL (489–544) binds to DOT1L (826–1095) with KD of 2,800 nm and ENL (523–559) binds with KD of 14,567 nm (Fig. 4B), demonstrating 10- and 30-fold decreased binding affinity respectively in comparison with the intact ENL C-terminal domain (Table 1). These results clearly demonstrate that the entire C-terminal domain from ENL is required for optimal interaction with DOT1L, which is consistent with the reports that helical segments are essential for the transformation potential of MLL-ENL fusion protein.
FIGURE 4.

Analyzing the C-terminal domain in ENL protein and its binding to DOT1L. A, alignment of ENL and AF9 C-terminal domain. The three α-helices are indicated, and conserved residues are marked with asterisks. B, binding sensorgrams of immobilized Mocr-DOT1L (826–1095) with ENL (489–544) and ENL (523–559), tested in concentration ranges from 1 to 16 μm and from 5 to 35 μm, respectively. The kon, koff, and KD were calculated by simultaneous nonlinear regression using 1:1 binding model and BIAevaluation 3.1 software. The experimental data are shown in black, whereas the global fit analyses are shown in red. RU, response units.
Homology Modeling of DOT1L-ENL Complex
Recently the structure of AF9-AF4 complex was determined using NMR spectroscopy (Protein Data Bank code 2LM0) (22). Knowing that AF9 and ENL are highly homologous proteins and to take advantage of the reported structure, we performed template (AF9-AF4)-based homology modeling approach of generating target, ENL, in complex with DOT1L 7-mer peptide.
The accuracy of the structures generated by homology modeling is highly dependent on the sequence identity between the target and the template protein. The C-terminal hydrophobic domain of AF9 (497–568), which was used as a template for modeling the ENL domain (489–559), shared high level of identity with 79.2% (Fig. 4A). Thus the generated homology model is accurate and reliable and can be used for structure-based studies. Furthermore, the alignment of the AF4 template peptide (761–767) and DOT1L target peptide (865–871) (Fig. 2A) shows that they also share sequence homology with three conserved and one similar residues. These residues were determined as essential for the PPI between ENL and DOT1L (Fig. 3). The quality of the obtained model was further confirmed with quantitative comparison between the template and target complex structures and the estimated root mean square deviation of 0.32 Å. The generated homology model showed that the peptide docking region cleft in ENL is extensively hydrophobic (Fig. 5A). All residues except Ser868 of the DOT1L peptide, LPISIPL, have hydrophobic interactions with the protein, especially Ile867 and Ile869, which are buried in the two deep subpockets of the ENL hydrophobic core. This model is consistent and supported by our alanine mutagenesis studies, which demonstrated that mutation of hydrophobic residues to alanine abolished the binding of DOT1L peptide to ENL protein (Fig. 3A). ENL-DOT1L complex involves five hydrogen bonds: two between ENL Phe537 and Ile867 from DOT1L peptide, two between ENL Phe535 and Ile869 DOT1L, and one between ENL His503 and Ser868 DOT1L (Fig. 5B).
FIGURE 5.
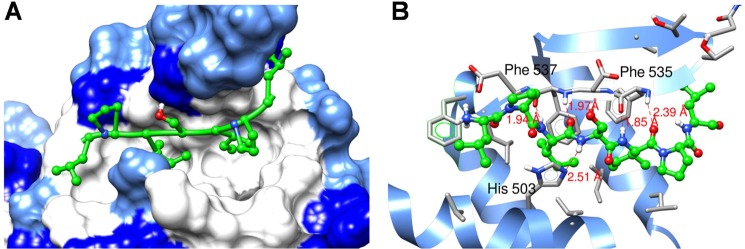
Homology modeling of the ENL-DOT1L complex. A, molecular surface representation of the ENL-DOT1L complex. ENL surface is colored according to electrostatic potential: charged residues (dark blue), polar residues (light blue), and hydrophobic (white). The Cα trace of DOT1L peptide, LPISIPL, is shown as a green ribbon together with the side chains of the residues. B, five hydrogen bonds can be found between the complex of ENL-DOT1L shown with red dashed lines. DOT1L peptide is presented as a ball and stick model.
This model will allow further understanding of this PPI on the structural level, gaining knowledge about other PPI that AF9 and ENL are involved, as well as critical insights toward developing peptidomimetics as one of the known strategies for targeting PPI.
DOT1L 10-mer Peptide Disrupts in Vivo Interactions between Full-length DOT1L and MLL-AF9 Fusion Protein
To assess whether DOT1L 10-mer peptide can bind and block the interaction between full-length DOT1L and MLL fusion proteins, we have transiently co-transfected HEK293 cells with FLAG-tagged human DOT1L protein and Myc-tagged CXXC-AF9 protein. The whole cell lysate was preincubated with different concentrations of DOT1L 10-mer peptide, and co-immunoprecipitation experiments were performed. The obtained results demonstrate that the DOT1L 10-mer peptide blocks the interaction of cellular DOT1L with CXXC-AF9 in a dose-dependent manner (Fig. 6A). To further probe and confirm the cellular target of DOT1L 10-mer peptide, we have synthesized a biotinylated DOT1L 10-mer peptide. Binding studies demonstrate that this peptide has similar binding affinity as corresponding unlabeled DOT1L 10-mer peptide, confirming that biotin labeling did not affect the binding to AF9 protein. Using this peptide as a tool, we performed streptavidin-biotin pulldown experiments in the HEK293 cell lysates and showed that the biotinylated DOT1L 10-mer peptide recognizes and binds to the cellular CXXC-AF9 protein in a dose-dependent manner (Fig. 6B). Together these experiments demonstrate that DOT1L 10-mer peptide binds to the cellular AF9 protein and blocks its interaction with DOT1L, consistent with our in vitro binding data using recombinant MLL fusion proteins.
FIGURE 6.
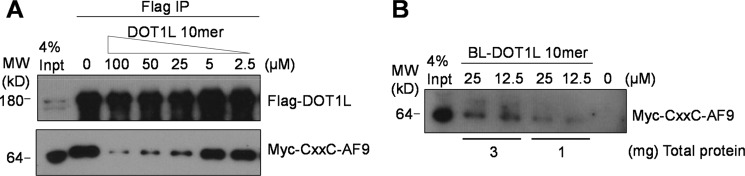
DOT1L 10-mer peptide binds cellular MLL-AF9 fusion protein and disrupts its interaction with DOT1L. A, DOT1L 10-mer peptide disrupts the interaction between DOT1L and MLL-AF9 in cells. FLAG-DOT1L and Myc-CXXC-AF9 were co-transfected in HEK293 cells, and co-immunoprecipitation (IP) was performed. B, pulldown assay using biotin-labeled DOT1L 10-mer peptide. MW, molecular mass.
Identified 10 Residues Are Essential for DOT1L Recruitment and MLL-AF9 Leukemic Transformation
To assess further the functional importance of the recruitment of DOT1L by MLL fusion proteins and determine whether the identified 10-residue segment of DOT1L is required for MLL-AF9 transformation, we performed CFU assays. In these experiments MSCV-based vectors were used to transduce mouse bone marrow cells with leukemogenic MLL-AF9 fusion protein from floxed Dot1l mice generated as described previously (18). HA-tagged wild type mouse DOT1L, methyltransferase inactive full-length mDOT1L (RCR) with GSG to RCR mutation in the S-adenosylmethionine-binding domain lacking enzymatic activity (8, 29), full-length mDOT1L 10aaΔ lacking 10 amino acid AF9 interacting residues (residues 863–872, which are conserved and correspond to the human DOT1L 865–874; Fig. 2B), or neomycin vector control was introduced into endogenous DOT1L lacking MLL-AF9 transformed cells (Fig. 7A). Endogenous Dot1l excision was confirmed by PCR, and expression of exogenous DOT1L was confirmed by quantitative PCR of HA tag sequences (Fig. 7, B and C). As was expected, wild type mDOT1L and mDOT1L 10aaΔ with histone methyltransferase domain intact were able to restore H3K79 methylation when studied after the first round of plating, whereas mDOT1L (RCR) failed to restore H3K79 methylation (Fig. 7D). Consistent with the reported findings, the colony-forming potential of MLL-AF9-immortalized cells was completely abolished by introducing mDOT1L (RCR) construct after Dot1l deletion, whereas introduction of exogenous wild type DOT1L was able to rescue the transformation capability and restore CFU formation (Fig. 7, E and F). Importantly, mDOT1L 10aaΔ construct, despite being able to restore H3K79 methylation level in a similar way as the wild type DOT1L construct, failed to restore CFU formation (Fig. 7, E and F). These results strongly suggest that DOT1L interaction with MLL-AF9 and its recruitment are required for transformation by MLL-AF9, and full-length DOT1L lacking the AF9 interacting residues could not rescue CFU formation.
FIGURE 7.

AF9 binding site in DOT1L is essential for MLL-AF9 leukemic transformation. A, schematic presentation of the CFU assay. B, genotyping of transduced bone marrow cells. PCR showed high excision efficiency of endogenous DOT1L with 4-hydroxytamoxifen (4-OHT) treatment of all cells. C, quantitative PCR of exogenous DOT1L expression. All constructs showed expression compared with Neo vector alone. D, Western blot of H3K79me2 global level. Histone 3 blot was used as loading control. E and F, colony formation on methocult plates. Pictures of iodonitrotetrazolium chloride staining (E) and bar graph of colony counts (F) after second round.
DISCUSSION
In this work, we have identified and mapped the protein-protein interaction site between DOT1L and MLL fusion proteins, AF9 and ENL. Using SPR- and FP-based binding assays, ITC, immunoprecipitation, and pulldown experiments, we determined the binding affinity of these PPI and defined the AF9/ENL interaction site to the region corresponding to DOT1L 865LPISIPLSTV874. The importance of this binding motif is indicated by its conservation in different species. Our in vitro and in vivo findings demonstrated that the corresponding synthetic DOT1L 10-mer peptide can competitively interfere with the interaction between MLL fusion proteins, AF9 and ENL, and DOT1L. The alanine scanning mutagenesis studies showed the critical importance of several conserved residues, Leu865, Ile867, Ile869, and Leu871, for binding to AF9 and ENL. Binding studies confirmed a direct interaction between AF9/ENL and DOT1L and showed that for optimal interaction an intact C-terminal domain in MLL fusion proteins is critical. These binding results, together with reported functional studies, are further demonstrating a high correlation between the DOT1L recruitment and the transforming potential of MLL-ENL. Several groups have demonstrated that DOT1L is required for MLL-AF4 and MLL-AF9 fusion protein-mediated leukemic transformation (17–20, 30), establishing the crucial role of DOT1L in MLL-rearranged leukemia (31). Consistent with these findings, our functional studies using colony-forming unit assays provide strong evidence that the identified DOT1L865–874 fragment is essential for the DOT1L recruitment and MLL-AF9 leukemic transformation. The colony forming potential of the MLL-AF9 immortalized cells was completely abolished by introduction of a DOT1L construct lacking the 10-amino acid AF9 interacting residues. Taken together, in addition to H3K79 methylation, these results highlight the importance of the DOT1L recruitment and its interaction with MLL-AF9 for the transforming activity of this MLL fusion oncogene.
The recent development of a specific small molecule inhibitor of DOT1L, EPZ004777, which is a competitive inhibitor of the methyl donor S-adenosyl-methionine provide proof of principle for the development of DOT1L inhibitors as targeted therapeutics for MLL-rearranged leukemia (30). However, constitutive and conditional knock-out studies of DOT1L, which is the only known H3K79 methyltransferase, have shown it is essential for embryonic development, prenatal and postnatal hematopoiesis, and cardiac function (17, 18, 20, 32, 33). This universal and essential function of DOT1L in multiple cell types (34) suggests that directly inhibiting DOT1L histone methyltransferase activity might be toxic. Consequently, development of therapeutic strategies allowing selective inhibition of DOT1L function is important and necessary. In this study, we validated the PPI between DOT1L and AF9 or ENL, respectively, as a promising therapeutic target and potential strategy for pharmacological targeting of DOT1L. Our results indicate that disruption of AF9-DOT1L interaction abolishes MLL-AF9 leukemia transformation, without affecting the global level of H3K79 methylation level. More importantly, they also suggest that selective disruption of this PPI is a promising therapeutic strategy with potentially fewer adverse effects than enzymatic inhibition of DOT1L for MLL fusion protein-associated leukemia.
Furthermore, our binding studies clearly demonstrate that DOT1L and AF4 proteins, as well as the DOT1L 10-mer and AF4 14-mer peptides, bind to the same C-terminal hydrophobic domain of AF9 and ENL (Fig. 8A). These results are consistent with biochemical, structural, and functional analyses of protein complexes associated with MLL fusion proteins, which showed that AF9 and ENL exist in two mutually exclusive complexes, AF9/ENL-DOT1L and AF9/ENL-AF4-pTEFb (12, 13, 22). Based on our results, we propose that small molecules that bind the conserved hydrophobic domain of AF9 and ENL will abolish the AF9/ENL interactions with both AF4 and DOT1L, disrupting the higher order complex AEP and the complex with DOT1L and thereby blocking the MLL fusion-mediated leukemogenesis (Fig. 8B). Taken together, these results support the hypothesis that DOT1L recruitment is a potential therapeutic target in MLL-rearranged leukemia and forms the basis toward future studies on targeting these PPI.
FIGURE 8.
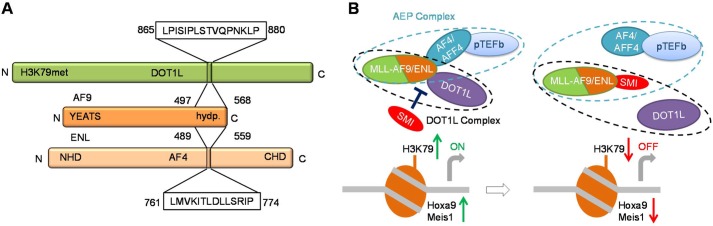
Schematic of proposed model for targeting DOT1L and MLL fusion protein-protein interactions. A, AF9/ENL-binding sites mapped in DOT1L and AF4 proteins. B, small molecule inhibitor (SMI) that binds to C-terminal domain of AF9/ENL will disrupt the MLL fusion protein complexes involved in mixed lineage leukemia, the DOT1L, and AEP complex.
Acknowledgments
We thank Dr. Y. Zhang (University of North Carolina) for providing us with the pcDNA3-FLAG-DOT1L plasmid. We are grateful to Dr. S. Wang, who generously allowed us to use the peptide synthesizer; Dr. J. Stuckey for providing us the access to the ITC; Dr. A. Gafni for the use of the dichroism spectrophotometer; and Dr. W. C. Brown for the help with DOT1L constructs (all from University of Michigan).
This work was supported, in whole or in part, by a National Institutes of Health grant (to J. L. H.) and the Ruth Kirschstein Award (to S. Y. J.). This work was also supported by funds from the Cancer Research Committee Fund of the University of Michigan Comprehensive Cancer Center (to Z. N. C.) and the Lymphoma and Leukemia Society (to J. L. H.).
- MLL
- mixed lineage leukemia
- PPI
- protein-protein interactions
- MBP
- maltose-binding protein
- LIC
- ligation-independent cloning
- MSCV
- murine stem cell virus
- SPR
- surface plasmon resonance
- FP
- fluorescence polarization
- ITC
- isothermal titration calorimetry
- CFU
- colony-forming unit.
REFERENCES
- 1. Krivtsov A. V., Armstrong S. A. (2007) MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 7, 823–833 [DOI] [PubMed] [Google Scholar]
- 2. Daser A., Rabbitts T. H. (2004) Extending the repertoire of the mixed-lineage leukemia gene MLL in leukemogenesis. Genes Dev. 18, 965–974 [DOI] [PubMed] [Google Scholar]
- 3. Eguchi M., Eguchi-Ishimae M., Greaves M. (2003) The role of the MLL gene in infant leukemia. Int. J. Hematol 78, 390–401 [DOI] [PubMed] [Google Scholar]
- 4. Ayton P. M., Cleary M. L. (2001) Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene 20, 5695–5707 [DOI] [PubMed] [Google Scholar]
- 5. Biondi A., Cimino G., Pieters R., Pui C. H. (2000) Biological and therapeutic aspects of infant leukemia. Blood 96, 24–33 [PubMed] [Google Scholar]
- 6. Huret J. L., Dessen P., Bernheim A. (2001) An atlas of chromosomes in hematological malignancies. Example. 11q23 and MLL partners. Leukemia 15, 987–989 [DOI] [PubMed] [Google Scholar]
- 7. Slany R. K. (2005) When epigenetics kills. MLL fusion proteins in leukemia. Hematol. Oncol. 23, 1–9 [DOI] [PubMed] [Google Scholar]
- 8. Okada Y., Feng Q., Lin Y., Jiang Q., Li Y., Coffield V. M., Su L., Xu G., Zhang Y. (2005) hDOT1L links histone methylation to leukemogenesis. Cell 121, 167–178 [DOI] [PubMed] [Google Scholar]
- 9. Zeisig D. T., Bittner C. B., Zeisig B. B., García-Cuéllar M. P., Hess J. L., Slany R. K. (2005) The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene 24, 5525–5532 [DOI] [PubMed] [Google Scholar]
- 10. Mueller D., Bach C., Zeisig D., Garcia-Cuellar M. P., Monroe S., Sreekumar A., Zhou R., Nesvizhskii A., Chinnaiyan A., Hess J. L., Slany R. K. (2007) A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 110, 4445–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bitoun E., Oliver P. L., Davies K. E. (2007) The mixed-lineage leukemia fusion partner AF4 stimulates RNA polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum. Mol. Genet. 16, 92–106 [DOI] [PubMed] [Google Scholar]
- 12. Yokoyama A., Lin M., Naresh A., Kitabayashi I., Cleary M. L. (2010) A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 17, 198–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biswas D., Milne T. A., Basrur V., Kim J., Elenitoba-Johnson K. S., Allis C. D., Roeder R. G. (2011) Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc. Natl. Acad. Sci. U.S.A. 108, 15751–15756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin C., Smith E. R., Takahashi H., Lai K. C., Martin-Brown S., Florens L., Washburn M. P., Conaway J. W., Conaway R. C., Shilatifard A. (2010) AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell 37, 429–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Milne T. A., Martin M. E., Brock H. W., Slany R. K., Hess J. L. (2005) Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 65, 11367–11374 [DOI] [PubMed] [Google Scholar]
- 16. Krivtsov A. V., Feng Z., Lemieux M. E., Faber J., Vempati S., Sinha A. U., Xia X., Jesneck J., Bracken A. P., Silverman L. B., Kutok J. L., Kung A. L., Armstrong S. A. (2008) H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14, 355–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bernt K. M., Zhu N., Sinha A. U., Vempati S., Faber J., Krivtsov A. V., Feng Z., Punt N., Daigle A., Bullinger L., Pollock R. M., Richon V. M., Kung A. L., Armstrong S. A. (2011) MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20, 66–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jo S. Y., Granowicz E. M., Maillard I., Thomas D., Hess J. L. (2011) Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood 117, 4759–4768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chang M. J., Wu H., Achille N. J., Reisenauer M. R., Chou C. W., Zeleznik-Le N. J., Hemenway C. S., Zhang W. (2010) Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by MLL oncogenes. Cancer Res. 70, 10234–10242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nguyen A. T., Taranova O., He J., Zhang Y. (2011) DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood 117, 6912–6922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. DelProposto J., Majmudar C. Y., Smith J. L., Brown W. C. (2009) Mocr. A novel fusion tag for enhancing solubility that is compatible with structural biology applications. Protein Expr. Purif. 63, 40–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leach B. I., Kuntimaddi A., Schmidt C. R., Cierpicki T., Johnson S. A., Bushweller J. H. (2013) Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure 21, 176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schulze J. M., Wang A. Y., Kobor M. S. (2009) YEATS domain proteins. A diverse family with many links to chromatin modification and transcription. Biochem. Cell Biol. 87, 65–75 [DOI] [PubMed] [Google Scholar]
- 24. Zhang W., Xia X., Reisenauer M. R., Rieg T., Lang F., Kuhl D., Vallon V., Kone B. C. (2007) Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel α. J. Clin. Invest. 117, 773–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dobson C. L., Warren A. J., Pannell R., Forster A., Lavenir I., Corral J., Smith A. J., Rabbitts T. H. (1999) The mll-AF9 gene fusion in mice controls myeloproliferation and specifies acute myeloid leukaemogenesis. EMBO J. 18, 3564–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Slany R. K., Lavau C., Cleary M. L. (1998) The oncogenic capacity of HRX-ENL requires the transcriptional transactivation activity of ENL and the DNA binding motifs of HRX. Mol. Cell Biol. 18, 122–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang W., Xia X., Reisenauer M. R., Hemenway C. S., Kone B. C. (2006) Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCα in an aldosterone-sensitive manner. J. Biol. Chem. 281, 18059–18068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Srinivasan R. S., Nesbit J. B., Marrero L., Erfurth F., LaRussa V. F., Hemenway C. S. (2004) The synthetic peptide PFWT disrupts AF4-AF9 protein complexes and induces apoptosis in t(4;11) leukemia cells. Leukemia 18, 1364–1372 [DOI] [PubMed] [Google Scholar]
- 29. Min J., Feng Q., Li Z., Zhang Y., Xu R. M. (2003) Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 112, 711–723 [DOI] [PubMed] [Google Scholar]
- 30. Daigle S. R., Olhava E. J., Therkelsen C. A., Majer C. R., Sneeringer C. J., Song J., Johnston L. D., Scott M. P., Smith J. J., Xiao Y., Jin L., Kuntz K. W., Chesworth R., Moyer M. P., Bernt K. M., Tseng J. C., Kung A. L., Armstrong S. A., Copeland R. A., Richon V. M., Pollock R. M. (2011) Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20, 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bernt K. M., Armstrong S. A. (2011) A role for DOT1L in MLL-rearranged leukemias. Epigenomics 3, 667–670 [DOI] [PubMed] [Google Scholar]
- 32. Feng Y., Yang Y., Ortega M. M., Copeland J. N., Zhang M., Jacob J. B., Fields T. A., Vivian J. L., Fields P. E. (2010) Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood 116, 4483–4491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones B., Su H., Bhat A., Lei H., Bajko J., Hevi S., Baltus G. A., Kadam S., Zhai H., Valdez R., Gonzalo S., Zhang Y., Li E., Chen T. (2008) The histone H3K79 methyltransferase Dot1L is essential for mammalian development and heterochromatin structure. PLoS Genet. 4, e1000190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nguyen A. T., Zhang Y. (2011) The diverse functions of Dot1 and H3K79 methylation. Genes Dev. 25, 1345–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]