

Background: Lysine carbamylation facilitates metal coordination for enzymatic activities.
Results: Structures of dihydropyrimidinase as the apo- and holoenzyme with one and two metals and its substrate/product complexes are determined.
Conclusion: The structures reveal four steps in the assembly of the holoprotein with the carbamylated lysine and two metal ions.
Significance: The results illustrate how proteins exploit lysines and metals to accomplish lysine carbamylation and enzymatic functions.
Keywords: Crystal Structure, Crystallography, Metalloenzymes, Post-translational Modification, Protein Carboxylation, Protein Complexes
Abstract
Lysine carbamylation, a post-translational modification, facilitates metal coordination for specific enzymatic activities. We have determined structures of the vertebrate dihydropyrimidinase from Tetraodon nigroviridis (TnDhp) in various states: the apoenzyme as well as two forms of the holoenzyme with one and two metals at the catalytic site. The essential active-site structural requirements have been identified for the possible existence of four metal-mediated stages of lysine carbamylation. Only one metal is sufficient for stabilizing lysine carbamylation; however, the post-translational lysine carbamylation facilitates additional metal coordination for the regulation of specific enzymatic activities through controlling the conformations of two dynamic loops, Ala69–Arg74 and Met158–Met165, located in the tunnel for the substrate entrance. The substrate/product tunnel is in the “open form” in the apo-TnDhp, in the “intermediate state” in the monometal TnDhp, and in the “closed form” in the dimetal TnDhp structure, respectively. Structural comparison also suggests that the C-terminal tail plays a role in the enzymatic function through interactions with the Ala69–Arg74 dynamic loop. In addition, the structures of the dimetal TnDhp in complexes with hydantoin, N-carbamyl-β-alanine, and N-carbamyl-β-amino isobutyrate as well as apo-TnDhp in complex with a product analog, N-(2-acetamido)-iminodiacetic acid, have been determined. These structural results illustrate how a protein exploits unique lysines and the metal distribution to accomplish lysine carbamylation as well as subsequent enzymatic functions.
Introduction
Post-translational modification of proteins increases the diversity in the proteome (1–3). Carbamylation on lysine extends the residue length by ∼2 Å and changes the side chain from a positive to negative charge at neutral pH. This unique modification enables the carbamylated lysine to play multifunctional roles in proteins, especially critical for many physiologically important enzymes to exhibit the proper activities (4, 5). In humans, the proteins involved with lysine carbamylation are found to be related to the human diseases, such as type 2 diabetes, developmental delay, metabolic acidosis, mental retardation, hypotonia, and seizures (6–9). In plants, to convert the atmospheric carbon dioxide to energy-rich molecules, the enzyme Rubisco3 catalyzes the CO2 fixation in photosynthesis (10). Rubisco has no enzymatic function until the key lysine residue is carbamylated. The roles of the carbamylated lysine are for metal binding, such as the magnesium ion, as well as to serve as a base for proton abstraction in the catalytic cycle. To counter antibiotics, bacteria have developed the β-lactamase resistance system to destroy the β-lactam antibiotics for survival (11–13). The function of the carbamylated lysine residue in class D β-lactamases is not only to promote the acylation step of the active-site serine but also to activate hydrolytic water for the second catalytic step of the deacylation event (13). So far, there are about 250 structures containing carbamylated lysine residues found in the Protein Data Bank). Although carbamylated lysines exist in a broad range of organisms and have been considered as a residue of catalytic capabilities in nature among the 20 canonical amino acids (14), much less is known about the carbamylation, biochemically and structurally, on the ϵ-amino group of lysine.
Lysine carbamylation does not need an additional enzyme to achieve the covalent modification, a process that has been reported to be a spontaneous and reversible (15, 16). By comparison, additional specific enzymes or catalysts, such as methyltransferases and acetyltransferases, are required to catalyze methylation and acylation reactions, respectively (17, 18), both with high kinetic barriers for the covalent bond formation. Thus, the spontaneous and reversible features of lysine carbamylation are unique in post-translational modifications. In fact, lysine carbamylation is difficult to establish. Most of the carbamylated lysines in proteins are identified by x-ray crystal structures.
In addition to the catalytic role, lysine carbamylation is also involved in the coordination with metal ions. For example, metal ions are found in the amidohydrolase superfamily (19), such as dihydropyrimidinase (DHP; EC 3.5.2.2) in animals. The physiological function of DHP is to hydrolyze dihydrouracil (DHU) or dihydrothymine (DHT) to N-carbamyl-β-alanine (NCβA) or N-carbamyl-β-amino isobutyrate (NCβI), respectively (20), in the second step of the pyrimidine degradative pathway. The bacterial counterpart of DHP, also called hydantoinase (HYD), is not involved in the pyrimidine catabolism, but in industry, it is used for the production of enantiomerically active N-carbamyl amino acids, which are further converted to non-proteinogenic amino acids for synthesis of antibiotics, peptide hormones, and pesticides (21, 22). The active sites of HYD in the bacterial enzymes usually contain dimetal ions with the carbamylated lysine based on their crystal structures (23, 24). However, animal DHP contains only a single divalent-metal ion in the active site according to several independent studies (25–27), despite the fact that a dimetal binding site potentially exists in animal DHP based on the multiple sequence alignments. In fact, before this work, there has been no structural information on the active-site environment in DHP with the single divalent metal co-existing with carbamylated lysine.
Here we describe the crystal structures of the vertebrate dihydropyrimidinase from Tetraodon nigroviridis (TnDhp) in three states: the apoenzyme as well as the holoenzyme with one divalent metal and two divalent metal ions, respectively. In addition, the apo-TnDhp in complex with a product analog, N-(2-acetamido)-iminodiacetic acid (ADA), and the dimetal TnDhp in complexes with hydantoin, NCβA, and NCβI have also been determined. These structures have enabled us to correlate the metal distribution within this enzyme with lysine carbamylation as well as subsequent enzymatic functions and mechanisms.
EXPERIMENTAL PROCEDURES
Molecular Cloning and Protein Purification
Healthy green spot pufferfish, T. nigroviridis, were sacrificed. Total RNA was isolated from fish liver using the RNeasy® Protect minikit (Qiagen) according to the manufacturer's protocol. The RNA was transcribed into the single-stranded cDNA using the SuperScript III first-strand synthesis system for RT-PCR kit (Invitrogen). The cDNA of the recombinant TnDhp was amplified by PCR using the sense primer 5′-GCGCGGATCCATGGCAGAAGCCGGCGAG-3′ and the antisense primer 5′-GCGCGAATTCTCAGTCAGACGTTCCCAGAGCAAC-3′ (with the BamHI and EcoRI cutting sites underlined). After digestion by restriction enzymes, a fragment with BamHI and EcoRI sticky ends was purified from agarose gel using the Gene-SpinTM 1-4-3 DNA Purification Kit-V2 (PROTECH) and subcloned into the BamHI/EcoRI precut pET44a(+) (Novagen) plasmid by T4 DNA ligase. pET 44a(+) harboring the TnDhp cDNA was subsequently transformed into Escherichia coli BL21 (DE3) cells for recombinant protein expression and purification. The bacteria were grown in Luria-Bertani (LB) medium, and protein expression was induced with isopropyl-β-d-thiogalactopyranoside at 25 °C for 30 h. To improve the solubility of the recombinant TnDhp, the fusion protein with native E. coli NusA (Nus-tag) attached to the C terminus (28) was first prepared and then purified and eluted with an imidazole gradient through an Ni2+ column. After thrombin cleavage of the Nus-tag, the pure recombinant TnDhp (non-amended enzyme) was eluted again through the Ni2+ column.
Preparation of the Apo- and Metal-amended TnDhps
The apoenzyme was prepared by dialyzing non-amended TnDhp against the chelating buffer (2.5 liters) containing MES (50 mm, pH 6.5), EDTA (1 mm), and 8-hydroxy-5-quinolinesulfonic acid (15 mm) at 25 °C for 4 days with one change of the fresh chelating buffer (2.5 liters). The apo-TnDhp was then desalted through a desalting column (HiTrap; 1.6 × 2.5 cm) with HEPES (50 mm, pH 7.0). The metal-amended TnDhp was prepared by adding zinc chloride (1 mm) or cobalt chloride (1 mm) into the LB medium during isopropyl-β-d-thiogalactopyranoside induction to produce the zinc-amended (mono-Zn TnDhp) or the cobalt-amended TnDhp (mono-Co TnDhp), respectively.
Inductively Coupled Plasma Mass Spectrometry (ICP-MS) Assays
The metal ion compositions of the various ThDhps were determined by ICP-MS. The Centricon (molecular weight cut-off 30,000; Amicon) was used to replace the buffer solution of each sample with potassium phosphate buffer (10 mm, pH 7.0), which served as the background solution for the determination of the metal ions. The measurement on each sample was independently repeated three times with calculated S.D. values.
Activity Assays
The specific activity measurements were performed by a rapid spectrophotometric assay (27). The TnDhps activity was determined by measuring the decrease of the absorbance at 298 nm upon the hydrolysis of phthalimide as the substrate at 25 °C. For routine assays, the reaction mixture (1 ml) contained phthalimide (1 mm) and BisTris propane (100 mm) at pH 7.0. Under the reaction conditions, a change in A298 of 2.26 corresponded to the hydrolysis of 1 μmol of the substrate. Substrate hydrolysis was monitored with a UV-visible spectrophotometer (Hitachi U 3300).
Crystallization of the TnDhps
All crystallizations were performed with the hanging-drop vapor diffusion method in a 48-well ADX plate (Hampton Research) at 291 K. 1-μl drops of the protein solution (∼10 mg ml−1) in Tris buffer (20 mm, pH 7.0) were mixed with 1-μl drops of the reservoir solution and equilibrated against the reservoir solution (150 μl). Crystals of the apo-TnDhp could be obtained when the solution contained lithium sulfate monohydrate (100 mm), PEG 4000 (12%, w/v) and isopropyl alcohol (2%, v/v) in ADA buffer (100 mm, pH 6.5). Crystals of the mono-Zn TnDhp, which were purified naturally from E. coli with the addition of zinc chloride during isopropyl-β-d-thiogalactopyranoside induction, were grown from a solution containing lithium sulfate monohydrate (100 mm), PEG 4000 (12%, w/v), and trisodium citrate dihydrate (100 mm, pH 6.5). Crystals of the di-Zn TnDhp were prepared by soaking zinc chloride (0.1 mm) into the crystals of the mono-Zn TnDhp in the crystallization buffer. Complexes of the di-Zn TnDhp were co-crystallized using a molar ratio of 3 (substrates/protein). Suitable single crystals (0.2 × 0.1 × 0.1 mm3) were selected and transferred to the crystallization solution containing glycerol (20%) as the cryoprotectant and fast-cooled in liquid nitrogen for further x-ray diffraction experiments.
Data Collection and Processing
X-ray diffraction data from frozen crystals of apo-TnDhp were collected at 110 K using a CCD detector (Q-4R, ADSC) at wavelength 1.0 Å on the Taiwan contracted beamline BL12B2 at SPring-8 in Japan. A 240º rotation with 1.0º oscillation was measured with an exposure duration of 10 s and distance of 180 mm from the crystal to the detector, at 110 K in a nitrogen stream using a cryosystem (X-Stream, Rigaku/MSC, Inc.). The mono-Zn TnDhp data were collected on the BL44XU beamline at SPring-8 with a similar procedure as for the apo-TnDhp and recorded by a SMART-6500 CCD detector (Bruker). The x-ray diffraction data of di-Zn TnDhp as well as their substrate complexes were collected at 110 K on beamline BL13B1 equipped with a CCD detector (Q315, ADSC) at the National Synchrotron Radiation Research Center in Taiwan. All data sets were indexed, integrated, and scaled using HKL2000 (29). Details of the data statistics appear in Table 2.
TABLE 2.
Data collection and refinement statistics
| Apo-TnDhp + ADA | Mono-Zn TnDhp | Di-Zn TnDhp | Di-Zn TnDhp + NCβI | Di-Zn TnDhp + NCβA | Di-Zn TnDhp + hydantoina | |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Wavelength (Å) | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| Temperature (K) | 110 | 110 | 110 | 110 | 110 | 110 |
| Space group | I4122 | I4122 | I4122 | I4122 | I4122 | I4122 |
| Cell dimensions (Å) | ||||||
| a | 160.82 | 160.99 | 160.48 | 161.20 | 161.08 | 161.03 |
| b | 160.82 | 160.99 | 160.48 | 161.20 | 161.08 | 161.03 |
| c | 94.55 | 93.4 | 93.30 | 93.80 | 94.53 | 93.44 |
| Resolution (Å)a | 30–1.95 (2.1–1.95) | 30–2.0 (2.03–2.00) | 30–1.9 (1.97–1.90) | 30–1.8 (1.86–1.80) | 30–2.0 (2.07–2.00) | 30–2.2 (2.28–2.00) |
| Completeness (%)a | 99.6 (100) | 99.2 (99.6) | 99.7 (99.9) | 99.8 (100) | 100 (100) | 97.6 (99.4) |
| Redundancya | 9.7 (9.5) | 3.3 (3.2) | 8.4 (8.3) | 4.6 (4.5) | 5.0 (5.0) | 4.5 (4.4) |
| 〈I/σI〉a | 25.6 (11.8) | 18.8 (2.2) | 17.4 (3.9) | 18.7 (4.4) | 20.6 (5.5) | 18.0 (6.1) |
| Rsym (%)a,ba | 4.5 (30.2) | 5.6 (33.5) | 8.1 (49.9) | 6.9 (34.1) | 7.6 (33.9) | 7.6 (38.0) |
| Refinement | ||||||
| Resolution (Å) | 30–2.0 | 30–2.0 | 30–2.0 | 30–1.8 | 30–2.0 | 30–2.2 |
| Rworkc/Rfreed (%) | 15.9/19.5 | 17.0/20.4 | 17.6/21.1 | 17.5/20.0 | 17.2/20.3 | 16.6/20.2 |
| No. of atoms | ||||||
| Protein | 3805 | 3759 | 3759 | 3759 | 3759 | 3759 |
| Ligand | 39 (ADA)a | 10 | 9 | 7 | ||
| Zinc | 1 | 2 | 2 | 2 | 2 | |
| Water | 232 | 206 | 216 | 247 | 200 | 170 |
| B-factors (Å2) | ||||||
| Protein | 18.92 | 28.69 | 18.99 | 24.97 | 25.18 | 29.67 |
| Ligand | 39.1 | 41.62 | 57.90 | 73.9 | ||
| Zinc | 37.93 | 17.19/20.02 | 26.34/26.86 | 33.7/39.6 | 39.3/50.1 | |
| Water | 23.21 | 32.13 | 22.92 | 30.30 | 30.14 | 33.50 |
| Root mean square deviations | ||||||
| Bond lengths (Å) | 0.024 | 0.020 | 0.020 | 0.024 | 0.021 | 0.020 |
| Bond angles (degrees) | 2.041 | 1.927 | 1.763 | 2.143 | 1.867 | 1.729 |
a Values in parentheses are for the highest resolution shell.
b Rsym = Σh Σi [Ii (h) - <I(h)>/Σh ΣiIi (h)], where Ii is the ith measurement and <I(h)> is the weighted mean of all measurements of I(h).
c Rwork = Σh|Fo − Fc|/ΣhFo, where Fo and Fc are the observed and calculated structure factor amplitudes of reflection h.
d Rfree is as Rwork but calculated with 10% of randomly chosen reflections omitted from refinement.
Determination and Refinement of Structures
Assuming one monomer structure in the asymmetric unit, the Matthew's coefficient was estimated to be 2.82 Å3/Da, which corresponds to a solvent content of ∼56%. The structure of apo-TnDhp was first solved by the molecular replacement method using the structure of DHP from Dictyostelium discoideum (Protein Data Bank code 2FTW) (5) (sequence identity 58%). This attempt generated initial phases with an R factor of ∼45% by the program MOLREP (30). Throughout the refinement, a random selection (10%) of the data were set aside as a “free data set,” and the model was refined against the remaining data with F ≥ 0 as a working data set (31). After the rigid body refinement and simulated annealing with CNS version 1.3 (32), several rounds of model building with Coot (33) were performed. The refinement then proceeded through another cycle of individual B-factor refinement and restrained refinement by CNS and Refmac5 (34), respectively, to generate a model with R-factor of ∼25%. Subsequently, the water molecules were located at peaks of the difference maps automatically, using the program ARP/warp (35). After the assignment of 232 waters, the refinement converged to a final R-factor of 15.9% and Rfree of 19.5% for all data at a resolution of 30 to 2.0 Å. The structures of the mono- and di-Zn TnDhp and complexes with various substrates were determined by molecular replacement with the program MOLREP using the structure of apo-TnDhp as the search model. The structures were refined with the procedures described above. The correctness of the stereochemistry of the models was verified using MolProbity (36). All main-chain dihedral angles of residues were in the most favored and additionally allowed regions with only glycine exceptions in the Ramachandran plots. All refinement statistics are summarized in Table 2.
RESULTS
Characterizations of TnDhp; ICP-MS and Reactivation Assays
The ICP-MS data show that the non-amended TnDhp monomer contains various metals with different compositions (Table 1), of which iron is the major metal ion, implying that the reactive metal ion(s) at the active site can be largely replaced with iron(s) during protein expression in E. coli. Previously, it was found that the iron incorporated into the DHP might result from iron contamination of the LB medium (37). In fact, the enzyme containing iron as the abundant metal ion is nearly inactive, exhibiting only a low level of activity.
TABLE 1.
The metal contents and specific activities of TnDhp
| Cell cultures | Mol of metal/mol of monomera |
Relative activity | ||
|---|---|---|---|---|
| Iron | Cobalt | Zinc | ||
| mol/mol | % | |||
| Cobalt-amendeda | 0.08 ± 0.01 | 0.83 ± 0.01 | 0.07 ± 0.00 | 100 |
| Zinc-amendedb | 0.10 ± 0.01 | 0.03 ± 0.00 | 0.88 ± 0.01 | 72.4 |
| Non-amended | 0.77 ± 0.01 | 0.01 ± 0.00 | 0.14 ± 0.00 | 4.3 |
| Apo-TnDhpc | 0.03 ± 0.01 | 0.02 ± 0.00 | 0.07 ± 0.00 | 0.2 |
a The molar ratios of metal to protein were determined by ICP-MS.
b Metal ions at 1 mm concentration were amended in the cell cultures.
c Apo-TnDhp was prepared as described under “Experimental Procedures.”
The metal compositions in both the zinc-amended (mono-Zn TnDhp) and the cobalt-amended TnDhp (mono-Co TnDhp) are also analyzed by ICP-MS (Table 1). These measurements are repeated three times, with similar results obtained for each metal. The metal contents suggest that the total metal capacity (metal/enzyme monomer) is ∼1 (mol/mol), implying that the active site of TnDhp contains only one tightly bound metal ion in its functional form. The specific activity of the cobalt-amended TnDhp is about 1.4- and 23-fold higher than those of the zinc-amended and the non-amended (iron-containing) TnDhp, respectively. Despite the higher activity of the cobalt-amended TnDhp, we have used the zinc-amended TnDhp as the model for discussion here because the DHP in vertebrate is a natural zinc-metalloenzyme (25).
The above metal stoichiometry was confirmed by reactivity assays. Various concentrations of zinc ions were added to a solution of the apo-TnDhp, and the activity was measured. The reactivation experiment clearly indicates that the addition and incorporation of zinc ions into the apo-TnDhp could restore the enzymatic activity (Fig. 1). However, the activity curve also shows that concentrations of zinc ions significantly higher than the 1:1 stoichiometry caused a slight inhibitory effect on the protein activity. At a concentration of 25 μm, the activity decreased to 60% of the maximum value (data not shown).
FIGURE 1.
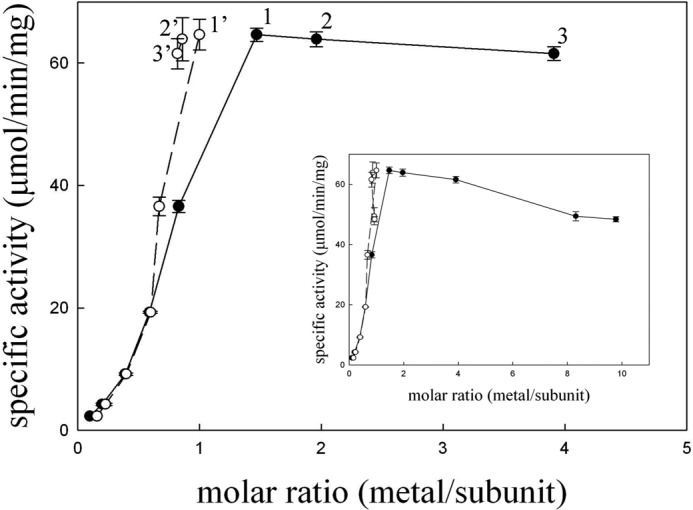
Reactivation of the apo-TnDhp with zinc ions. Variable amounts of Zn2+ were added into a solution of the apo-TnDhp at pH 7.0 (10 mm potassium phosphate) and incubated at 4 °C for 3 days. The specific activities were determined by the standard assay (●). In parallel experiments, the same samples were further desalted by passage over a HiTrap desalting column with 10 mm potassium phosphate (pH 7.0), and the metal contents in the reconstituted enzymes were analyzed by ICP-MS. The ICP-MS data points (○) 1′, 2′, and 3′ correspond to the samples 1, 2, and 3 after desalting column, respectively. The inset presents higher molar ratio (up to 8 and 10) data with similar desalting results. Error bars, S.D.
To verify that the binding of zinc ions is specific, we titrated samples at various metal/protein ratios and determined the zinc contents by ICP-MS after the solutions were passed through the desalting column to remove zinc ions that were non-specifically bound (Fig. 1). The ICP-MS data show that all samples contain only one zinc ion per protein monomer, confirming that the maximum activity was obtained with only one metal ion occupying a specific site in the protein.
Overall Structure and Molecular Packing of TnDhp
The crystal structures of TnDhp in various states were determined and summarized in Table 2. The structure of apo-TnDhp was first determined. It exhibits a monomer per asymmetric unit in crystals of the space group I4122, with 492 amino acids from Ala4 to Ala495 out of a total of 500 residues (Fig. 2, A and B). The molecular dimensions of TnDhp are about 70 × 55 × 50 Å3, with a surface area of ∼18,912 Å2 accessible to the solvent. The overall structure comprises 18 α-helices and 20 β-strands and folds into one minor (Ala4–Gly58, Phe384–Lys405, and Pro431–Gly447) and one major domain (Gly59–Asn383 and Thr406–His430). After the last α-helix (Glu466–Glu477), the C-terminal fragment (Val478–Ala495) forms a long tail of 18 amino acids. As is commonly found in DHPs, the major domain folds as a distorted (α/β)8 TIM barrel, whereas the minor domain forms a sandwich-like structure with 10 mixed anti-parallel/parallel β-strands. The electrostatic surface reveals a cleft of about 10 × 10 × 20 Å3 in the center of the major domain that facilitates the entry of substrates to the active site.
FIGURE 2.

The overall structure of the apo-TnDhp protein. A, monomer TnDhp structure could be divided into a major domain (green), minor domain (red), and C-terminal tail (yellow). B, electrostatic potential surface shows the positive (blue) and negative (red) charge distributions. The active site is located in the major domain with a tunnel for transport of substrates and products. C, the C-terminal tail not only contributes to the main interactions for maintaining the dimeric state of the protein molecules (green, monomer A; pink, monomer B) but also extends to the dynamic loop (yellow) near the active site (blue). D, the tetramer structure is formed by two dimers. The bottom scheme in each panel shows the simplified dimer and tetramer structures. Monomers are shown in different colors, and the red squares represent the locations of the minor domains.
The TnDhp exhibits a monomer in the asymmetric unit of the crystals; however, the crystallographic symmetry-related dimer and tetramer structures could be generated with the space group I4122 (Fig. 2, C and D), which exhibits molecular packing similar to that of other previously reported tetramers (39). The results of the gel filtration during protein purification suggest that the TnDhp is a tetramer in the solution state. Interestingly, the dimeric interface of TnDhp shows that the long C-terminal tail (Val478–Ala495) from one monomer winds onto the surfaces of another monomer and reaches close to its active site. Several hydrogen bonds stabilize the dimeric interaction through the C terminus.
The Metal-binding Site and Post-translational Modification of Lysine by Carbamylation
The metal-free TnDhp (apo-TnDhp) was generated, and the metal content (∼0) was confirmed by ICP-MS (Table 1). The active site of apo-TnDhp contains four histidines (His63, His65, His188, and His244), one aspartic acid (Asp322), and one lysine (Lys155) in the catalytic pocket (Fig. 3A). In the structure of the apo-TnDhp, the residue of Lys155 is not carbamylated at the ϵ-amino group. Two water molecules, but no metals, occupy the potential metal-binding sites.
FIGURE 3.

Structures of the active site. The composite omit maps at 1.0 σ are shown in blue mesh, and the zinc anomalous difference maps at 5.0 σ are shown in red mesh. A, the apo-TnDhp contains two waters and a non-carbamylated Lys155 at the active site. B, the mono-Zn TnDhp contains one zinc atom at the Mα site. The electron density clearly indicates that Lys155 is carbamylated. C, two zinc atoms are present in the di-Zn TnDhp. D, structural superimposition of the apo- (light green), mono-Zn (cyan), and di-Zn TnDhp (yellow) shows conformational differences. Displacements of the carbamylated Lys155 in the mono-Zn to the di-Zn TnDhp are highlighted by dark red arrows.
The mono-Zn TnDhp contains one metal at the active site, as confirmed by ICP-MS assays (Table 1). The zinc-anomalous difference Fourier map clearly indicates only one zinc atom in the active site (defined as the Mα site) (Fig. 3B). This zinc ion is coordinated with residues His63, His65, and Asp322, with zinc to nitrogen distances of 2.21, 2.16, and 2.13 Å, respectively. Two water molecules are also observed. One water molecule is loosely associated with the zinc atom at a distance of 4.4 Å. The other water is directly coordinated (2.2 Å) to Tyr160, which is the proposed key residue participating in the catalysis (39). The composite omit map shows that the electron density between the Mα metal and Lys155 is connected. This electron density is consistent with the carbamate of a carbamylated lysine with a reasonable distance of 2.14 Å between one of the oxygens of the carbamate and the Mα metal. An inspection of 10 data sets, which were collected independently, reveals the same result as described above. The structure of mono-Zn TnDhp shows that one metal (Mα) is sufficient to promote the carbamylation process. Evidently, Lys155 is chemically modified, with one metal ion at the active site of the protein.
Sequence alignment as well as the structural features of the active site uncovered above suggest that the catalytic pocket of TnDhp could potentially accommodate two metal ions. To generate the di-Zn TnDhp, we soaked crystals of the mono-Zn TnDhp in the crystallization milieu containing zinc ions at high concentrations (0.1 mm). The structure of the di-Zn TnDhp shows two zinc ions at the active site (Fig. 3C), of which one zinc atom is located at the same Mα site as in the mono-Zn structure, and the other, dubbed the Mβ site, is bound to the two histidine residues His188 and His244 with zinc to nitrogen distances of 2.15 and 2.13 Å, respectively. The distance between the two zinc atoms (i.e. Mα and Mβ) is 3.74 Å in the di-zinc TnDhp. The existence and position of the second zinc was confirmed by zinc-anomalous diffraction. These results indicate that the active site could accommodate a total of two zinc atoms. A water molecule is also found at the position between the two zinc atoms (Fig. 3C). In the dimetal TnDhp, each of the zinc ions is coordinated with one of the carbamate oxygens of the carbamylated Lys155 with a zinc to oxygen distance of 1.97 Å.
Superimposed structures of the apo-TnDhp and the mono- and di-Zn TnDhp reveal conformational changes upon lysine carbamylation and metal binding (Fig. 3D). In the surrounding environment of the active site, the side-chain conformations of His63, His65, and His244 are relatively stable, whereas those of Tyr160, His188, and Asp322 are altered. Relative to their positions in the apo-TnDhp structure, we find that Tyr160, His188, and Asp322 have shifted their side-chain orientations by distances of ∼1.9, 0.5, and 0.3 Å, respectively, upon binding of one metal and carbamylation in the mono-Zn structure. After carbamylation and the binding of the two zinc ions, these residues move closer to the metal sites, and the movements increase to 2.0, 0.8, and 0.6 Å, respectively.
The conformation of the Lys155 is also greatly affected by the metal binding and carbamylation (Fig. 3D). In apo-TnDhp, Lys155 exhibits the general non-carbamylated conformation. Upon zinc binding to the Mα site, the post-modification process takes place by stabilizing CO2 already on the ϵ-amino group of Lys155. It is of interest to note that the free oxygen atom of the carbamate in the mono-Zn TnDhp structure is located at the Mβ position coordinating with His244 (2.4 Å). Moreover, the side chain of the carbamylated Lys155 has shifted away 1.4 Å to yield space for the second zinc atom to fit into the Mβ site in the structure of the di-Zn TnDhp.
It has been reported that cobalt atoms could substitute for zinc at the active site (40). We have confirmed that the structures of the mono-Co and di-Co TnDhp are similar to the zinc structures described above.
Comparison of Active Sites of Carbamylated Proteins
A protein generally contains several lysine residues, but carbamylation is typically found at special positions, which is extremely interesting and important. The propensity toward carbamylation must depend on the environment of the lysine, including the surrounding residues. The carbamylated lysines are usually buried within the protein structures with a small solvent-accessible surface area, ∼6.2 Å2, as in apo-TnDhp, and are surrounded by hydrophobic residues, such as Val and Phe. At present, there are about 250 structures in the Protein Data Bank containing carbamylated lysines required for their functions. These structures could be classified into three groups based on the metal content: 1) no metal; 2) carbamylated lysine with one metal in “half-occupied” geometry; and 3) carbamylated lysine with two metals in full geometry (Scheme 1).
SCHEME 1.
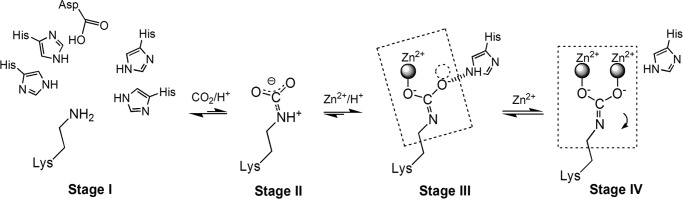
Four stages are involved in lysine carbamylation. Stage I, the structural requirements for lysine carbamylation. “Half-occupied” geometry (at least two histidines) is needed for coordination with one metal, whereas the full geometry is employed for two-metal coordination. Stage II, the carbon dioxide interacts weakly with the lysine in a resonance structure. Stage III, the state with the carbamylated lysine coordinated to one metal. The free oxygen of the carbamate containing the electron lone pair is stabilized by a histidine. The dashed circle indicates the binding site for the second metal. The six atoms in the dashed square are coplanar. Stage IV, the carbamylated Lys155 moves by 1.4 Å, as indicated by the arrow, upon binding of the second metal. In this stage, the seven atoms are coplanar (dashed square).
The structures of TnDhp reveal a common structural motif for lysine carbamylation with both Mα and Mβ sites. This motif has been demonstrated to be important for maintaining the carbamate structure according to site-directed mutagenesis and activity rescue experiments (40). One metal is bound to the Mα site sharing two histidines. Apparently, this metal coordination is sufficient to fix CO2 to the lysine. Interestingly, there are several other systems, such as transcarboxylase (Protein Data Bank entry 1RQB) (41), pyruvate carboxylase (2QF7) (6), and pyruvate carboxylase (3BG3) (7), with the same “half-occupied” geometry utilized by the respective lysine to coordinate one cobalt, zinc, and manganese ion, respectively (Fig. 4). A carbamylated lysine with one metal has also been reported in Rubisco (4). The two highly conserved histidine residues near the carbamylated lysine exist also in Rubisco, although the metal is bound to the ribulose 1,5-bisphosphate substrate instead of the histidines. Thus, our mono-Zn TnDhp provides a new structural feature for enzymes that contain only one tightly bound metal in full geometry, differing from those structures in the Protein Data Bank.
FIGURE 4.
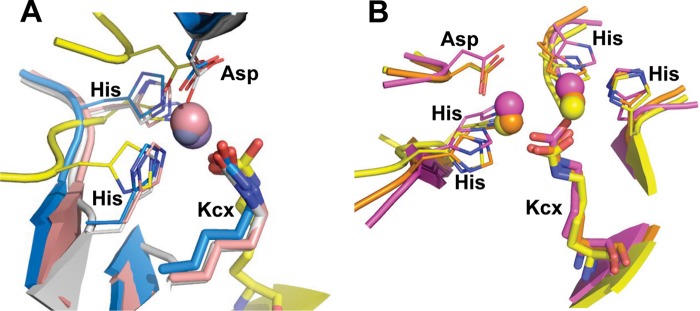
Structural comparisons. A, representative structures from various proteins with the carbamylated lysine coordinated with one metal by half-occupied geometry. Yellow, TnDhp; blue, transcarboxylase (1RQB); pink, pyruvate carboxylase (2QF7); white, pyruvate carboxylase (3BG3). B, the carbamylated lysine coordinated with two metals shares a similar geometry of the key residues as well as the same environment. Yellow, TnDhp; magenta, urease (1EJX); orange, dihydroorotase (2Z26).
A ligand structure comprising four histidines and one aspartic acid utilizing both the Mα and Mβ sites is required for lysine carbamylation and other biological functions mediated by this family of proteins (Fig. 4B). Examples include the amidohydrolase superfamily (e.g. urease (42), allantoinase (43), HYD (44), and DHP (5)) and other proteins, such as arginine carboxypeptidase (45).
Lysine Carbamylation and Metal Numbers Regulate the Structure and Function of the Protein
The post-translational lysine carbamylation facilitates additional metal coordination to trigger the observed structural alteration seen in our structures for specific enzymatic activities. The molecular structure of apo-TnDhp also reveals a tunnel from the surface of the protein to the active site suitable for the substrate entrance or product release (Fig. 5B). Notably, two loops located at the entrance of the tunnel and near the active site are highly dynamic (Fig. 5A). One loop from Ala69 to Arg74 exhibits the largest variation among three structures (apo-, mono-Zn, and di-Zn TnDhp). In particular, Phe70 and Met71 swing with a maximum range of 6.9 and 7.5 Å, respectively. The other loop from Met158 to Met165 also exhibits a similar structural disparity, allowing Tyr160 to move closer to the active site in the dimetal structure with a displacement of 2 Å.
FIGURE 5.

Dynamic loops and the C-terminal tail. A, upon the metal binding, two dynamic loops (Ala69–Arg74 and Met158–Met165) move toward the active site (light green, apo-TnDhp; yellow, di-Zn TnDhp). In particular, Phe70 and Met71 on the loop Ala69–Arg74 swing with maximum distances of 6.9 and 7.5 Å, respectively. The C-terminal tail from another monomer (orange) is shown, and the most dynamic residues are labeled. The carbamylated lysine 155 is labeled as Kcx155. B, the structure of apo-TnDhp shows that the dynamic loops (green) in the “open form” expose the active site (blue) to the molecular surface. The C-terminal tail from another monomer (orange) is shown in close proximity to the dynamic loop. C, the structure of di-Zn TnDhp shows that dynamic loops (yellow) in the “closed form” obstruct the active site. In this state, the C-terminal tail (orange) is too flexible to be modeled, because there is no electron density seen for the last seven residues. D, a side view of the active site with the dynamic loops in the open form. The cross-section bottleneck of the tunnel is about 10 Å (light green), which is wider than the average size of substrates (6 Å). The active site pocket surrounded by key residues is shown in blue. E, a side view of the active site with the dynamic loops in the closed form. The cross-section bottleneck is reduced dramatically to ∼3 Å (yellow). The active site pocket is shown in blue. The two zinc ions are depicted as spheres.
In the structure of apo-TnDhp, the two loops are positioned away from the active site with distances of 12.5 and 5.7 Å measured from Phe70 (on the first loop) and Tyr160 (on the second loop) to Mα and Mβ, respectively. We surmise that this is the “open form” of the enzyme because there is no apparent barrier restricting the transport of the substrate from the surface to the active site (Fig. 5B). The bottleneck dimension of the tunnel in an open form is about 10 Å (Fig. 5D), which is greater than the average size (∼6 Å) of the TnDhp substrates, such as DHU and DHT. However, the two dynamic loops in di-Zn TnDhp move closer to the active site with distances of 8.5 and 4.5 Å from Phe70 and Tyr160 to Mα and Mβ, respectively, and significantly reduce the bottleneck dimension to 3 Å (Fig. 5, C and E), resulting in a “closed form” of TnDhp and limiting the substrate entry into or product release from the active site.
Interestingly, in the structure of the mono-Zn TnDhp, the orientation of the (Ala69–Arg74) loop is in the “intermediate state” between the conformations of apo- and di-Zn TnDhp. The dynamic loops in the mono-Zn structure are about 40% in the open form and 60% in the closed form based on the electron density analysis. The averaged temperature B-factor of this loop (Ala69–Arg74) in the mono-Zn TnDhp (∼60.2 Å2) is higher than that of either the apo-TnDhp (∼32.2 Å2) or the di-Zn TnDhp (∼26.0 Å2), suggesting that the (Ala69–Arg74) loop has the propensity to undergo structural fluctuations even in the crystalline state.
It has been reported that the d-HYD from Bacillus stearothermophilus is equipped with three stereochemistry gate loops (SGL) located in the tunnel gate to regulate the substrate specificity and activity (46, 47). The first (Ala69–Arg74) and second (Met158–Met165) dynamic loops found in TnDhp are structurally related to the gate loops SGL1 and SGL3, respectively, in B. stearothermophilus d-HYD. In this study, we have provided evidence for structural or conformational fluctuations of similar SGLs in vertebrate dihydropyrimidase through a comparison of the apo- and metal-binding structures.
The Flexible C-terminal Tail
The role of the C terminus in DHP has been investigated for a long time, but its function is still a mystery. It has been suggested that the C terminus may play a role in maintaining a multimer form of proteins. It is known that the deletion of the C-terminal fragment in d-HYD from B. stearothermophilus results in dissociation of the protein from dimers to monomers (48). In our structure of TnDhp, the C-terminal tail also maintains the dimeric form, with its terminus reaching near the dynamic (Ala69–Arg74) loop of the active site (Figs. 2C and 5B). An inspection and comparison of the C-terminal tails in apo-, mono-Zn, and di-Zn TnDhp structures show that the stability of the C-terminal tail, especially the fragment from Pro488 to Ala495, affects the dynamic (Ala69–Arg74) loop (Fig. 5A). The structure of the C-terminal fragment (Pro488–Ala495) is rigid, with well defined electron densities in the apo-TnDhp structure (the open form) (Fig. 5B), whereas it is flexible in the monometal structure (an intermediate state). In the latter, the electron density of this region is diminished so that model building could only be allowed with the density map at a lower contour level (0.7 σ). Finally, in the di-Zn TnDhp structure (the closed form), the electron density of the C-terminal tail is too indistinct to allow model building in this region of the protein structure (Fig. 5C). Thus, the flexibility of the C-terminal tail and the conformation of the dynamic loops appear to be correlated. Note that a small C terminus fragment of five residues after Ala495 is too flexible to be identified in all of the structures, so we could not exclude the possibility that these five residues are also interacting with the first dynamic loop.
Structures of TnDhp in Complexes with Hydantoin, NCβI, and NCβA
To explore the substrate-binding site and probe the interactions between the substrates and the residues essential for catalysis, we have co-crystallized TnDhp in complexes with hydantoin, DHT, and DHU, separately. Although both mono-Zn and di-Zn TnDhp are prepared for co-crystallization, only the structures of the complexes with two zincs in the active site could be successfully obtained (Fig. 6). This interesting result might reflect the fact that the substrate channel of the intermediate state in mono-Zn TnDhp does not contain barriers to restrict the diffusion of substrate and products to and from the active site. The hydantoin is found in the catalytic pocket (Fig. 6A) and interacts with two zinc atoms (with distances of 2.8 and 3.0 Å), the main chain of residue Gly294 (∼3.8 Å), and the side chains of Tyr160 (∼3.8 Å) and Asp322 (∼3.4 Å). Phe70, which is located in the dynamic loop (Ala69–Arg74), described above, is in van der Waals contact with the hydantoin (∼3.8 Å) (Fig. 5A) and stabilizes the substrate via π-π stacking interactions.
FIGURE 6.

The di-Zn TnDhp complexed with various substrates. The magenta mesh (2Fo − Fc map, 1σ) shows that the substrates are well fitted. The dashed lines denote the interactions between the substrates and the protein residues. A, structure of the hydantoin complex. The circular electron density is found at the active site. B, the electron density clearly locates the putative product, N-carbamyl-β-amino isobutyrate, in the active site, although the circular substrate, DHT, is used for the co-crystal experiment. C, a similar situation is found with the substrate DHU. The putative product, N-carbamyl-β-alanine, is found in the active site. D, superposition of the substrates. The cleavage site (blue arrow) of hydantoin is shifted about 2.5 Å from the cleavage site of DHT and DHU (blue dashed arrows). Yellow, di-Zn TnDhp; cyan, hydantoin; light green, N-carbamyl-β-amino isobutyrate; pink, N-carbamyl-β-alanine.
From the co-crystallization experiments with the DHT and DHU, it is clear from the electron densities that the C4–N3 bonds in these substrates have been broken to open up the dihydropyrimidine ring and generate the corresponding products NCβI and NCβA, respectively. These observations indicate that catalytic cleavage of the substrates has taken place, but the products could not be released with the two zinc atoms present in the active site (Fig. 6, B and C). Several residues are utilized for stabilizing the NCβI as well as the NCβA, as listed in Table 3. The distances from Tyr160 to the N3 and O4 atoms of NCβI are 3.0 and 2.7 Å, and the distances to the N3 and O4 atoms of NCβA are 3.1 and 2.3 Å, respectively. From the structure of the NCβI in complex with the TnDhp, we conclude that the stereo-selectivity of the catalysis is the l-conformation of the substrate.
TABLE 3.
The interactions between substrates and residues of TnDhp
| Substrate | Protein residues |
|---|---|
| NCβI | His65, Tyr160, His188, Gly249a, Asp322, Asn343a |
| NCβA | Tyr160, His188, Gly249a, Asp322 |
| Hydantoin | Tyr160, Gly249a, Asp322, Phe70 |
| ADA1 | Gly294a, Asp322 |
| ADA2 | Tyr160, Thr339a, Ile341a, Asn343, Asn343a |
| ADA3 | His213, Phe338a, Thr339a, Ans343 |
a The main-chain interaction.
Activity assays indicate that the TnDhp processes hydantoin with a low activity (∼330-fold lower compared with DHT or DHU),4 in agreement with our structural results indicating that the hydantoin is maintained in the cyclic form, but both DHT and DHU have been hydrolyzed in the structures of the complexes. The positions of the NCβI and NCβA are similar in the structures (Fig. 6D), with the cutting sites pointing toward Tyr160. However, the cutting site of hydantoin is displaced ∼2.5 Å away from Tyr160 compared with DHT or DHU, which may explain the low activity of the di-Zn TnDhp toward hydantoin.
The Structure of Apo-TnDhp in Complex with the Substrate Analog ADA
Crystals of the apo-TnDhp have been also grown in the crystallization buffer containing ADA, which could be regarded as a substrate analog (Scheme 2). In this apo-TnDhp structure, the electron density clearly reveals that three ADA molecules are lined up in a channel from the entrance surface to the active site (Fig. 7), highlighting the potential pathway(s) for substrate entry and product release. One ADA molecule (ADA3) is coordinated with the side chains of His231 and Asn343 and the main chains of Phe338 and Thr339. A second ADA (ADA2) is located in the middle position and interacts with the side chains of Tyr160 and Asn343 and the main chains of Thr339, Ile341, and Asn343. The residue Tyr160 in TnDhp is structurally equivalent to Tyr152 in Sinorhizobium meliloti DHP, which has been proposed to be associated with the catalysis process by a mutagenesis study (39). The third ADA (ADA1) at the position close to the active site is coordinated with the side chain of Asp322 and main chain of Gly294. Notably, this ADA1 molecule is located at the same position with an orientation comparable with that in the substrates hydantoin, NCβI, and NCβA described earlier in the structures of the substrate complexes (Fig. 7B). In addition, several hydrogen bonds mediated by water molecules are found to stabilize ADA molecules between residues and ADA molecules. The detailed interactions with the corresponding distances are listed in Table 3.
SCHEME 2.
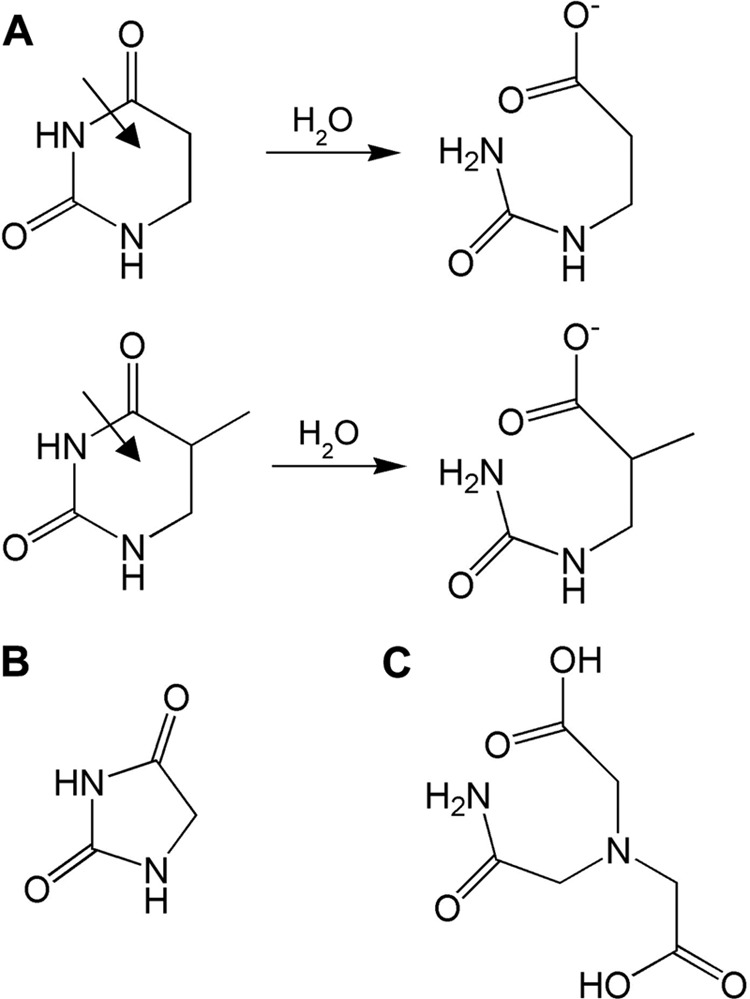
A, the reactions of dihydrouracil (top) and dihydrothymine (bottom). B, structure of hydantoin. C, structure of ADA.
FIGURE 7.
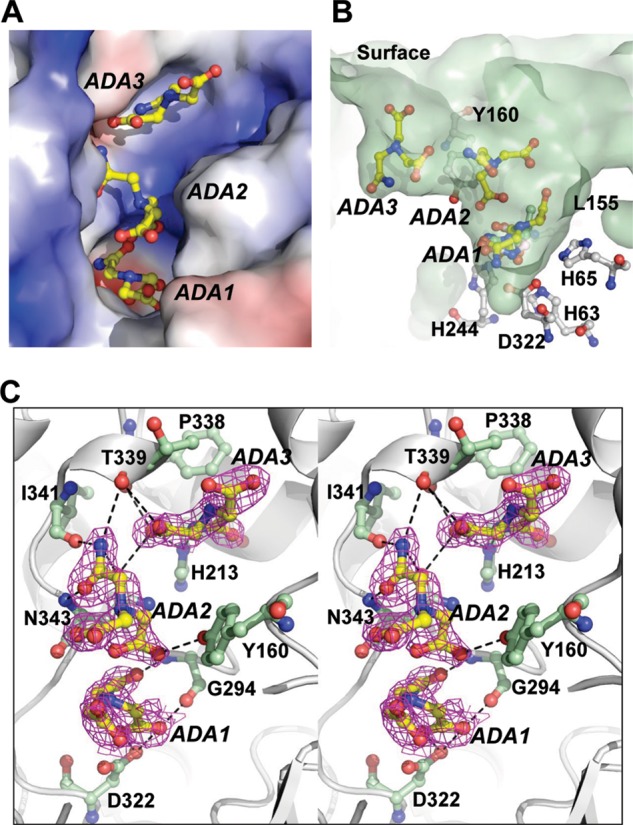
The ADAs in complex with apo-TnDhp. A, there are three ADAs (yellow), labeled ADA1, ADA2, and ADA3, bound in the tunnel of the active site. The electrostatic potential surface shows the charge distribution within the tunnel (blue, positive; red, negative). B, the surface shows the side view of the tunnel (light green). Three ADAs are lined up from the active site to the surface. The ADA1 is in the same location as the substrates in the active site. ADA3 is the closest to the surface. Tyr160 is located between ADA2 and ADA3. C, stereoview of the residues (light green) interacting with the ADAs (yellow). The electron density (2Fo − Fc map 1.0 σ) is shown in magenta.
DISCUSSION
Structural Insights into the Stages of Lysine Carbamylation
The structures with varying metal contents provide insights into the different stages of lysine carbamylation (Scheme 1). The first stage is depicted by apo-TnDhp, where Lys155 is not carbamylated. The second stage (precarbamylation) might involve the formation of an intermediate with a carbon dioxide weakly associated with the ϵ-amino group of the lysine (49). This structure is expected to be quite labile, so that the process is reversible. The third stage corresponds to the carbamylation of the lysine, culminating in the structure of the mono-Zn TnDhp, in which the lysine has been carbamylated at the active site with only one metal. Although carbon dioxide exists at a concentration of ∼50 ppm in the solution, and reversible carbamylation on Lys155 (Stage II of Scheme 1) can be expected, one zinc or cobalt ion must be positioned at the active site for CO2 fixation (Stage III of Scheme 1). The final stage is represented by the structure of the di-Zn TnDhp, where a second metal ion has bound to the active site to stabilize the carbamylated lysine.
The Potential Role of the Mβ Metal Ion
Beside the catalytic metal ion at the Mα site, the active site of TnDhp allows binding of a second metal ion at the Mβ site. Enzymes that equip the active site with full geometry as well as a second metal ion for regulatory function are observed in the amidohydrolase superfamily (50). It has also been reported that zinc ions at high concentrations could enhance the thermal stability of DHP (51). A structural comparison of apo-TnDhp and mono- and dimetal TnDhp suggests that the metal ion at the Mβ site regulates the size of the substrate tunnel (Fig. 5). Presumably, the Mβ metal site plays the role of controlling the two dynamic loops (Ala69–Arg74 and Met158–Met165) for substrate entrance, substrate locking, and product release. Upon the substrate entering the active-site pocket, the Mβ metal ion is simultaneously positioned at the Mβ site to alter the conformations of the dynamic loops to the “closed form” and potentially lock the substrate at the right position with Phe70 for catalysis. After initiating the catalytic turnover, the Mβ metal ion would of course ultimately have to leave the Mβ site to generate an “open-form” substrate tunnel for the product release and new substrate entrance. Thus, the weak metal binding exhibited by the Mβ site is obligatory for its regulatory function, making the occupancy of the Mβ site by a metal ion observable only upon soaking crystals of the mono-Zn TnDhp with zinc ions at a high concentration. This phenomenon might resolve the puzzle as to why only one metal was detected previously in vertebrate DHPs with the full geometry (26, 52). In the normal enzymatic state, the Mα metal ion is tightly bound for catalysis, but the binding of metal to the Mβ site is weak in order to provide the protein with dynamic features to control the “open/closed” conformations of the two dynamic loops. It has been proposed that the metal ion at the Mβ site is loosely bound in DHP (5).
The Importance of Protein Oligomerization
The dimerization of TnDhp results in the C-terminal tail from one monomer reaching near the dynamic (Ala69–Arg74) loop of another monomer (Figs. 2C and 5). This feature of the structure allows us to probe the function of the C terminus. The structural analysis reveals that the conformational stability of the C-terminal tail is in the following order: apo-TnDhp > mono-Zn TnDhp > di-Zn TnDhp. It seems that the flexibility of the C-terminal tail is correlated with the conformations of the dynamic (Ala69–Arg74) loop. The open form of the dynamic loop is linked to the greater stability (low flexibility) of the C-terminal tail, whereas the low stability (high flexibility) of the C-terminal tail results in a closed form of the dynamic loop, implying that the protein is exploiting the flexibility of the C-terminal tail to control the size of the substrate tunnel. Therefore, our structural analysis indicates that the C-terminal tail dictates not only the dimerization of the protein but also the enzymatic activity. It is possible that dimerization of TnDhp by the C-terminal tail assists the opening of the two dynamic loops (SGL1 and SGL3) for the enzyme function.
Previous reports have shown that modifications to the C termini of DHP/HYD enzymes could decrease the enzyme activity (9, 48, 53–55). In the d-HYD from B. stearothermophilus and Bacillus thermocatenulatus, the truncated enzymes with a deletion of 40 amino acids on the C terminus have lost 64% of their enzymatic activities (48). In Pseudomonas putida YZ-26, a series of deletions were performed in the d-HYD and all of the truncated enzymes exhibited either low or non-detectable activities (53, 54). However, CD spectroscopy showed that these deletions of the C-terminal fragment did not alter the secondary structures of the overall folding (48, 54). More recently, deletions on the C terminus of DHP from human, a vertebrate similar to T. nigroviridis, have been undertaken, and these mutants also show no detectable activity (9). Evidently, the C-terminal tail plays an important role for the activity of the enzyme.
Proposed Structure-based Mechanism; Monometal Active Site
According to our structures of apo-, monometal, and dimetal TnDhp, substrate complexes, and the ADA complex, we have gained important insights into the mechanism of action of the DHP family of enzymes. The first step of the catalytic turnover is to get the substrate into the active site. This state is highlighted by the apo-TnDhp and monometal TnDhp structures with the open form of the tunnel, which provides a suitable cross-section for the substrate to diffuse into the active site (Figs. 5D and 8A).
FIGURE 8.

Proposed reaction mechanism of TnDhp. A, the open form of the tunnel enables the substrate to diffuse into the active site. Dashed circle, Mβ binding site. Oblongs, the dynamic loops Ala69–Arg74 and Met158–Met165, in which Phe70 and Tyr160 are labeled. Dashed lines, interactions. B, two dynamic loops shift to the closed form (dashed oblong to the solid line) and lock the substrate in the specific position. The carbamylated lysine 155 (Kcx155) may assist the substrate binding and catalysis. C, the water undergoes nucleophilic attack and generates putative products. The interactions between product and Tyr160 observed in the structures of the complex may indicate the involvement of Tyr160 in the stabilization of the intermediate state (dashed line). D, the C terminus from another monomer helps to open the tunnel, and the substrate diffuses out of the active site.
In order to maintain the substrate in the specific position for catalysis, some structural changes have occurred. Two dynamic loops (Ala69–Arg74 and Met158–Met165) with aromatic residues Phe70 and Tyr160 have moved closer to the active site, locking the substrates in the active site, as seen in the dimetal TnDhp with the tunnel characteristic of the closed form (Figs. 5E and 8B).
While the substrate is bound to the active site, some residues, such as Gly294 and Asn343, are employed to stabilize the substrate. For substrates, such as DHT, that belong to 5-monosubstituted dihydropyrimidines, the stereo-selectivity of DHP is the l-configuration, not the d-configuration, as seen in the structure of the NCβI complex (Fig. 6B).
Catalytic mechanisms involving both one zinc and two zinc ions have been proposed for the DHP proteins (5, 37, 55). The mechanism with one zinc ion was first proposed for the DHP from calf and pig liver (55). In this proposed mechanism, the zinc ion acts as a Lewis acid, and an amino acid residue behaves both as a general base and acid to mediate the catalysis. The substrate is bound to the metal ion (Lewis acid) through the 4-oxo group; the general base, most likely the aspartic acid (Asp322 in TnDhp), activates a water molecule for the nucleophilic attack at the C-4 carbon of the substrate, generating a tetrahedral intermediate, which is stabilized by a tyrosine residue (Tyr160 in TnDhp) (5). The ring of the substrate in the tetrahedral intermediate then opens up to yield the products upon general acid protonation of the ring nitrogen using the same aspartic acid residue. On the other hand, the two-zinc mechanism of DHP proposed is based on the mechanism of dihydroorotase (24, 38), because it is generally believed that these two protein systems share the similar catalytic mechanism (5, 39). In this scheme, the Mβ zinc interacts with the carbonyl oxygen (O4) of the substrate. This is followed by nucleophilic attack through the hydroxyl group of the water molecule and assisted by the carboxylate of the aspartic acid. Both the Mα and the Mβ zinc ions as well as a tyrosine residue (Tyr160 in TnDhp) (5) are used to stabilize the tetrahedral intermediate state. After protonation of the amide nitrogen of the substrate assisted by the same aspartic acid, the product is generated and coordinates with the two zinc ions.
Indeed, our activity and structure analyses of TnDhp described earlier show that one stable metal in the active site could achieve the maximum activity. Thus, it seems that the one-metal mechanism is operative in the catalysis mediated by the vertebrate DHP. However, according to our results, the metal in the Mβ site is only loosely bound in the absence of substrate. The regulatory role we discussed earlier for this metal could well be related to the binding of substrate and subsequent catalysis (Fig. 8C). Hence, it might be that only one metal is detected in the active site in vertebrate DHPs, but for the biochemistry, the second metal is essential, and it is conceivable that the binding of the second metal and of the substrate reinforce each other to promote the catalysis. On the other hand, as a ligand of the Mα metal, the role of Asp322 as a general base/acid is diminished. Instead, Tyr160 stabilizing the intermediate state may be important in the catalysis turnover.
After the catalytic reaction, the product can only be released through the same tunnel. To change the tunnel from the closed to the open form, extra forces will be needed to entice the dynamic loops to return to the open state. According to our structures, the C-terminal tail from the other monomer is positioned near the dynamic loop and the active site. This C-terminal region has high temperature B-factors in the x-ray structure, so the dynamic loop could potentially open via thermal fluctuations (Fig. 5, B and C). While the dynamic loop is open, the product (and possibly the second metal as well) could diffuse out of the active site, following the pathway(s) of the three ADA molecules in the tunnel of the apo-TnDhp structure (Figs. 7 and 8D). Subsequently, another cycle of the reaction can be repeated with a new substrate entering into the active site.
In summary, we have demonstrated the possible existence of four stages of the metal-mediated lysine carbamylation with various conformational changes at the active site for the biological function of a vertebrate DHP. Lysine carbamylation results in a tightly bound metal ion at the Mα site, which participates in both CO2 fixation and catalysis, and a weakly bound metal ion at the Mβ site introduces specific dynamic properties into the protein structure to allow the regulation of the size of the substrate/product tunnel. This information provides a basis to explain the longstanding question in the literature on the varying number of metal ions associated with the carbamylated lysine in this class of enzymes.
Acknowledgments
We are indebted to the supporting staffs at beamlines BL13B1 and BL13C1 of the National Synchrotron Radiation Research Center and to Masato Yoshimura and Hirofumi Ishii at Beamlines BL12B2 and BL44XU of SPring-8.
This work was supported in part by National Science Council (NSC) Grants NSC 98-2311-B-213-MY3 and NSC 101-2628-B-213-MY4 and National Synchrotron Radiation Center Grants 1003RSB02 and 1013RSB02 (to C. J. C.) and NSC Grant NSC 99-2311-B-009-004-MY3 and the Aiming for the Top University and Elite Research Center Development Plan of Ministry of Education (MOE-ATU Plan) (to Y. S. Y.).
The atomic coordinates and structure factors (codes 4GZ7, 4H00, 4H01, 4LCQ, 4LCR, and 4LCS) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Y.-C. Hsieh, M.-C. Chen, C.-C. Hsu, S. I. Chan, Y.-S. Yang, and C.-J. Chen, unpublished data.
- Rubisco
- ribulose-bisphosphate carboxylase/oxygenase
- DHP
- dihydropyrimidinase
- TnDhp
- dihydropyrimidinase from T. nigoviridis
- HYD
- hydantoinase
- ADA
- N-(2-acetamido)-iminodiacetic acid
- NCβI
- N-carbamyl-β-amino isobutyrate
- NCβA
- N-carbamyl-β-alanine
- BisTris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- DHU
- dihydrouracil
- DHT
- dihydrothymine
- ICP
- inductively coupled plasma.
REFERENCES
- 1. Zhang K., Zheng S., Yang J. S., Chen Y., Cheng Z. (2013) Comprehensive profiling of protein lysine acetylation in Escherichia coli. J. Proteome Res. 12, 844–851 [DOI] [PubMed] [Google Scholar]
- 2. Huq M. D., Ha S. G., Barcelona H., Wei L. N. (2009) Lysine methylation of nuclear co-repressor receptor interacting protein 140. J. Proteome Res. 8, 1156–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gaucher S. P., Redding A. M., Mukhopadhyay A., Keasling J. D., Singh A. K. (2008) Post-translational modifications of Desulfovibrio vulgaris Hildenborough sulfate reduction pathway proteins. J. Proteome Res. 7, 2320–2331 [DOI] [PubMed] [Google Scholar]
- 4. Taylor T. C., Backlund A., Bjorhall K., Spreitzer R. J., Andersson I. (2001) First crystal structure of Rubisco from a green alga, Chlamydomonas reinhardtii. J. Biol. Chem. 276, 48159–48164 [DOI] [PubMed] [Google Scholar]
- 5. Lohkamp B., Andersen B., Piskur J., Dobritzsch D. (2006) The crystal structures of dihydropyrimidinases reaffirm the close relationship between cyclic amidohydrolases and explain their substrate specificity. J. Biol. Chem. 281, 13762–13776 [DOI] [PubMed] [Google Scholar]
- 6. St Maurice M., Reinhardt L., Surinya K. H., Attwood P. V., Wallace J. C., Cleland W. W., Rayment I. (2007) Domain architecture of pyruvate carboxylase, a biotin-dependent multifunctional enzyme. Science 317, 1076–1079 [DOI] [PubMed] [Google Scholar]
- 7. Xiang S., Tong L. (2008) Crystal structure of human and Staphylococcus aureus pyruvate carboxylase and molecular insights into the carboxyltransfer reaction. Nat. Struct. Mol. Biol. 15, 295–302 [DOI] [PubMed] [Google Scholar]
- 8. Wang D., De Vivo D. (2009) Pyruvate carboxylase deficiency. in GeneReviewsTM (Pagon R. A., Bird T. D., Dolan C. T., Stephens K., Adam M. P., eds) www.ncbi.nlm.nih.gov/books/NBK6852/, University of Washington, Seattle, WA: [PubMed] [Google Scholar]
- 9. van Kuilenburg A. B., Dobritzsch D., Meijer J., Meinsma R., Benoist J. F., Assmann B., Schubert S., Hoffmann G. F., Duran M., de Vries M. C., Kurlemann G., Eyskens F. J., Greed L., Sass J. O., Schwab K. O., Sewell A. C., Walter J., Hahn A., Zoetekouw L., Ribes A., Lind S., Hennekam R. C. (2010) Dihydropyrimidinase deficiency. Phenotype, genotype and structural consequences in 17 patients. Biochim. Biophys. Acta 1802, 639–648 [DOI] [PubMed] [Google Scholar]
- 10. Cleland W. W., Andrews T. J., Gutteridge S., Hartman F. C., Lorimer G. H. (1998) Mechanism of rubisco. The carbamate as general base. Chem. Rev. 98, 549–562 [DOI] [PubMed] [Google Scholar]
- 11. Bush K., Mobashery S. (1998) How β-lactamases have driven pharmaceutical drug discovery. Form mechanistic knowledge to clinical circumvention. Adv. Exp. Med. Biol. 456, 71–98 [PubMed] [Google Scholar]
- 12. Kotra L P., Samama J. P., Mobashery S. (2002) Structural aspects of β-lactamase evolution. in Bacterial Resistance to Antimicrobials, Mechanisms, Genetics, Medical Practice and Public Health (Lews A., Salyers A. A., Haber H., Wax R. G., eds) pp. 123–159, Marcel Dekker, Inc., New York [Google Scholar]
- 13. Bush K. (2001) New β-lactamases in Gram-negative bacteria. A diversity and impact on the selection of antimicrobial therapy. Clin. Infect. Dis. 32, 1085–1089 [DOI] [PubMed] [Google Scholar]
- 14. Golemi D., Maveyraud L., Vakulenko S., Samama J. P., Mobashery S. (2001) Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc. Natl. Acad. Sci. U.S.A. 98, 14280–14285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ewing S. P., Lockshon D., Jencks W. (1980) Mechanism of cleavage of carbamate anions. J. Am. Chem. Soc. 102, 3071–3084 [Google Scholar]
- 16. Lorimer G. H. (1983) Carbon dioxide and carbamate formation. The makings of a biochemical control system. Trends Biochem. Sci. 8, 65–68 [Google Scholar]
- 17. Walsh C. T., Garneau-Tsodikova S., Gatto G. J., Jr. (2005) Protein posttranslational modifications. The chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 44, 7342–7372 [DOI] [PubMed] [Google Scholar]
- 18. An W. (2007) Histone acetylation and methylation. Combinatorial players for transcriptional regulation. Subcell. Biochem. 41, 351–369 [PubMed] [Google Scholar]
- 19. Seibert C. M., Raushel F. M. (2005) Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry 44, 6383–6391 [DOI] [PubMed] [Google Scholar]
- 20. Wallach D. P., Grisolia S. (1957) The purification and properties of hydropyrimidine hydrase. J. Biol. Chem. 226, 277–288 [PubMed] [Google Scholar]
- 21. Syldatk C., May O., Altenbuchner J., Mattes R., Siemann M. (1999) Microbial hydantoinases-industrial enzymes from the origin of life? Appl. Microbiol. Biotechnol. 51, 293–309 [DOI] [PubMed] [Google Scholar]
- 22. Altenbuchner J., Siemann-Herzberg M., Syldatk C. (2001) Hydantoinases and related enzymes as biocatalysts for the synthesis of unnatural chiral amino acids. Curr. Opin. Biotechnol. 12, 559–563 [DOI] [PubMed] [Google Scholar]
- 23. Radha Kishan K. V., Vohra R. M., Ganesan K., Agrawal V., Sharma V. M., Sharma R. (2005) Molecular structure of d-hydantoinase from Bacillus sp. AR9. Evidence for mercury inhibition. J. Mol. Biol. 347, 95–105 [DOI] [PubMed] [Google Scholar]
- 24. Thoden J. B., Phillips G. N., Jr., Neal T. M., Raushel F. M., Holden H. M. (2001) Molecular structure of dihydroorotase. A paradigm for catalysis through the use of a binuclear metal center. Biochemistry 40, 6989–6997 [DOI] [PubMed] [Google Scholar]
- 25. Brooks K. P., Jones E. A., Kim B. D., Sander E. G. (1983) Bovine liver dihydropyrimidine amidohydrolase. Purification, properties, and characterization as a zinc metalloenzyme. Arch. Biochem. Biophys. 226, 469–483 [DOI] [PubMed] [Google Scholar]
- 26. Kikugawa M., Kaneko M., Fujimoto-Sakata S., Maeda M., Kawasaki K., Takagi T., Tamaki N. (1994) Purification, characterization and inhibition of dihydropyrimidinase from rat liver. Eur. J. Biochem. 219, 393–399 [DOI] [PubMed] [Google Scholar]
- 27. Su T. M., Yang Y. S. (2000) Identification, purification, and characterization of a thermophilic imidase from pig liver. Protein Expr. Purif. 19, 289–297 [DOI] [PubMed] [Google Scholar]
- 28. Davis G. D., Elisee C., Newham D. M., Harrison R. G. (1999) New fusion protein systems designed to give soluble expression in Escherichia coli. Biotechnol. Bioeng. 65, 382–388 [PubMed] [Google Scholar]
- 29. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 30. Vagin J., Teplyakov A. (1997) Molrep. An automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 31. Brünger A. T. (1992) Free R value. A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 [DOI] [PubMed] [Google Scholar]
- 32. Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system. A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 33. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 34. Winn M. D., Isupov M. N., Murshudov G. N. (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 57, 122–133 [DOI] [PubMed] [Google Scholar]
- 35. Lamzin V. S., Wilson K. S. (1993) Automated refinement of protein models. Acta Crystallogr. D Biol. Crystallogr. 49, 129–147 [DOI] [PubMed] [Google Scholar]
- 36. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gojkovic Z., Rislund L., Andersen B., Sandrini M. P., Cook P. F., Schnackerz K. D., Piskur J. (2003) Dihydropyrimidine amidohydrolases and dihydroorotases share the same origin and several enzymatic properties. Nucleic Acids Res. 31, 1683–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Benning M. M., Kuo J. M., Raushel F. M., Holden H. M. (1995) Three-dimensional structure of the binuclear metal center of phosphotriesterase. Biochemistry 34, 7973–7978 [DOI] [PubMed] [Google Scholar]
- 39. Martínez-Rodríguez S., Martínez-Gómez A. I., Clemente-Jiménez J. M., Rodríguez-Vico F., García-Ruíz J. M., Las Heras-Vázquez F. J., Gavira J. A. (2010) Structure of dihydropyrimidinase from Sinorhizobium meliloti CECT4114. New features in an amidohydrolase family member. J. Struct. Biol. 169, 200–208 [DOI] [PubMed] [Google Scholar]
- 40. Huang C. Y., Hsu C. C., Chen M. C., Yang Y. S. (2009) Effect of metal binding and posttranslational lysine carboxylation on the activity of recombinant hydantoinase. J. Biol. Inorg. Chem. 14, 111–121 [DOI] [PubMed] [Google Scholar]
- 41. Hall P. R., Zheng R., Antony L., Pusztai-Carey M., Carey P. R., Yee V. C. (2004) Transcarboxylase 5S structures. Assembly and catalytic mechanism of a multienzyme complex subunit. EMBO J. 23, 3621–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ha N. C., Oh S. T., Sung J. Y., Cha K. A., Lee M. H., Oh B. H. (2001) Supramolecular assembly and acid resistance of Helicobacter pylori urease. Nat. Struct. Biol. 8, 505–509 [DOI] [PubMed] [Google Scholar]
- 43. Kim K., Kim M. I., Chung J., Ahn J. H., Rhee S. (2009) Crystal structure of metal-dependent allantoinase from Escherichia coli. J. Mol. Biol. 387, 1067–1074 [DOI] [PubMed] [Google Scholar]
- 44. Lee M., Maher M. J., Christopherson R. I., Guss J. M. (2007) Kinetic and structural analysis of mutant Escherichia coli dihydroorotases. A flexible loop stabilized the transition state. Biochemistry 46, 10538–10550 [DOI] [PubMed] [Google Scholar]
- 45. Xiang D. F., Patskovsky Y., Xu C., Meyer A. J., Sauder J. M., Burley S. K., Almo S. C., Raushel F. M. (2009) Functional identification of incorrectly annotated prolidases from the amidohydrolase superfamily of exzymes. Biochemistry 48, 3730–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cheon Y. H., Kim H. S., Han K. H., Abendroth J., Niefind K., Schomburg D., Wang J., Kim Y. (2002) Crystal structure of d-hydantoinase from Bacillus stearothermophilus. Insight into the stereochemistry of enantioselectivity. Biochemistry 41, 9410–9417 [DOI] [PubMed] [Google Scholar]
- 47. Cheon Y. H., Park H. S., Kim J. H., Kim Y., Kim H. S. (2004) Manipulation of the active site loops of d-hydantoinase, a (β/α)8-barrel protein, for modulation of the substrate specificity. Biochemistry 43, 7413–7420 [DOI] [PubMed] [Google Scholar]
- 48. Kim G. J., Kim H. S. (1998) C-terminal regions of d-hydantoinases are nonessential for catalysis, but affect the oligomeric structure. Biochem. Biophys. Res. Commun. 243, 96–100 [DOI] [PubMed] [Google Scholar]
- 49. Che T., Bonomo R. A., Shanmugam S., Bethel C. R., Pusztai-Carey M., Buynak J. D., Carey P. R. (2012) Carboxylation and decarboxylation of active site Lys 84 controls the activity of OXA-24 β-lactamase of Acinetobacter baumannii. Raman crystallographic and solution evidence. J. Am. Chem. Soc. 134, 11206–11215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Liaw S. H., Chen S. J., Ko T. P., Hsu C. S., Chen C. J., Wang A. H., Tsai Y. C. (2003) Crystal structure of d-aminoacylase from Alcaligenes faecalis DA1. A novel subset of amidohydrolases and insights into the enzyme mechanism. J. Biol. Chem. 278, 4957–4962 [DOI] [PubMed] [Google Scholar]
- 51. Martínez-Rodríguez S., Encinar J. A., Hurtado-Gómez E., Prieto J., Clemente-Jiménez J. M., Heras-Vázquez F. J. L., Rodríguez-Vico F., Neira J. L. (2009) Metal-triggered changes in the stability and secondary structure of a tetrameric dihydropyrimidinase. A biophysical characterization. Biophys. Chem. 139, 42–52 [DOI] [PubMed] [Google Scholar]
- 52. Kautz J., Schnackerz K. D. (1989) Purification and properties of 5,6-dihydropyrimidine amidohydrolase from calf liver. Eur. J. Biochem. 181, 431–435 [DOI] [PubMed] [Google Scholar]
- 53. Zhang X. Y., Niu L. X., Shi Y. W., Yuan J. M. (2008) The flexibility of the non-conservative region at C terminus of d-hydantoinase from Pseudomonas putida YZ-26 is extremely limited. Appl. Biochem. Biotechnol. 144, 237–247 [DOI] [PubMed] [Google Scholar]
- 54. Niu L., Zhang X., Shi Y., Yuan J. (2007) Subunit dissociation and stability alteration of d-hydantoinase deleted at the terminal amino acid residue. Biotechnol. Lett. 29, 303–308 [DOI] [PubMed] [Google Scholar]
- 55. Jahnke K., Podschun B., Schnackerz K. D., Kautz J., Cook P. F. (1993) Acid-base catalytic mechanism of dihydropyrimidinase from pH studies. Biochemistry 32, 5160–5166 [DOI] [PubMed] [Google Scholar]