

Background: Matrix metalloproteinase-1 (MMP-1) collagenolysis relies on interdomain flexibility.
Results: In all high maximum occurrence conformations, the MMP-1 hemopexin-like domain residues reported responsible for binding to the collagen triple-helix are solvent exposed.
Conclusion: MMP-1 in solution is poised to interact with collagen and proceed along the steps of collagenolysis.
Significance: The maximum occurrence approach can evaluate the predominant domain conformations for numerous multidomain enzymes.
Keywords: Chemical Biology, Collagen, Matrix Metalloproteinase (MMP), Metalloprotease, Structural Biology, Collagenolysis, Maximum Occurrence
Abstract
Catalysis of collagen degradation by matrix metalloproteinase 1 (MMP-1) has been proposed to critically rely on flexibility between the catalytic (CAT) and hemopexin-like (HPX) domains. A rigorous assessment of the most readily accessed conformations in solution is required to explain the onset of substrate recognition and collagenolysis. The present study utilized paramagnetic NMR spectroscopy and small angle x-ray scattering (SAXS) to calculate the maximum occurrence (MO) of MMP-1 conformations. The MMP-1 conformations with large MO values (up to 47%) are restricted into a relatively small conformational region. All conformations with high MO values differ largely from the closed MMP-1 structures obtained by x-ray crystallography. The MO of the latter is ∼20%, which represents the upper limit for the presence of this conformation in the ensemble sampled by the protein in solution. In all the high MO conformations, the CAT and HPX domains are not in tight contact, and the residues of the HPX domain reported to be responsible for the binding to the collagen triple-helix are solvent exposed. Thus, overall analysis of the highest MO conformations indicated that MMP-1 in solution was poised to interact with collagen and then could readily proceed along the steps of collagenolysis.
Introduction
Matrix metalloproteinases (MMPs)3 are a family of proteases with the striking feature of hydrolyzing structurally unrelated substrates (1, 2). This broad proteolytic specificity, together with tight regulation of enzyme activation and localization, has been achieved by an evolutionary process where specialization of protein domains and protein flexibility interplay to facilitate recognition and hydrolysis of a variety of substrates (3). In particular, several active MMPs, including MMP-1, are two-domain (catalytic (CAT) and hemopexin-like (HPX)) enzymes capable of catalyzing the hydrolysis of highly structured substrates such as triple-helical, interstitial (types I–III) collagen (4). Interdomain flexibility appears particularly important for allowing movement of the MMP along collagen fibrils and for unwinding/perturbation of the collagen and accommodation of a single, otherwise inaccessible, peptide chain into the active site (3, 5–11).
The steps contributing to the collagenolytic process are becoming better understood (Fig. 1). A range of solution and crystal state conformations has been described for full-length MMP-1 (Fig. 1B) (8, 9, 12–16). MMP-1 has been experimentally found to interact with the collagen triple-helix through specific residues in blades I and II of the HPX domain (Fig. 1A) (9, 16, 17). CAT domain binding is guided by the association of the HPX domain with collagen and the interdomain flexibility is crucial; for example, if one superimposes the MMP-1 x-ray crystallographic structure with the HPX domain in the experimentally determined position, the CAT domain collides with the triple-helical peptide (THP) (Fig. 1C). Once both MMP domains interact (Fig. 1D), the triple-helix is destabilized, allowing insertion of a single-strand into the active site. Hydrolysis of the first strand is presumably followed by rapid hydrolysis of the other two strands. The initial interaction of MMP-1 with collagen is controversial, and depends upon which structure is favored by MMP-1 in solution prior to binding the substrate (9, 16). Overall, assessment of the most easily accessed enzyme conformations within the ensemble of all sterically possible conformations in solution can be critical to understanding substrate recognition.
FIGURE 1.

Interaction of MMP-1 with the collagen triple-helix. A, the HPX domain of MMP-1 binds the collagen triple-helix through specific residues in blades I and II (highlighted in magenta; the Gly-Ile cleavage site within the triple-helical peptide is shown in blue). B, experimentally determined regions of CAT and HPX domains involved in binding of the triple-helix are highlighted in magenta. The conformation of MMP-1 is based on the x-ray crystallographic structure 2CLT (active, full-length MMP-1). C, if the 2CLT structure is maintained, binding of the HPX domain to the triple-helix results in the collision of the CAT domain with the triple-helix (residues highlighted in red). D, interdomain flexibility is required for the MMP-1 to correctly approach the substrate, as described for the first step of collagenolysis (9).
When a system rapidly samples multiple conformations, the experimental data are a weighted average relative to each conformation. Various methods (18–29) have been proposed to reconstruct ensembles consistent with the experimental data. To advance from simply obtaining many “plausible” ensembles to identifying specific conformations within these ensembles that are more likely sampled by the system, maximum allowed probability was proposed (30), later extended to the concept of maximum occurrence (MO) (31, 32). The MO of a given conformation is defined, and numerically calculated, as the maximum weight that this conformation can have in any suitable ensemble while still maintaining the ability of the ensemble to reproduce the experimental data. Paramagnetic NMR spectroscopic and small angle x-ray scattering (SAXS) data can be used as experimental restraints to calculate the MO of conformations of two-domain proteins, as previously demonstrated for calmodulin (CaM) alone (31, 33, 34) and its complexes with target peptides (30, 35). The paramagnetic restraints originate from the presence of paramagnetic metals, incorporated either in an existing metal binding site (36) or in a tag covalently bound to the protein (33). In the present case a lanthanide binding tag was used. Remarkably, this is the first case in which a paramagnetic thiol-reactive tag is attached to a protein bearing structural disulfides.
MMP-1 was analyzed herein using the MO approach. Many of the MMP-1 conformations with the highest MO value were found to have interdomain orientations and positions that can be clearly grouped into a cluster. Remarkably, in the conformations belonging to this cluster, (i) the collagen binding residues of the HPX domain were solvent exposed and (ii) the CAT domain was already correctly positioned for its subsequent interaction with the collagen. A structural rearrangement involving a ∼50° rotation around a single axis of the CAT domain with respect to the HPX domain was sufficient to position the CAT domain right in front of the preferred cleavage site in triple-helical collagen. The conformations belonging to this cluster thus defined the antecedent step of collagenolysis.
EXPERIMENTAL PROCEDURES
Protein Preparation
The MMP-1 E219A construct (residues Asn106 to Asn469) was prepared as described previously. E219A mutation was performed to prevent self-proteolysis (8). The MMP-1 mutations H132C and K136C were engineered to attach (Ln)CLaNP-5 to the protein through disulfide bonds. The residues mutated were on the rigid amphipathic helix (hA), far enough from the active site cleft and the HPX domain to avoid steric clashes that could affect the conformational heterogeneity of the protein. The double mutation H132C/K136C was obtained during a single PCR step using the QuikChange Site-directed Mutagenesis Kit (Stratagene): 5′-GCC AAG AGC AGA TGT GGA CTG TGC CAT TGA GTG TGC CTT CCA ACT CTG GAG-3′; 5′-CTC CAG AGT TGG AAG GCA CAC TCA ATG GCA CAG TCC ACA TCT GCT CTT GGC-3′. The mutations were confirmed by nucleotide sequencing. The expression vector was inserted into the competent Escherichia coli BL21(DE3) CodonPlus RIPL strain, and the colonies were selected for ampicillin and chloramphenicol resistance. Monolabeled 15N protein was expressed using minimal medium containing 15N-enriched (NH4)2SO4. Cell growth occurred at 37 °C with induction at 0.6 optical density with 500 μm isopropyl 1-thio-β-d-galactopyranoside and harvesting after 5 h expression. Triple mutant MMP-1 (H132C/K136C/E219A) precipitated into inclusion bodies, and these were solubilized, after lysis of the cells, in a solution of 8 m urea, 20 mm dithiothreitol, and 20 mm Sigma Trizma (Tris base), pH 8, and stored at −20 °C. The refolding of triple mutant MMP-1 consisted of decreasing the urea concentration according to the following steps, performed at 4 °C. The desired amount of protein was diluted in a 500-ml solution containing 6 m urea, 50 mm Trizma, 10 mm CaCl2, 0.1 mm ZnCl2, and 20 mm cysteamine, pH 8.0. The solution was then dialyzed against (a) 4 liters of 4 m urea, 50 mm Trizma, pH 8.0, 10 mm CaCl2, 0.1 mm ZnCl2, 5 mm 2-mercaptoethanol, and 1 mm hydroxyethyl disulfide (overnight dialysis); (b) 4 liters of 2 m urea, 50 mm Trizma, pH 7.2, 10 mm CaCl2, 0.1 mm ZnCl2, and 0.3 m NaCl; and (c) three steps of 20 mm Trizma, pH 7.2, 10 mm CaCl2, 0.1 mm ZnCl2, and 0.3 m NaCl. The resulting 500-ml protein sample was concentrated down to 100 ml using MiniKros Modules (Spectrumlabs). H132C/K136C/E219A MMP-1 was purified using HiLoad 26/60 Superdex 75 pg (Amersham Biosciences) in 20 mm Trizma, pH 7.2, 10 mm CaCl2, 0.1 mm ZnCl2, and 0.3 m NaCl buffer. Protein pure stocks were stored at 4 °C.
(Ln)CLaNP-5-Protein Ligation
CLaNP-5 was synthesized and functionalized with the different lanthanides as previously described (37). 2 mg of purified H132C/K136C/E219A MMP-1 was concentrated down to 1 ml in 2 m Trizma, pH 7.2, 10 mm CaCl2, 0.1 mm ZnCl2, and 0.3 m NaCl buffer. 6–10 equivalents of (Ln)CLaNP-5 (where the lanthanide ions were Lu3+, Tb3+, Dy3+, and Tm3+) from N,N-dimethylformamide stock (about 3–6 μl) were added to the protein solution. The triple mutant MMP-1/(Ln)CLaNP-5 mixture was left on mild stirring overnight. Some protein precipitation was observed after reaction. Contrary to the procedure for the single MMP-1 CAT domain (37), no DTT or reductant of any kind was added to the protein at any stage of the (Ln)CLaNP-5-protein ligation to avoid reduction of the structurally important and solvent-exposed disulfide bridge present in the HPX domain between Cys278 and Cys466. After reaction with (Ln)CLaNP-5, ∼10–20% of unreacted MMP-1 remained, as estimated from the 1H-15N heteronuclear single quantum coherence spectra acquired on these samples. The signals of the unreacted fraction were easily distinguishable from the paramagnetic ones. The presence of a significant amount of protein bound to the tag with a single bond can be excluded because it would have resulted in some protein molecules bearing a mobile tag, with consequent doubling of some resonances in the CAT domain due to a significant difference in the pseudocontact shifts (PCS) (and residual dipolar couplings (RDC)) of nuclei in this domain. No double peaks of this type were observed. The overall yield of obtained paramagnetic (Ln)CLaNP-5-MMP-1, considering precipitation occurring during CLaNP-5 reaction and efficiency of MMP-1 functionalization, was estimated to be ∼60–70%.
NMR Measurements
All experiments were performed on samples of triple mutant (H132C/K136C/E219A), full-length MMP-1 functionalized with (Ln)CLaNP-5 (Ln = Lu3+, Tb3+, Dy3+, Tm3+), at concentrations ranging between 0.10 and 0.20 mm in water buffer solution (20 mm Tris, pH 7.2, 0.15 m NaCl, 0.1 mm ZnCl2, 10 mm CaCl2, and 200 mm l-azidohomoalanine). All NMR experiments were performed at 310 K and acquired on a Bruker AVANCE 700 spectrometer equipped with triple resonance cryo-probe. All spectra were processed with the Bruker TOPSPIN software packages and analyzed by the program CARA (Computer Aided Resonance Assignment, ETH Zurich). The two-dimensional 1H-15N heteronuclear single quantum coherence spectrum of (Lu3+)CLaNP-5-MMP-1 was recorded as the diamagnetic reference to evaluate the PCSs. The assignment of the protein functionalized with (Lu3+)CLaNP-5 was based on the assignment previously reported for MMP-1 (37); the spectrum was easily reassigned because no meaningful shifts with respect to the non-functionalized protein were observed, indicating that the presence of the (Ln3+)CLaNP-5 does not alter the structure of the protein. The assignment of MMP-1 in the presence of the paramagnetic lanthanides was performed by comparison with the assigned spectra obtained for the isolated CAT domain in the presence of the same metal ions. 1H-15N RDCs were measured for the MMP-1 functionalized with (Tb3+)CLaNP-5, (Dy3+)CLaNP-5, and (Tm3+)CLaNP-5, by using the IPAP-heteronuclear single quantum coherence method.
SAXS Measurements and Data Processing
The synchrotron x-ray scattering data were collected on the X33 beamline of the EMBL (storage ring DORIS-III, DESY, Hamburg) (38) using a MAR345 image plate detector as described (8). The scattering patterns were measured with a 2-min exposure time for several solute concentrations in the range from 0.8 to 8.3 mg/ml. The data were reduced by standard procedures, processed and extrapolated to infinite dilution by PRIMUS (39). The scattering from the high resolution models was computed using CRYSOL (40), and the agreement with the experimental data were characterized by a discrepancy function provided by Equation 1.
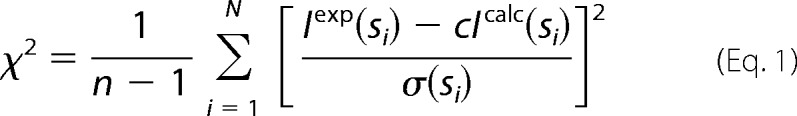 |
Here, n is the number of experimental points, c is a scaling factor, Iexp(si) and Icalc(si) are the experimental and calculated intensities, respectively, and σ(si) is the experimental error at the momentum transfer si. The latter is defined as s = 4π sin(θ)/λ, where 2θ is the scattering angle, and λ = 1.5 Å is the x-ray wavelength.
MO Calculations
Information on the conformations that can be experienced by a flexible multidomain protein is recovered by calculating the maximum weight that each of them can have in all possible structural ensembles, and still provide agreement between predicted and experimental data. This was done by generating a large pool (composed in this case of 50,000 conformations) of sterically possible protein structures, and then calculating the MO value of a subset of structures (1,000 conformations herein), which monitor how the MO changes throughout the conformational space. The pool of 50,000 conformations was randomly generated with the program RanCh, using a flexible linker of 13 residues (from Arg262 to Thr274) to connect the rigid structures of the previously refined CAT (37) and HPX (PDB entry 1SU3) domains. The MO value of each of the selected 1,000 MMP-1 conformations was obtained from the largest weight that the conformation can have when included in any ensemble able to reproduce the experimental data together with 50 other conformations (each of them with its own weight) freely chosen from the entire pool of 50,000 structures (20, 31, 41). In practice, the MO of each of the selected conformations was calculated by taking the conformation with a given weight and selecting the 50 other conformations needed to complete the ensemble to reproduce the experimental data. The fact that a good fit was obtained indicated that the selected conformation could be sampled for the given weight. Such a weight was then increased until no ensemble could be recovered providing a good fit of the experimental data. The MO of this selected conformation was thus fixed (by definition) to the largest weight that still provided a good fit of the data.
The selection of the 50 conformations used to reproduce the experimental data, as well as their weight, was performed by minimizing a target function (TF) defined as a measure of the disagreement from the experimental data and the data calculated according to the ensemble itself,
where the weighting coefficients aPCS and aRDC were 1.0 and aSAXS was 0.1 (these weights were chosen to obtain a reasonable balance between the different restraints).
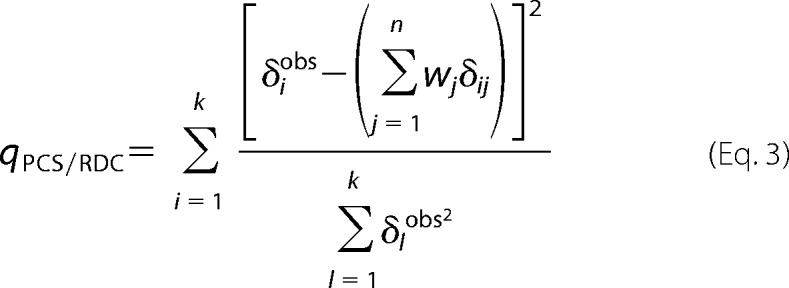 |
δiobs were the experimental PCS/RDC values, δij were the PCS/RDC values calculated for the jth conformation, with weight wj, k was the number of PCS/RDC, n was the number of conformations in the family, χ2 was calculated using the average scattering intensity, Icalc, calculated as,
 |
and c was a scaling coefficient calculated as,
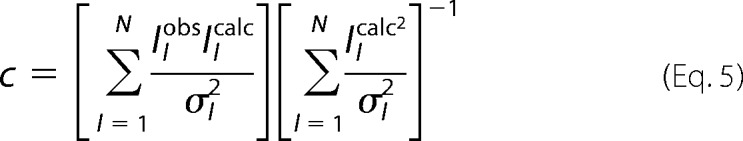 |
PCS and RDC values (δij) were calculated according to,
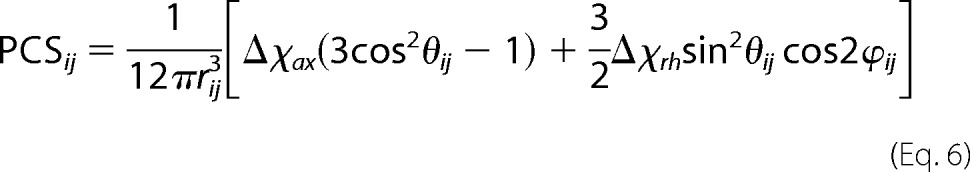 |
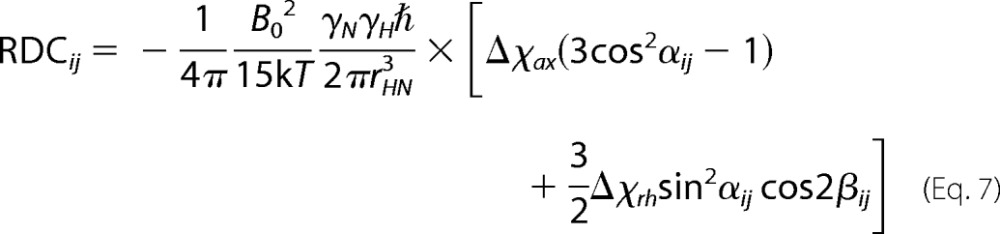 |
where the symbols have the following meaning: rij, θij and ϕij are the spherical coordinates defining the position of the nucleus corresponding to the ith PCS value, in the jth conformation, expressed in the frame of the magnetic susceptibility anisotropy tensor, rHN is the distance between the two coupled nuclei N and NH (set to 1.02 Å), the αij and βij angles define the orientation of the vector connecting the coupled N and NH nuclei corresponding to the ith RDC value, in the jth conformation, expressed in the frame of the magnetic susceptibility anisotropy tensor, Δcax and Δcrh are the axial and rhombic magnetic susceptibility anisotropy parameters, B0 is the magnetic field, T the absolute temperature, k the Boltzmann constant, γH and γN the magnetogyric ratios of proton and nitrogen, respectively, and ℏ the Planck constant divided by 2π.
The TF was defined dimensionless because it was calculated as the squared difference between experimental and calculated values normalized by the squared experimental value for PCS and RDC, and as the squared difference between experimental and calculated values divided by the squared error for SAXS data.
During the minimization, the weight of each of the selected 1,000 MMP-1 conformations was set to different values ranging from 0 to 50% in 5% steps. The MO of such a conformation was set to the largest weight providing a TF smaller than a given threshold. The value of this threshold was fixed in relationship with the absolute minimum of the TF, computed by determining the best fit against structural ensembles generated without any fixed conformation. In the present case the minimum of the TF was equal to ∼0.253. The threshold was defined 10% larger than this lowest value (0.278). This TF value reflected the agreement between calculated and experimental data for SAXS (Fig. 2A), PCS (Fig. 2B), and RDC (Fig. 2C).
FIGURE 2.
A, agreement between the experimental SAXS data (red dots with error bars) and the averaged values (black line) calculated from the best fit ensemble of conformations. The discrepancy between the experimental and calculated data provides a χ2 of 1.0, indicating an excellent fit. Agreement between the experimental (B) PCS and (C) RDC values of backbone amide protons belonging to the HPX domain at 310 K and the averaged values calculated from the best fit ensembles of conformations.
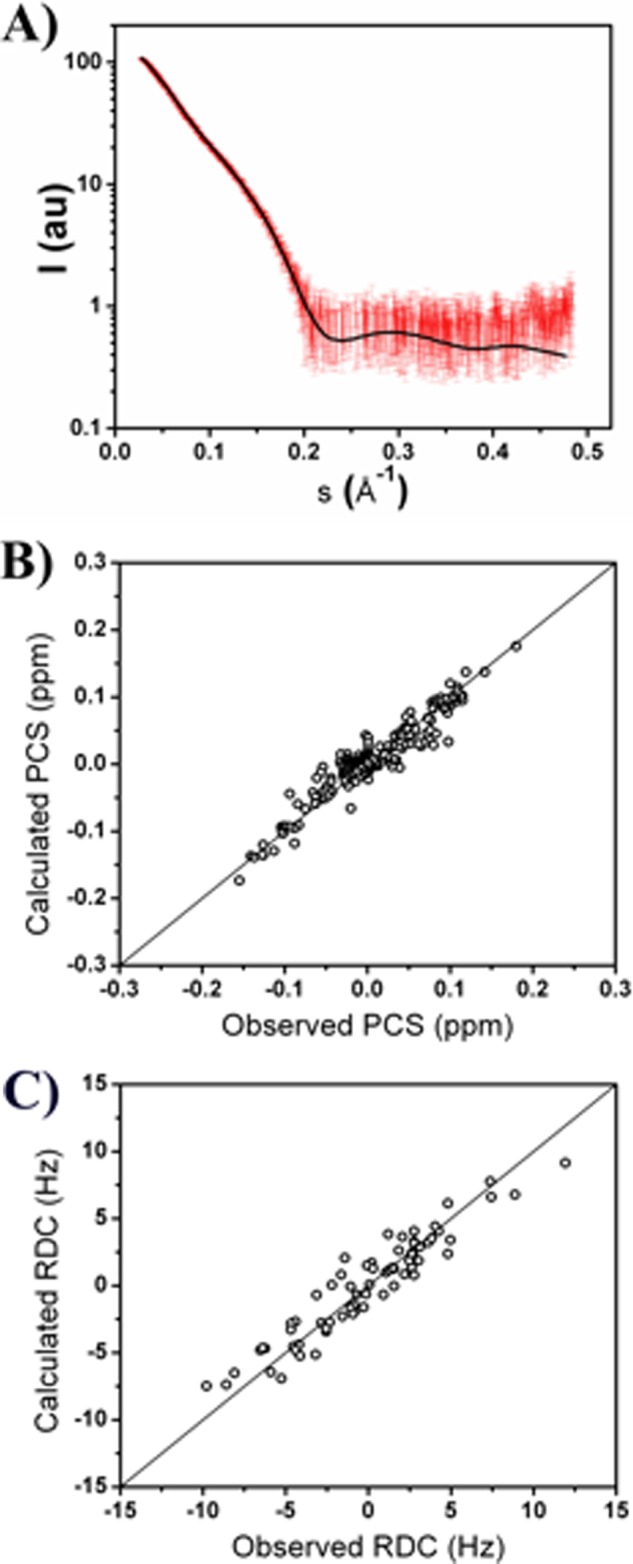
Some of the selected structures (Fig. 3; each of them corresponding to a different line) allowed for small TF values only if their weight was low, whereas other structures allowed for TF values below the given threshold even for much larger weights. This indicated that the former cannot be included in any ensemble in agreement with the experimental data with a relatively large weight, whereas the latter can be included in a best-fit ensemble even for such a weight. This difference is quantitatively described by the MO, which was thus able to discriminate among the different conformations depending on the maximum weight that they can have whatever the best-fit ensemble was among the many possible existing ensembles in good agreement with the data. Calculations were performed through Maxocc, accessible to WeNMR registered users (42).
FIGURE 3.
Progression of the TFs as a function of the weight given to the selected structure within the minimized ensemble of 50 structures. The dashed line represents the threshold at which the MO value was assigned when intercepted by the TF. The TF for the 1,000 calculated structures is represented.
To visualize the results of MO calculations for the selected 1,000 MMP-1 conformations, the following approach was applied: either the CAT (Fig. 4B, left panels) or HPX (Fig. 4B, right panels) domain was superimposed to a reference structure (PDB entry 2CLT). The other domain (i.e. the HPX and the CAT domains in the left and right panels, respectively, of Fig. 4B) was replaced by a “reference frame” composed of a triad of vectors pointing along the axes of a Cartesian coordinate system. The reference frame was oriented to reflect the orientation of the non-superimposed domain, and positioned in the center of mass of the domain. Each reference frame thus corresponded to a different conformation, and indicated the relative position and orientation of the HPX and CAT domains. These reference frames were colored blue, green, yellow, orange, or red depending on the MO value of the conformation that it represented. The lowest MO values were shown in blue, whereas the largest MO values were in red. Two conformations have been selected (Fig. 4A) to show where the reference frame is positioned for these two structures among all the selected 1,000 conformations for which the MO was calculated (Fig. 4B).
FIGURE 4.

Visualization of the results of MO calculations for 1,000 randomly selected MMP-1 conformations. The conformations are displayed superimposed on the CAT domain (left) and on the HPX domain (right). The catalytic metal was represented as an orange sphere. Within the CAT domain (left), the residues in pink are those mutated to Cys to incorporate (Ln)CLaNP-5. In the HPX domain (right) the residues that were experimentally found to interact with a triple-helical peptide (9) are shown in magenta. B, simplified representation of 1000 conformations selected randomly with the allowed conformational space. Each conformation was represented, for graphical simplicity, as a color-coded 3-axes system, positioned in the center of mass of the HPX (left column) or CAT (right column) domain. Colors from blue (<5%) to red (47%) represent the MO values of the various structures. A, the structure with the highest MO (47%) and the x-ray crystallographic structure 2CLT are colored according to their MO values. In the center of mass, the 3-axes system associated with the structure is shown. Both the perspective of the CAT (left) and HPX domains (right) are represented.
RESULTS
Paramagnetism-based NMR Data
PCS and self-orientation of RDC (43) from at least three metals ions with large paramagnetic susceptibility anisotropy are needed as paramagnetic NMR spectroscopic restraints to provide sufficient average data and minimize degeneracy (44). It has been shown that the paramagnetic metal ion cobalt(II) can be introduced in the place of the catalytically active zinc(II) ion (45, 46), and this could complement paramagnetic NMR data. However, the magnetic susceptibility anisotropy of cobalt(II) in the MMP-1 CAT domain is not large enough to provide measurable effects as far away as needed to reach the HPX domain nuclei. The introduction of covalently bound lanthanide chelators has been widely exploited to introduce paramagnetic centers in diamagnetic proteins (47–49); the rigid lanthanide chelator CLaNP-5 (50) does so by covalently binding two neighboring Cys residues in a rigid fashion. CLaNP-5 was incorporated into the MMP-1 CAT domain via Cys132 and Cys136. The correspondence of the chemical shifts in the diamagnetic (Lu3+)-tagged and untagged protein indicated that the presence of (Ln)CLaNP-5 does not affect the CAT structure (37). Furthermore, CLaNP-5 is positioned far away from the HPX domain, so that it is outside of the sterically accessible conformations of the full-length protein.
The magnetic susceptibility anisotropy tensors were determined from the best fit of the PCSs of the CAT domain to the previously refined protein structure (Table 1). The averaged anisotropy tensors obtained from the best fit of the RDCs of the amide protons of the HPX domain to the available x-ray structure of full-length proMMP-1 (PDB entry 1SU3) (13) were also evaluated (Table 1). Sizable motions of the HN vectors could be excluded from the amide relaxation measurements performed on the isolated HPX domain (8). Because the RDCs induced by one paramagnetic center can always be described by a single averaged anisotropy tensor in the case of rigid domains, independently of the fact that they originate from a weighted average of a number of conformations, the good quality of the fits (Fig. 5) reflected good agreement of the data with the x-ray structure of the HPX domain. Thus, the HPX domain moves essentially as a rigid body with respect to the CAT domain. On the basis of these observations, comparison of the two sets of tensors for the CAT and HPX domains reports on the interdomain mobility. For a rigid system, the two sets of tensors should be similar to one another, whereas the tensors of a moving domain are expected to decrease with increased mobility, up to the extreme situation that all sterically allowed conformations are sampled to the same extent and the resulting tensor is dramatically reduced. In the present case, the relative magnitude and orientations of the tensors obtained for the HPX domain with respect to those of the CAT domain (to which (Ln)CLaNP-5 is attached) reveals the presence of sizable interdomain mobility and reflects the conformational heterogeneity experienced by the system in solution (see later). The spreading in the actual distribution from the RDC tensors (Fig. 6C) is sizably smaller (by a factor of 3–4) than expected for a rigid system (Fig. 6A), indicating considerable mobility, but also much larger than the uniform sampling case (Fig. 6B), indicating the occurrence of preferred conformations in solution.
TABLE 1.
Tensors obtained by FANTASIAN software package implemented in the WeNMR Portal med
The PCS measured on the CAT domain and the RDC measured on the HPX domain for 1H-15N couplings were used. Errors were calculated with a Monte Carlo analysis.
| Tb3+ |
Dy3+ |
Tm3+ |
||||
|---|---|---|---|---|---|---|
| Δχax | Δχrh | Δχax | Δχrh | Δχax | Δχrh | |
| 10−32 m3 | 10−32 m3 | 10−32 m3 | ||||
| PCS tensor of CAT | −45.4 (±1%) | 16.5 (±23%) | −40.4 (±1%) | −13.2 (±21%) | 51.9 (±1%) | −9.9 (±12%) |
| RDC tensor of HPX | −12.7 (±3%) | 7.7 (±4%) | −10.9 (±4%) | −2.3 (±28%) | 15.0 (±3%) | −2.3 (±19%) |
FIGURE 5.
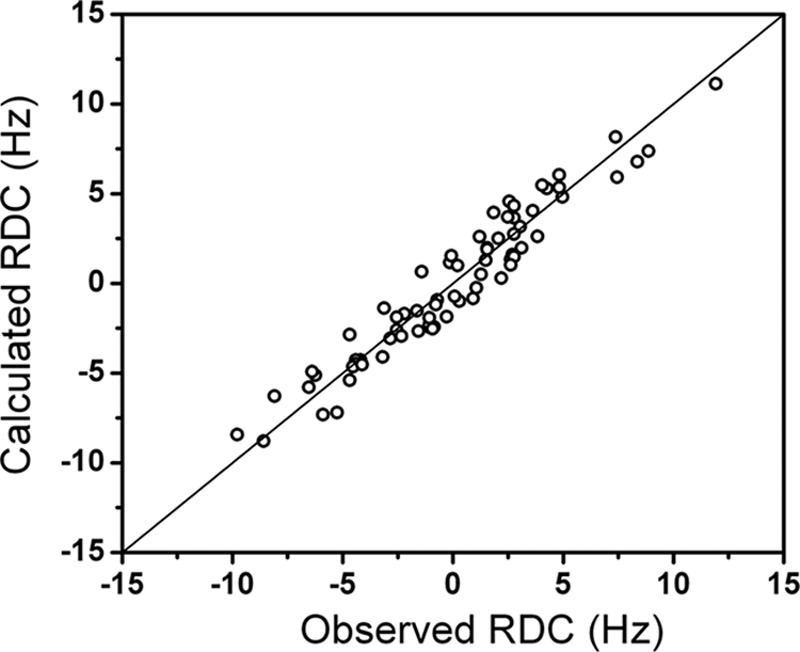
Quality of the fit of calculated versus observed RDC values of the HPX domain. The RDC were calculated for the HPX domain in the x-ray structure 1SU3 by using a single averaged anisotropy tensor. The quality of the fit indicates that the HPX domain moves as a rigid body.
FIGURE 6.
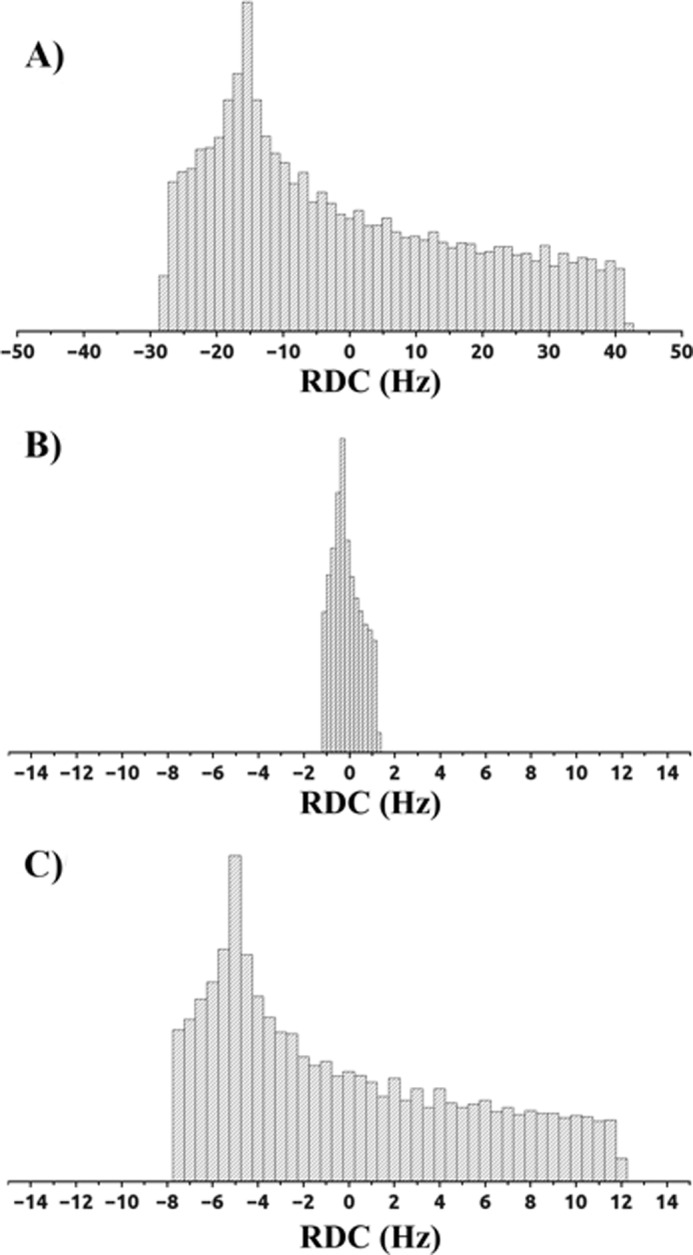
Distribution of the RDC values (Hz) of the (Tm3+)CLaNP-5 MMP-1 HPX domain based on the magnetic susceptibility tensors obtained from (A) experimental PCS of the CAT domain, (B) average RDC of the HPX domain obtained from sterically allowed uniformly sampled conformations, and (C) experimental RDC of the HPX domain.
The ratio of the spreading between the real RDC distribution and the RDC distribution calculated in the assumption of no motion can be taken as a generalized order parameter reflecting the interdomain mobility (25, 51). The generalized order parameters for MMP-1 are 0.28, 0.27, and 0.29 for Tb3+, Dy3+, and Tm3+, respectively. Different generalized order parameters as well as different scaling factors of the components of the anisotropy tensor indicated that the HPX domain motion caused different motional averaging for the different metals, because of the different rhombicity and directions of the principal axes of the anisotropy tensors. SAXS data, previously measured for MMP-1 in solution under the same experimental conditions as utilized here (8, 15), also indicated that the structure of the protein cannot be described by the crystallographic conformation alone, but that ensembles with closed and more extended conformations must be considered, further indicating that the protein experiences noticeable flexibility.
MO Analysis
Recovering the ensemble of structures experienced by a two-domain protein with interdomain flexibility from average data is not possible because an infinite number of equally good solutions exist. The MO approach was developed to determine the maximum weight for a conformation throughout all possible structural ensembles. This provides ranking of all sterically possible conformations according to the maximum weight that they can have within any ensemble that reproduces the experimental data. The conformations with low MO values will thus likely not be representative of the structural ensemble sampled by the protein, whereas conformations with the largest MO values, which better reflect the structural content of the average experimental data, can be more representative of the components of the protein structural ensemble.
A MO analysis was performed using as restraints the motionally averaged PCSs and RDCs collected for the HPX domain, the metal anisotropy tensors determined from the PCSs of the CAT domain, and the SAXS data. The latter data provided restraints complementary to those of the NMR data (52), and were recently demonstrated to be very useful to make the overall dataset more stringent in characterizing the different conformations through their MO values (31). The SAXS profile provides information on the overall shape of the protein in solution. In the presence of conformational heterogeneity, the experimental profile is the weighted average of the profiles resulting from all structures in the conformational ensemble. SAXS is strongly sensitive to the elongation of the molecule, i.e. to domain translations, and much less to domain rotations, differently from RDCs, which are sensitive to rotations and not translations. PCSs depend on both rotations and translations, but due to their small values, their sensitivity to translations is limited. Therefore, SAXS restraints provide useful complementary information for discriminating among protein structures with different shapes. In the calculation of the SAXS term for target function, the experimental data in the angular range extending to 0.2 Å−1 were used, providing a χ2 value of 1.0. This range is responsible for the overall structure of the protein and such fitting allowed us to diminish the influence of the configuration of the long linker, less relevant for the modeling. However, the final model provides a good fit to the SAXS data in the entire experimental range (Fig. 2A).
A number of possible conformations, generated with the program RanCh, were analyzed through MO calculations (see “Experimental Procedures”). The substantial differences in the maximum weight that a conformation can have when included in any ensemble in good agreement with the experimental PCS, RDC, and SAXS restraints (see Fig. 3) resulted in markedly different MOs. Only 6% of the 1000 analyzed conformations were found to have a MO smaller than 5%, whereas most of the conformations (80%) had a MO smaller than 20%. Only 3% of the conformations had a MO larger than 30%, and only 0.3% had a MO larger than 40%. To illustrate the results of the calculations for 1,000 randomly selected MMP-1 conformations (Fig. 4) the HPX domains of all of the structures were superimposed and the position of the CAT domain was schematized by a triad of vectors oriented depending on the orientation of the CAT domain with respect to the HPX domain and centered in the center of mass of the CAT domain. Different orientations and positions of the Cartesian axes system thus reflect different orientations and positions of the CAT domain with respect to the HPX domain. The triads of vectors (one for each selected MMP-1 conformation) were color-coded with respect to the MO of the corresponding conformation, from blue (MO lower than 5%) to red (highest MO, 47%).
The conformations having the HPX domain in the region proximal to (Ln)CLaNP-5 (and distal to the catalytic site cleft) were found to have a negligible weight in solution, with MO values below 5% (blue tensors in Fig. 4B). Thus, these conformations were not sampled significantly by the protein. A striking finding is that most of the conformations with the highest MO (orange-red tensors in Fig. 4) were clustered in a well defined region of the distribution, corresponding to relatively elongated structures. A second region comprising high MO conformations with lower density of structures, and more spread in the conformational space, was present. To increase the resolution of the regions populated by the structures with the highest MO, additional conformations near these high MO structures were selected from the pool of 50,000 generated by RanCh (20). In this way, the MO values of 281 additional conformations were evaluated. All conformations with MO larger than 35% were examined.
MO values can be represented as a function of the translational and rotational parameters of the corresponding structures with respect to the structure with the highest MO (Fig. 7). The translations were reported with respect to the center of mass of the reference structure. To simplify distance calculations, rotations were represented through the corresponding 4-component complex number (quaternion) and distances were calculated as the projection of one quaternion to the reference one (53). There was continuity in the MO values as a function of these structural parameters, thus indicating a correlation between position/orientation and MO. A reasonably well defined peak encompassing the conformations with the largest MO value was observed as well as another region with somewhat smaller MO values (Fig. 7). These latter conformations were likely “ghost” solutions, arising from the quadratic form of the RDC equation (44), which neither PCSs nor SAXS were able to remove (54–56). The ghost solutions were verified by performing two sets of calculations using, pairwise, only two of the three datasets, showing that only the conformations in the main cluster consistently preserved the largest MO values. From the shape of the three-dimensional plot (Fig. 7B), it thus appeared that the highest MO conformations could be clearly identified independently from the generation probability of RanCh.
FIGURE 7.
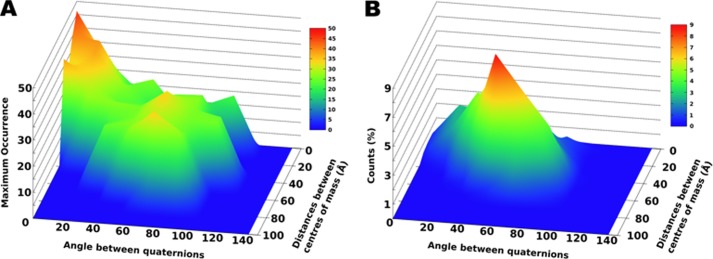
Representation of MO values as a function of translational and rotational parameters. Both plots are centered at the conformation with the highest MO value. A, three-dimensional plot representation of the MO value as a function of the distance between the centers of mass of the different HPX domain conformations and the angle between their quaternion representations. B, the probability distribution in space of the conformations generated by RanCh.
All available x-ray structures of human full-length MMP-1 (PDB entries 1SU3 (13), 2CLT (14), and 4AUO (16)) displayed relatively closed conformations. It is crucial to understand how much these structures are represented in the ensemble sampled by the protein in solution. To calculate their MO values, these structures were included in the pool of structures to be analyzed. The MO values obtained for x-ray structures 1SU3 (proMMP-1) and 2CLT (active MMP-1) were 20 and 19%, respectively. 2CLT (active MMP-1) was highly similar to the x-ray crystallographic structure of porcine full-length MMP-1 (12). These two structures are most relevant to the present study of MMP-1 in solution. The recently reported x-ray crystallographic structure of an MMP-1·THP complex (PDB entry 4AUO) has a more closed structure than 2CLT and has a MO of 18%.
The radii of gyration (Rg) of PDB 1SU3 and 2CLT crystallographic structures were 25.5 and 25.7 Å, respectively, whereas the structures with highest MO (>35%) had Rg of 29 ± 1.3 Å. Similar Rg values were also obtained for the highest MO structures without inclusion of the SAXS restraints in the calculations. This range of Rg is in better agreement with the experimentally determined values from the SAXS data alone (28.5–29.0 Å) (8, 15), indicating that x-ray structures were more compact than the average solution conformation. Furthermore, the relative orientations of HPX and CAT domains in the structures with the highest MO were different from those in the x-ray crystallographic structures.
The MMP-1 conformations that may be more relevant in solution can be examined by comparing the structures with the highest MO values (40–47%) among themselves, after having superimposed their HPX domains. The reciprocal orientation of the CAT domain has been evaluated by considering the differences in the orientation of the hA and hC helices of the CAT domain (defined by residues 130–141 and 250–258, respectively), which are almost perpendicular to one another. The angles for the first and second helix, among these highest MO structures, change up to a maximum of 26° and 18°, respectively, with respect to the mean orientation. This indicates that all of the highest MO structures are characterized by an interdomain orientation and position that can be defined relatively well.
DISCUSSION
Conformational selection or induced fit are often invoked to explain the mechanism by which proteins constituted of multiple domains and connected by flexible linkers recognize binding partners or substrates. Although detailed structural characterization of the bound-state conformation is often possible, much more difficult is the analysis of the conformations sampled by multidomain proteins before the interaction. However, analysis of the conformational space experienced by the free protein is useful not only to investigate the mechanism of binding, but also to determine the role of the different domains in the identification of substrates or partners, to predict new possible substrates or partners, and to investigate natural and new mechanisms of inhibition (3, 22, 24, 29, 35, 57–60).
Full-length MMP-1 was observed by NMR spectroscopy and SAXS to experience a sizable interdomain flexibility and an open-closed equilibrium (8, 15). The compact arrangements of the MMP-1 CAT and HPX domains observed in the x-ray crystallographic structures are not fully representative of the conformations sampled by the protein in solution, as for at least one-third of the time the enzyme exists with the CAT and HPX domains in an extended arrangement (8). Moreover, it has been hypothesized that the interface of the CAT and HPX domains may conceal secondary binding sites (exosites) involved in the recognition of collagenous substrates (15).
Although there is experimental evidence for the formation of the initial MMP-1·THP complex, the relative positioning of the enzyme domains prior to the interaction with substrate is still unclear. The MO analysis performed for MMP-1 can shed light on this conformation. The use of MO is well justified, as other methods that calculate average structures from, for example, several sets of RDC data, cannot be used in cases such as the present one (61) because the presence of large conformational rearrangement makes any obtained average structure devoid of a realistic physical meaning. Indeed, in our case, the magnetic susceptibility anisotropy tensor components calculated from the HPX domain are reduced by as much as a factor of 4 with respect to the components calculated from the CAT domain. Although (by definition) it cannot be demonstrated that the highest MO conformations correspond to the conformations with highest weight, it has been amply demonstrated through several simulations that the highest MO conformations do point to the conformations with the highest weight in synthetic ensembles (30, 31, 55, 56).
In the highest MO structures, the residues of the HPX domain essential for the binding to collagen were not buried between the CAT and HPX domains, and the open space between the two domains was wider than in the x-ray crystallographic structures. Furthermore, and more importantly, the secondary binding sites (exosites) of the HPX domain responsible for collagen interaction and the active site of the CAT domain face the same side. If triple-helical collagen was modeled in its experimentally determined bound position to the HPX domain (9), the CAT domain closely faces the collagen cleavage site, and in about half of the highest MO structures it sterically overlaps with the triple-helical substrate. In fact, all of the high MO conformations (MO >35%) of MMP-1 fall along the boundary between sterically overlapping and non-overlapping conformations.
Overall, the highest MO conformations sampled by MMP-1 when free in solution appeared to be much more poised for interaction with collagen than the compact x-ray crystallographic structures. Comparison of the non-overlapping structures with high MO values with the structural models corresponding to the different steps of the catalytic mechanism (9) indicated that the protein in solution has a marked tendency to assume “catalytically prone” conformations; once the HPX domain is bound to triple-helical collagen, the CAT domain can effectively search within a restricted and productive subset of binding modes that face the collagen hydrolysis site, and can start collagen unwinding/perturbation and cleavage. Therefore, the high MO conformations that are not colliding can be seen as a possible antecedent step for the recently proposed mechanism of collagenolysis (9).
To evaluate whether the protein can easily rearrange from the highest MO conformations to the conformation assumed when interacting with the substrate, a morphing between these two conformations was performed with the programs Climber (62) and FATCAT (63). Rearrangement from one conformation to the other involves only one twist in the hinge region, and the angle that the CAT domain has to cover to reorient itself on the cleavage site of collagen, once the HPX domain is attached, is about 50° along one single axis (Fig. 8). The transition seems to be feasible at the physiological temperature as the difference in free energy between these steps in the pathway is favorable (−0.133 kcal/mol, as calculated with the program Climber from the difference between the potential energy of the conformation assumed when the protein is interacting with the substrate and that of the structure with highest MO). The conformational rearrangement can therefore reasonably occur through a small energetic barrier, and the entropy loss is compensated by the enthalpy gain associated with the new interaction between the CAT domain and the triple-helix.
FIGURE 8.

Climber calculations of MMP-1 conformations. From top to bottom: structure with the highest MO, two morphing intermediate steps, and the previously proposed first step of collagenolysis (9). Structures in the right column are rotated 180° about the vertical axis with respect to the left column. The highest MO structure and morphing results were aligned to the HPX domain of the MMP-1·THP complex structure obtained previously (9). In yellow is the surface representation of MMP-1, in blue is the MMP consensus sequence HEXXHXXGXXH, in orange is the MMP-1 catalytic Zn2+, in green is the surface of the THP, and in blue is the THP cleavage site (Gly-Ile) of the first chain. The blue and red arrows indicate the directions of helices hA and hC, respectively, to facilitate visualizing the movement of the CAT domain with respect to the HPX domain and the THP. The THP sequence is (GPO)4-GPQGIAGQRGVVGLO-(GPO)4 (where O is 4-hydroxyproline), based on the human α1(I) collagen chain.
It has been previously reported that Gly271 is critical for the collagenolytic activity of MMP-1. In particular, it has been observed that replacement of this Gly residue with bulkier amino acids such as Asp drastically reduces the catalytic efficiency of the enzyme (64, 65). This effect has been explained as being due to an alteration of the linker mobility. Analysis performed with Climber on the G271D MMP-1 mutant showed that the conformational space sampled by the linker passing from the highest MO structures to the conformation in step 1 of the collagenolysis mechanism differs from that observed in the wild type protein, supporting a previous hypotheses that Gly271 is largely involved in the hinge bending motion.
The interaction of MMP-1 with a THP has been investigated utilizing NMR spectroscopy, leading to a plausible multistep mechanism for collagenolysis (9). In this mechanism, the initial binding of the HPX domain to the THP is followed by the interaction of the CAT domain with THP in front of the cleavage site, and by a subsequent back rotation of the CAT and HPX domains toward the closed conformation that drives the unwinding/perturbation of the triple-helix and causes the displacement of one peptide chain into the active site. Although x-ray crystallographic analysis of an MMP-1·THP complex has revealed binding of the THP to a closed form of MMP-1, it has been noted that the mode of binding in the MMP-1·THP structure was unproductive, in that the preferred collagen cleavage site was not correctly positioned for hydrolysis (16). The flexibility of MMP-1 domains, and particularly the highly favored extended conformation, also has a critical role in enzyme movement on collagen fibrils that occurs during the proteolytic process. MMPs are known to bind to numerous regions within the collagen triple-helix (66). MMPs then progressively move on collagen fibrils (67). Elongated MMP structures have been observed upon binding to collagen (3), from which an “inchworm” mechanism for MMP movement has been proposed (68). The application of mechanical stress facilitates collagen hydrolysis in the fibril (69). Both the MMP movement and the mechanical stress could be derived from the closing of an open MMP-1 conformation. As previously suggested (9), because collagen triple-helix does not fit into the CAT domain active site cavity, unwinding/perturbation of the triple-helix by an MMP is required to make one of the three-peptide chains able to fit the active site of MMP. The unwinding/perturbation can be achieved by back rotation of the CAT domain with respect to the HPX domain, so that the closed structure observed by x-ray crystallography is approached (9).
Conformational selection followed by induced fit can be thus invoked to describe the MMP-1/collagen binding process. In fact, among the many conformations sampled by MMP-1 where the residues of the HPX domain essential for collagen binding are not buried between the protein domains, the largest MO conformations have the CAT domain in an orientation that can easily access the collagen once the latter binds to the HPX domain. Therefore, conformational selection can play a role in this case to accelerate productive binding of MMP-1 to collagen. Once both domains are bound, subtle structural changes of the type previously proposed (9) would occur, essentially driven by an induced fit mechanism. The present study represents a striking example of the pathway followed by a multidomain protein with flexible linker(s) to perform its catalytic activity. In a broader context, the MO approach described here can evaluate the predominant domain conformations for numerous multidomain enzymes, including members of the protease and kinase superfamilies.
Acknowledgments
We thank the EU ESFRI Instruct Core Centre CERM, Italy. We acknowledge the support and assistance at the X33 beamline of the EMBL (synchrotron DESY, Hamburg) for SAXS data collection. We acknowledge discussion of these studies with Hashim Al-Hashimi, David Fushman, and Marcellus Ubbink at the XII Chianti-Instruct meeting and Homayoun Valafar and Konstantin Berlin at the Gordon Conference on Computational Aspects-Biomolecular NMR.
This work was supported, in whole or in part, by National Institutes of Health Grant CA98799, Ente Cassa di Risparmio, MIUR-FIRB contracts RBLA032ZM7 and RBRN07BMCT, European Commission contracts East-NMR number 228461, WeNMR number 261572, and Bio-NMR number 261863, and Fundação para a Ciência e Tecnologia (FCT), Portugal, Grant SFRH/BD/45928/2008 (to J. M. C. T.), and Instruct, part of the European Strategy Forum on Research Infrastructures (ESFRI) and supported by national member subscriptions.
Ivano Bertini passed away on July 7th, 2012, following completion of the work described herein. This study is dedicated to his memory, inspiration, and contribution to the research.

This article contains supplemental Tables S1 and S2.
- MMP
- matrix metalloproteinase
- CaM
- calmodulin
- CAT
- catalytic
- HPX
- hemopexin-like
- MO
- maximum occurrence
- PCS
- pseudocontact shifts
- RDC
- residual dipolar couplings
- SAXS
- small angle x-ray scattering
- TF
- target function
- THP
- triple-helical peptide
- PDB
- Protein Data Bank.
REFERENCES
- 1. Overall C. M. (2002) Molecular determinants of metalloproteinase substrate specificity. Mol. Biotechnol. 22, 51–86 [DOI] [PubMed] [Google Scholar]
- 2. Bode W. (2003) in Proteases and the Regulation of Biological Processes: Biochemical Society Symposia Volume 70 (Saklatvala J., Nagase H., Salvesen G. S., eds) pp. 1–14, Portland Press, Portland, OR [Google Scholar]
- 3. Rosenblum G., Van den Steen P. E., Cohen S. R., Grossmann J. G., Frenkel J., Sertchook R., Slack N., Strange R. W., Opdenakker G., Sagi I. (2007) Insights into the structure and domain flexibility of full-length pro-matrix metalloproteinase-9/gelatinase B. Structure 15, 1227–1236 [DOI] [PubMed] [Google Scholar]
- 4. Fields G. B. (2013) Interstitial collagen catabolism. J. Biol. Chem. 288, 8785–8793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chung L., Dinakarpandian D., Yoshida N., Lauer-Fields J. L., Fields G. B., Visse R., Nagase H. (2004) Collagenase unwinds triple-helical collagen prior to peptide bond hydrolysis. EMBO J. 23, 3020–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tam E. M., Moore T. R., Butler G. S., Overall C. M. (2004) Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP). The differential roles of the MMP hemopexin C domains and the MMP-2 fibronectin type II modules in collagen triple-helicase activities. J. Biol. Chem. 279, 43336–43344 [DOI] [PubMed] [Google Scholar]
- 7. Bertini I., Calderone V., Fragai M., Luchinat C., Maletta M., Yeo K. J. (2006) Snapshots of the reaction mechanism of matrix metalloproteinases. Angew. Chem. Int. Ed. Engl. 45, 7952–7955 [DOI] [PubMed] [Google Scholar]
- 8. Bertini I., Fragai M., Luchinat C., Melikian M., Mylonas E., Sarti N., Svergun D. I. (2009) Interdomain flexibility in full-length matrix metalloproteinase-1 (MMP-1). J. Biol. Chem. 284, 12821–12828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bertini I., Fragai M., Luchinat C., Melikian M., Toccafondi M., Lauer J. L., Fields G. B. (2012) Structural basis for matrix metalloproteinase 1 catalyzed collagenolysis. J. Am. Chem. Soc. 134, 2100–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Minond D., Lauer-Fields J. L., Cudic M., Overall C. M., Pei D., Brew K., Moss M. L., Fields G. B. (2007) Differentiation of secreted and membrane-type matrix metalloproteinase activities based on substitutions and interruptions of triple-helical sequences. Biochemistry 46, 3724–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lu K. G., Stultz C. M. (2013) Insight into the degradation of type-I collagen fibrils by MMP-8. J. Mol. Biol. 425, 1815–1825 [DOI] [PubMed] [Google Scholar]
- 12. Li J., Brick P., O'Hare M. C., Skarzynski T., Lloyd L. F., Curry V. A., Clark I. M., Bigg H. F., Hazleman B. L., Cawston T. E. (1995) Structure of full-length porcine synovial collagenase reveals a C-terminal domain containing a calcium-linked, four bladed β-propeller. Structure 3, 541–549 [DOI] [PubMed] [Google Scholar]
- 13. Jozic D., Bourenkov G., Lim N.-H., Visse R., Nagase H., Bode W., Maskos K. (2005) X-ray structure of human proMMP-1. New insights into procollagenase activation and collagen binding. J. Biol. Chem. 280, 9578–9585 [DOI] [PubMed] [Google Scholar]
- 14. Iyer S., Visse R., Nagase H., Acharya K. R. (2006) Crystal structure of an active form of human MMP-1. J. Mol. Biol. 362, 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arnold L. H., Butt L. E., Prior S. H., Read C. M., Fields G. B., Pickford A. R. (2011) The interface between catalytic and hemopexin domains in matrix metalloproteinase 1 conceals a collagen binding exosite. J. Biol. Chem. 286, 45073–45082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Manka S. W., Carafoli F., Visse R., Bihan D., Raynal N., Farndale R. W., Murphy G., Enghild J. J., Hohenester E., Nagase H. (2012) Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc. Natl. Acad. Sci. U.S.A. 109, 12461–12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lauer-Fields J. L., Chalmers M. J., Busby S. A., Minond D., Griffin P. R., Fields G. B. (2009) Identification of specific hemopexin-like domain residues that facilitate matrix metalloproteinase collagenolytic activity. J. Biol. Chem. 284, 24017–24024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Valafar H., Prestegard J. H. (2004) REDCAT. A residual dipolar coupling analysis tool. J. Magn Reson. 167, 228–241 [DOI] [PubMed] [Google Scholar]
- 19. Bernadó P., Blanchard L., Timmins P., Marion D., Ruigrok R. W., Blackledge M. (2005) A structural model for unfolded proteins from residual dipolar couplings and small-angle x-ray scattering. Proc. Natl. Acad. Sci. U.S.A. 102, 17002–17007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bernadó P., Mylonas E., Petoukhov M. V., Blackledge M., Svergun D. I. (2007) Structural characterization of flexible proteins using small-angle x-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 [DOI] [PubMed] [Google Scholar]
- 21. Berlin K., O'Leary D. P., Fushman D. (2009) Improvement and analysis of computational methods for prediction of residual dipolar couplings. J. Magn. Reson. 201, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Anthis N. J., Doucleff M., Clore G. M. (2011) Transient, sparsely populated compact states of apo and calcium-loaded calmodulin probed by paramagnetic relaxation enhancement. Interplay of conformational selection and induced fit. J. Am. Chem. Soc. 133, 18966–18974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lange O. F., Lakomek N.-A., Farès C., Schröder G. F., Walter K. F., Becker S., Meiler J., Grubmüller H., Griesinger C., de Groot B. L. (2008) Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 320, 1471–1475 [DOI] [PubMed] [Google Scholar]
- 24. Bashir Q., Volkov A. N., Ullmann G. M., Ubbink M. (2010) Visualization of the encounter ensemble of the transient electron transfer complex of cytochrome c and cytochrome c peroxidase. J. Am. Chem. Soc. 132, 241–247 [DOI] [PubMed] [Google Scholar]
- 25. Bertini I., Del Bianco C., Gelis I., Katsaros N., Luchinat C., Parigi G., Peana M., Provenzani A., Zoroddu M. A. (2004) Experimentally exploring the conformational space sampled by domain reorientation in calmodulin. Proc. Natl. Acad. Sci. U.S.A. 101, 6841–6846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lindorff-Larsen K., Best R. B., Depristo M. A., Dobson C. M., Vendruscolo M. (2005) Simultaneous determination of protein structure and dynamics. Nature 433, 128–132 [DOI] [PubMed] [Google Scholar]
- 27. Iwahara J., Clore G. M. (2006) Detecting transient intermediates in macromolecular binding by paramagnetic NMR. Nature 440, 1227–1230 [DOI] [PubMed] [Google Scholar]
- 28. Volkov A. N., Worrall J. A., Holtzmann E., Ubbink M. (2006) Solution structure and dynamics of the complex between cytochrome c and cytochrome c peroxidase determined by paramagnetic NMR. Proc. Natl. Acad. Sci. U.S.A. 103, 18945–18950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Q., Stelzer A. C., Fisher C. K., Al-Hashimi H. M. (2007) Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature 450, 1263–1267 [DOI] [PubMed] [Google Scholar]
- 30. Bertini I., Gupta Y. K., Luchinat C., Parigi G., Peana M., Sgheri L., Yuan J. (2007) Paramagnetism-based NMR restraints provide maximum allowed probabilities for the different conformations of partially independent protein domains. J. Am. Chem. Soc. 129, 12786–12794 [DOI] [PubMed] [Google Scholar]
- 31. Bertini I., Giachetti A., Luchinat C., Parigi G., Petoukhov M. V., Pierattelli R., Ravera E., Svergun D. I. (2010) Conformational space of flexible biological macromolecules from average data. J. Am. Chem. Soc. 132, 13553–13558 [DOI] [PubMed] [Google Scholar]
- 32. Fragai M., Luchinat C., Parigi G., Ravera E. (2013) Conformational freedom of metalloproteins revealed by paramagnetism-assisted NMR. Coord. Chem. Rev. 257, 2652–2667 [Google Scholar]
- 33. Dasgupta S., Hu X., Keizers P. H., Liu W.-M., Luchinat C., Nagulapalli M., Overhand M., Parigi G., Sgheri L., Ubbink M. (2011) Narrowing the conformational space sampled by two-domain proteins with paramagnetic probes in both domains. J. Biomol. NMR 51, 253–263 [DOI] [PubMed] [Google Scholar]
- 34. Bertini I., Luchinat C., Nagulapalli M., Parigi G., Ravera E. (2012) Paramagnetic relaxation enhancement for the characterization of the conformational heterogeneity in two-domain proteins. Phys. Chem. Chem. Phys. 14, 9149–9156 [DOI] [PubMed] [Google Scholar]
- 35. Nagulapalli M., Parigi G., Yuan J., Gsponer J., Deraos G., Bamm V. V., Harauz G., Matsoukas J., de Planque M. R., Gerothanassis I. P., Babu M. M., Luchinat C., Tzakos A. G. (2012) Recognition pliability is coupled to structural heterogeneity. A calmodulin intrinsically disordered binding region complex. Structure 20, 522–533 [DOI] [PubMed] [Google Scholar]
- 36. Bertini I., Gelis I., Katsaros N., Luchinat C., Provenzani A. (2003) Tuning the affinity for lanthanides of calcium binding proteins. Biochemistry 42, 8011–8021 [DOI] [PubMed] [Google Scholar]
- 37. Bertini I., Calderone V., Cerofolini L., Fragai M., Geraldes C. F., Hermann P., Luchinat C., Parigi G., Teixeira J. M. (2012) The catalytic domain of MMP-1 studied through tagged lanthanides. FEBS Lett. 586, 557–567 [DOI] [PubMed] [Google Scholar]
- 38. Roessle M. W., Klaering R., Ristau U., Robrahn B., Jahn D., Gehrmann T., Konarev P. V., Round A., Fiedler S., Hermes S., Svergun D. I. (2007) Upgrade of the small-angle x-ray scattering beamline X33 at the European Molecular Biology Laboratory, Hamburg. J. Appl. Crystallogr. 40, s190–s194 [Google Scholar]
- 39. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS. A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 40. Svergun D. I., Barberato C., Koch M. H. J. (1995) CRYSOL. A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 41. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikheney A. G., Gajda M., Gorba C., Mertens H. D. T., Konarev P. V., Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bertini I., Ferella L., Luchinat C., Parigi G., Petoukhov M. V., Ravera E., Rosato A., Svergun D. I. (2012) MaxOcc. A web portal for maximum occurrence analysis. J. Biomol. NMR 53, 271–280 [DOI] [PubMed] [Google Scholar]
- 43. Bertini I., Luchinat C., Parigi G. (2002) Magnetic susceptibility in paramagnetic NMR. Progr. NMR Spectrosc. 40, 249–273 [Google Scholar]
- 44. Longinetti M., Parigi G., Sgheri L. (2002) Uniqueness and degeneracy in the localization of rigid structural elements in paramagnetic proteins. J. Phys. A Math. Gen. 35, 8153–8169 [Google Scholar]
- 45. Bertini I., Fragai M., Lee Y. M., Luchinat C., Terni B. (2004) Paramagnetic metal ions in ligand screening. The Co(II) matrix metalloproteinase 12. Angew. Chem. Int. Ed. Engl. 43, 2254–2256 [DOI] [PubMed] [Google Scholar]
- 46. Bertini I., Calderone V., Fragai M., Jaiswal R., Luchinat C., Melikian M., Mylonas E., Svergun D. I. (2008) Evidence of reciprocal reorientation of the catalytic and hemopexin-like domains of full-length MMP-12. J. Am. Chem. Soc. 130, 7011–7021 [DOI] [PubMed] [Google Scholar]
- 47. Wöhnert J., Franz K. J., Nitz M., Imperiali B., Schwalbe H. (2003) Protein alignment by a coexpressed lanthanide-binding tag for the measurement of residual dipolar couplings. J. Am. Chem. Soc. 125, 13338–13339 [DOI] [PubMed] [Google Scholar]
- 48. Ikegami T., Verdier L., Sakhaii P., Grimme S., Pescatore B., Saxena K., Fiebig K. M., Griesinger C. (2004) Novel techniques for weak alignment of proteins in solution using chemical tags coordinating lanthanide ions. J. Biomol. NMR 29, 339–349 [DOI] [PubMed] [Google Scholar]
- 49. Su X. C., Man B., Beeren S., Liang H., Simonsen S., Schmitz C., Huber T., Messerle B. A., Otting G. (2008) A dipicolinic acid tag for rigid lanthanide tagging of proteins and paramagnetic NMR spectroscopy. J. Am. Chem. Soc. 130, 10486–10487 [DOI] [PubMed] [Google Scholar]
- 50. Keizers P. H., Saragliadis A., Hiruma Y., Overhand M., Ubbink M. (2008) Design, synthesis, and evaluation of a lanthanide chelating protein probe. CLaNP-5 yields predictable paramagnetic effects independent of environment. J. Am. Chem. Soc. 130, 14802–14812 [DOI] [PubMed] [Google Scholar]
- 51. Tolman J. R., Al-Hashimi H. M., Kay L. E., Prestegard J. H. (2001) Structural and dynamic analysis of residual dipolar coupling data for proteins. J. Am. Chem. Soc. 123, 1416–1424 [DOI] [PubMed] [Google Scholar]
- 52. Xu X., Reinle W., Hannemann F., Konarev P. V., Svergun D. I., Bernhardt R., Ubbink M. (2008) Dynamics in a pure encounter complex of two proteins studied by solution scattering and paramagnetic NMR spectroscopy. J. Am. Chem. Soc. 130, 6395–6403 [DOI] [PubMed] [Google Scholar]
- 53. Kuffner J. J. (2004) Effective sampling and distance metrics for 3D rigid body path planning. Proceedings of the IEEE International Conference on Robotics and Automation 4, 3993–3998 [Google Scholar]
- 54. Bertini I., Luchinat C., Parigi G. (2011) Moving the frontiers in solution and solid-state bioNMR. Coord. Chem. Rev. 255, 649–663 [Google Scholar]
- 55. Longinetti M., Luchinat C., Parigi G., Sgheri L. (2006) Efficient determination of the most favoured orientations of protein domains from paramagnetic NMR data. Inv. Probl. 22, 1485–1502 [Google Scholar]
- 56. Luchinat C., Nagulapalli M., Parigi G., Sgheri L. (2012) Maximum occurrence analysis of protein conformations for different distributions of paramagnetic metal ions within flexible two-domain proteins. J. Magn. Reson. 215, 85–93 [DOI] [PubMed] [Google Scholar]
- 57. Bertini I., Fragai M., Giachetti A., Luchinat C., Maletta M., Parigi G., Yeo K. J. (2005) Combining in silico tools and NMR data to validate protein-ligand structural models. Application to matrix metalloproteinases. J. Med. Chem. 48, 7544–7559 [DOI] [PubMed] [Google Scholar]
- 58. Tang C., Iwahara J., Clore G. M. (2006) Visualization of transient encounter complexes in protein-protein association. Nature 444, 383–386 [DOI] [PubMed] [Google Scholar]
- 59. Yuwen T., Post C. B., Skrynnikov N. R. (2011) Domain cooperativity in multidomain proteins. What can we learn from molecular alignment in anisotropic media? J. Biomol. NMR 51, 131–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ryabov Y. E., Fushman D. (2007) A model of interdomain mobility in a multidomain protein. J. Am. Chem. Soc. 129, 3315–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shealy P., Simin M., Park S. H., Opella S. J., Valafar H. (2010) Simultaneous structure and dynamics of a membrane protein using REDCRAFT. Membrane-bound form of Pf1 coat protein. J. Magn. Reson. 207, 8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weiss D. R., Levitt M. (2009) Can morphing methods predict intermediate structures? J. Mol. Biol. 385, 665–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ye Y., Godzik A. (2003) Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 19, ii246–ii255 [DOI] [PubMed] [Google Scholar]
- 64. Tsukada H., Pourmotabbed T. (2002) Unexpected crucial role of residue 272 in substrate specificity of fibroblast collagenase. J. Biol. Chem. 277, 27378–27384 [DOI] [PubMed] [Google Scholar]
- 65. Fasciglione G. F., Gioia M., Tsukada H., Liang J., Iundusi R., Tarantino U., Coletta M., Pourmotabbed T., Marini S. (2012) The collagenolytic action of MMP-1 is regulated by the interaction between the catalytic domain and the hinge region. J. Biol. Inorg. Chem. 17, 663–672 [DOI] [PubMed] [Google Scholar]
- 66. Sun H. B., Smith G. N., Jr., Hasty K. A., Yokota H. (2000) Atomic force microscopy-based detection of binding and cleavage site of matrix metalloproteinase on individual type II collagen helices. Anal. Biochem. 283, 153–158 [DOI] [PubMed] [Google Scholar]
- 67. Saffarian S., Collier I. E., Marmer B. L., Elson E. L., Goldberg G. (2004) Interstitial collagenase is a Brownian rachet driven by proteolysis of collagen. Science 306, 108–111 [DOI] [PubMed] [Google Scholar]
- 68. Overall C. M., Butler G. S. (2007) Protease yoga. Extreme flexibility of a matrix metalloproteinase. Structure 15, 1159–1161 [DOI] [PubMed] [Google Scholar]
- 69. Sarkar S. K., Marmer B., Goldberg G., Neuman K. C. (2012) Single-molecule tracking of collagenase on native type I collagen fibrils reveals degradation mechanism. Curr. Biol. 22, 1047–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]