

Background: Changes in integrin αIIbβ3 binding affinity occur in strongly stimulated platelets.
Results: Platelet mitochondrial permeability transition pore formation enhances calpain activity, which leads to integrin β3-associated proteolytic cleavage and integrin inactivation.
Conclusion: Mitochondrially mediated integrin αIIbβ3 inactivation limits platelet aggregation and thrombus growth.
Significance: Modulation of this pathway may offer a novel alternative for the prevention of thrombosis.
Keywords: Calpain, Integrin, Mitochondria, Platelets, Thrombosis
Abstract
When platelets are strongly stimulated, a procoagulant platelet subpopulation is formed that is characterized by phosphatidylserine (PS) exposure and epitope modulation of integrin αIIbβ3 or a loss of binding of activation-dependent antibodies. Mitochondrial permeability transition pore (mPTP) formation, which is essential for the formation of procoagulant platelets, is impaired in the absence of cyclophilin D (CypD). Here we investigate the mechanisms responsible for these procoagulant platelet-associated changes in integrin αIIbβ3 and the physiologic role of procoagulant platelet formation in the regulation of platelet aggregation. Among strongly stimulated adherent platelets, integrin αIIbβ3 epitope changes, mPTP formation, PS exposure, and platelet rounding were closely associated. Furthermore, platelet mPTP formation resulted in a decreased ability to recruit additional platelets. In the absence of CypD, integrin αIIbβ3 function was accentuated in both static and flow conditions, and, in vivo, a prothrombotic phenotype occurred in mice with a platelet-specific deficiency of CypD. CypD-dependent proteolytic events, including cleavage of the integrin β3 cytoplasmic domain, coincided closely with integrin αIIbβ3 inactivation. Calpain inhibition blocked integrin β3 cleavage and inactivation but not mPTP formation or PS exposure, indicating that integrin inactivation and PS exposure are mediated by distinct pathways subsequent to mPTP formation. mPTP-dependent alkalinization occurred in procoagulant platelets, suggesting a possible alternative mechanism for enhancement of calpain activity in procoagulant platelets. Together, these results indicate that, in strongly stimulated platelets, mPTP formation initiates the calpain-dependent cleavage of integrin β3 and associated regulatory proteins, resulting in integrin αIIbβ3 inactivation, and demonstrate a novel CypD-dependent negative feedback mechanism that limits platelet aggregation and thrombotic occlusion.
Introduction
Upon activation, platelets change shape, release granules, and firmly adhere and aggregate through activation of the adhesive receptor integrin αIIbβ3. In strongly stimulating conditions, additional morphologic and functional changes occur in a subpopulation of activated platelets. These platelets, to which we refer here as procoagulant platelets, vesiculate, expose high levels of PS2, and retain high amounts of α-granule proteins (1–3). Other names for this platelet subpopulation have included COAT (3), coated (4), balloon-like (5, 6), sustained calcium-induced platelet morphology (SCiP) (7), and highly activated platelets (8). Conditions that generate procoagulant platelets in vitro include adherence to collagen (5), stimulation with thrombin plus collagen (3), adherence to von Willebrand factor, particularly in the presence of the soluble agonists ADP or thrombin (7, 9, 10), and high shear stress (11).
In addition to these procoagulant and morphologic changes, alterations in integrin αIIbβ3 also occur in the procoagulant platelet subpopulation (1, 7, 12, 13). When activation-dependent antibodies such as PAC-1 or JON/A are used to interrogate the activation state of integrin αIIbβ3 after strong stimulation, these antibodies bind transiently. However, minutes after this initial activation, these activation-dependent epitopes are almost absent on the procoagulant platelet subpopulation. In contrast, the binding of antibodies to other extracellular integrin αIIbβ3 epitopes is unperturbed (1, 7, 12, 13). This integrin αIIbβ3 epitope modulation has alternatively been proposed to reflect either epitope inaccessibility because of α-granule protein retention (1) or a conformational change in integrin αIIbβ3 associated with loss of function (7, 14).
Mitochondrial permeability transition pore (mPTP) formation regulates procoagulant platelet formation (8, 15). The mPTP is an inducible inner mitochondrial membrane pore, the formation of which regulates necrotic cell death (16). A key regulator of mPTP formation is the mitochondrially localized peptidylprolylisomerase cyclophilin D (CypD), and in the absence of CypD, mPTP formation in response to calcium and oxidative stress is inhibited (17). In CypD-null platelets, agonist-induced mPTP formation, procoagulant platelet generation, and integrin αIIbβ3 epitope modulation are abrogated (8). Another potentially important regulator of integrin αIIbβ3 function in procoagulant platelets is calpain. However, the role of calpain in the regulation of integrin function remains controversial. Calpain activation has alternatively been proposed to activate integrin through talin cleavage (18), to increase outside-in signaling through distal cleavage of the integrin β3 cytoplasmic tail (19), and to disrupt inside-out mediated conformational changes in integrin αIIbβ3 through proximal cleavage of the integrin β3 cytoplasmic tail (19, 20).
Here we investigate the mechanisms regulating platelet integrin αIIbβ3 epitope modulation and its functional role in the regulation of platelet activation. Studies are also performed investigating the coordination of mPTP formation and calpain activation in procoagulant platelets. In strongly stimulated platelets, mPTP formation is shown to increase intracellular pH and enhance calpain-dependent cleavage of both talin and the cytoplasmic tail of integrin β3. These cleavage events are demonstrated to be closely associated with both integrin αIIbβ3 epitope modulation and down-regulation of integrin αIIbβ3 activity. Finally, using mice with platelet-specific deficiency of CypD, a role for platelet mPTP formation in limiting thrombotic occlusion in vivo is demonstrated conclusively.
EXPERIMENTAL PROCEDURES
Mice
CypD+/+ and CypD−/− mice were maintained on an SV129 background. PF4-Cre and Ppidtm1Mmos/J mice were obtained from The Jackson Laboratory (C57BL/6 background). PF4-Cre+/+/Ppidtm1Mmos+/− (CypDplt+/−) and PF4-Cre+/+/Ppidtm1Mmos−/− (CypDplt−/−), utilized for in vivo testing, were the F1 progeny of a PF4-Cre+/+/Ppidtm1Mmos+/− (CypDplt+/−) and PF4-Cre+/+/Ppidtm1Mmos−/− (CypDplt−/−) cross. β-actin-GFP transgenic mice (C57BL/6) were provided by Dr. David Archer (Emory University). All animal protocols were approved by the Emory University Institutional Animal Care and Use Committee under National Institutes of Health guidelines. Mice were housed in pathogen-free conditions at Emory University.
Preparation of Washed Platelets
Murine blood was collected in acid citrate dextrose anticoagulant or as indicated below. Washed platelets were isolated as described previously and resuspended in modified Tyrode buffer (134 mm NaCl, 2.9 mm KCl, 12 mm NaHCO3, 20 mm HEPES, 1 mm MgCl2, 0.34 mm Na2HPO4, and 5 mm glucose (pH 7.35)) for at least 20 min at room temperature before experiments (21).
Microfluidic Chamber Fabrication and Protein Patterning
Polydimethylsiloxane microfluidic chamber fabrication and collagen patterning were performed as described previously (22). In brief, a dry clean photoresist wafer was silanized with tridecaflouro-1,1,2,2-tetrahydrooctyl (Gelest Inc.) before fabrication. The Sylgard 184 silicone elastomer base and curing agent (Dow Corning) were mixed at 10:1 (v/v) and cured at 80 °C for 2 h. Prior to the experiment, the fibrillar type I collagen (250 μg/ml, Chrono-log Corp.) or fibrinogen (100 μg/ml, catalog no. F3879, Sigma) was immobilized on an acid-pretreated glass slide (catalog no. 2947, Corning) or cover slide (catalog no. 2940, Corning) for 1 h at room temperature. Then the chamber and patterned cover slide were blocked in HEPES-buffered saline (pH 7.4) with 3% BSA for another hour right before use.
Fluorescence Microscopy of Adherent Platelets
Washed platelets (1 × 109/ml) in modified Tyrode buffer containing 2 mm CaCl2 were allowed to adhere on a fibrinogen-coated (100 μg/ml) cover slide for 20 min in a polydimethylsiloxane microfluidic chamber. After removal of non-adherent platelets, PE-JON/A (catalog no. M023-2, Emfret Analytics), tetramethyl-rhodaminemethyl ester (TMRM, Invitrogen), or annexin V (Alexa Fluor 647-conjugated, Invitrogen) were preincubated with the adherent platelets for 10 min with or without pharmacologic inhibitors (i.e. MDL27180). Platelets were then stimulated with the indicated agonists, either thrombin (Hematologic Technologies) or convulxin (CenterChem, Inc.). Fluorescence and platelet morphology were visualized in real time using a Nikon eclipse Ti inverted microscope (Nikon Instruments Inc.) equipped with a high-resolution Cool Snap HQ2 camera (Photometrics) under a ×60 differential interference contrast objective. Images were recorded for off-line analysis using Nikon NIS Elements AR3.2 software.
Platelet Aggregation Assay
Washed platelets (45 μl, 1 × 109/ml) in a 96-well half-area plate (COSTAR) were incubated for 5 min in a microplate reader (Spectra Max Plus 384, Molecular Devices) at 37 °C before the addition of agonists and CaCl2. Measurements of optical density were performed at 500 nm every 15 s with mixing between every two readings.
Platelet Adhesion in Flow Conditions
To assess platelet recruitment, fibrinogen-adherent platelets were labeled with TMRM (0.1 μm) for 10 min and washed with mTyrode buffer. The adherent platelets were stimulated with thrombin (0.5 units/ml, Hematologic Technologies) and convulxin (100 ng/ml) for 5 min. After washing to remove these agonists, recalcified citrate/corn trypsin inhibitor (CTI)/apyrase-treated whole blood from β-actin-GFP transgenic mice was perfused over the pretreated platelets for 5 min at a 100 s−1 shear rate. Recruitment of GFP-expressing platelets by TMRM(+) platelets and TMRM(-) platelets was determined. For perfusion experiments at arterial shear rates (1500 s−1), murine whole blood was collected in 3.2% citrate with 50 μg/ml corn trypsin inhibitor (CTI, Fisher Scientific) and 0.5 units/ml apyrase and then preincubated with 1 μg/ml of PE-CD41 (eBioscience) for 10 min. Upon recalcification (2 mm CaCl2), the blood was immediately perfused in a microfluidics system across the collagen strip, and platelet accumulation was monitored continuously.
Mesenteric Arterial Thrombus Formation
Six- to eight-week-old littermate mice (F1 progeny of a PF4-Cre+/+/Ppidtm1Mmos+/− (CypDplt+/−) and PF4-Cre+/+/Ppidtm1Mmos−/− (CypDplt−/−) cross) were anesthetized with ketamine/xylazine, and the mesenteric vasculature was exteriorized. A fluorescently labeled anti-GP1b antibody (1 mg/kg, catalog no. X488, Emfret Analytics) and Rose Bengal (20 mg/kg, catalog no. 330000, Sigma) were injected retro-orbitally in 100 μl Tyrodes. Endothelial injury was initiated within 5 min of injection by exposing the tissue to TRITC bandwidth-filtered light at maximal intensity for 30 s. The FITC filter was then selected, and platelet accumulation and flow within the mesenteric vessel were visualized with continuous recording for a maximum of 30 min and analyzed using the NIS Elements software (Nikon). Videos were subsequently analyzed and the time of initial platelet adherence, start of thrombus embolization, and time of stable occlusion were determined (supplemental Fig. 1). Stable occlusion was defined as the absence of observed flow for a minimum of 5 min, a time point after which no recanalization was ever observed. The genotype remained blinded until video analysis was complete.
Tail Bleeding
Tails of anesthetized adult mice were transected 3 mm from the distal end and immediately placed into 37 °C prewarmed saline for 10 min. Time to cessation of bleeding and any rebleeding were recorded.
Western Blot Analysis
After preincubation with or without the indicated inhibitors, washed platelets (1 × 109/ml) were treated with the indicated agonists for 5 min at 37 °C with shaking. Platelets were then lysed at 4 °C in cell lysis buffer (catalog no. 9803, Cell Signaling Technology) with protease inhibitor mixture (Roche) and phenylmethanesulfonyl fluoride (1 mm, Sigma). Equal amounts of platelet lysate were run on a 10% SDS-PAGE gel (Bio-Rad). The target protein was evaluated by Western blot analysis using anti-integrin β3 C-terminal antibody (catalog no. sc6626, Santa Cruz Biotechnology), N-terminal antibody (catalog no. sc6627, Santa Cruz Biotechnology), and anti-talin antibody (catalog no. sc15336, Santa Cruz Biotechnology). Images were collected using the Odyssey infrared imaging system (LI-COR), and blot intensity was analyzed by Quantity One software (Bio-Rad).
Determination of Platelet Intracellular pH (pHi)
1 × 106/ml washed platelets suspended in PBS and the non-ionic detergent Pluronic® F-127 (5 μm, catalog no. P6866, Invitrogen) were incubated with the cell-permeant fluorescent pH indicator carboxyl seminaphtharhodafluor, acetoxymethyl ester, acetate 1 (SNARF-1 AM® (5 μm, catalog no. C-1272, Invitrogen) for 30 min at 37 °C and then washed and resuspended in PBS. For in situ calibration of pHi, dye-loaded platelets were resuspended in PBS (pH 6.9–8.2) and treated with the ionophore nigericin (2 μg/ml, catalog no. N-1495, Invitrogen) in the presence of 140 mm K+ to equilibrate the intracellular pH with the extracellular medium. For pHi determination, platelets were resuspended in Tyrodes and stimulated in the presence of the indicated agonist for 5 min. pHi was determined by the ratio of fluorescence intensities at 640 nm and 580 nm using flow cytometry.
Statistical Analysis
Results are presented as mean ± S.E. The statistical significance between two groups was determined by Student's t test (SPSS, Sigma). For in vivo thrombosis experiments, a Mann-Whitney U test was utilized.
RESULTS
Dependence of Platelet Shape Change and Integrin αIIbβ3 Epitope Modulation on mPTP Formation
Integrin αIIbβ3 epitope modulation has been described primarily in flow cytometry experiments (1, 12, 13) or in fixed platelets (7) at discrete time points. To allow uninterrupted visualization of procoagulant platelet formation and assess the coordination of adhesive and mitochondrial events, fibrinogen-adherent platelets were observed continuously following stimulation with thrombin and convulxin (Thr/Cvx). This agonist combination was chosen because it is particularly effective in generating procoagulant platelets (13). Within 1 min of stimulation, lamellipodia and filipodia formation occurred, associated with an increase in platelet diameter and JON/A binding (Fig. 1, A–C). Subsequently, in a platelet subpopulation, the previously extended lamellipodia and filipodia retracted, and the platelets assumed a rounded morphology that correlated with a decrease in platelet diameter and a decrease in JON/A binding (Fig. 1, A and B, and supplemental Fig. 2, B and C). Platelet rounding, decreased JON/A binding, and PS exposure were associated both in individual platelets and temporally (Fig. 1, A–C, and supplemental Fig. 2A). In contrast to the decreased binding of the activation-dependent epitope JON/A, no decrease was noted in the expression levels of non-activation-dependent integrin αIIbβ3 epitopes (supplemental Fig. 2, B and C).
FIGURE 1.

Dependence of platelet shape change and integrin αIibβ3 epitope modulation on mPTP formation. A, platelets were stimulated with thrombin (0.5 units/ml) and convulxin (100 ng/ml) and visualized continuously. Merged fluorescent (JON/A) and differential interference contrast (DIC) images were merged and are shown at the indicated time points. Arrows indicate platelets that assume a rounded morphology. B–D, platelet diameter (B), JON/A-positive (C), and TMRM-positive (D) platelets were assessed in CypD+/+ and CypD−/− platelets stimulated with Thr/Cvx. Platelet diameter was quantified relative to initial diameter. Up to 500 platelet diameters were counted in each condition. TMRM-positive platelets were quantified relative to the number present immediately prior to stimulation. JON/A+ platelets were quantified relative to peak levels (1 min after stimulation). E, platelet morphology and TMRM fluorescence were assessed at the indicated time points in Thr/Cvx-stimulated CypD+/+ and CypD−/− platelets. Arrows point to rounded platelets. Scale bar = 5 μm. *, p < 0.05. n = 3–6 mice in each experiment. The dashed line connecting two asterisks indicates same significance level in between these observations.
TMRM is a Δψm-sensitive cationic dye that labels intact and metabolically active mitochondria. Evaluation of mitochondrial transmembrane potential (Δψm) using TMRM revealed that platelet rounding was associated with loss of Δψm (Fig. 1, D and E). Consistent with the central role of mPTP in procoagulant platelet formation (8), platelet rounding, integrin αIIbβ3 epitope modulation, and PS exposure were all markedly abrogated in the absence of CypD (Fig. 1, B–E, and supplemental Fig. 2A , ii). Together, these results clearly demonstrate the close association of procoagulant platelet rounding with integrin αIIbβ3 epitope modulation and loss of Δψm and their dependence on the mPTP regulatory protein CypD.
Mitochondrially Dependent Events Limit Platelet Aggregation and Adhesion
To more directly ascertain the relationship of these changes in morphology and Δψm with integrin αIIbβ3 function, platelet adherence to TMRM-labeled platelets was assessed in a platelet recruitment assay (Fig. 2A and supplemental Fig. 3A). Whole blood from a GFP transgenic mouse was perfused over a mixed population of adherent TMRM(+) and TMRM(-) platelets generated by prior stimulation with Thr/Cvx. In these conditions, stable recruitment of perfused GFP(+) platelets was decreased significantly (6-fold) in platelets that had lost Δψm (TMRM(-)) relative to TMRM(+) platelets. These results demonstrate the close association of mitochondrial depolarization with a decreased ability to maintain stable platelet-platelet contacts in flow conditions.
FIGURE 2.

Mitochondrial-dependent events limit platelet aggregation and adhesion. A, assessment of platelet recruitment by TMRM(+) and TMRM(-) platelets. Recalcified CTI/apyrase-treated whole blood from β-actin GFP transgenic mice was perfused for 5 min at 100 s over washed adherent platelets stimulated previously with Thr/Cvx. The percentage of adherent platelets with stably associated GFP+ platelets was determined. *, p < 0.05. n = 3. B, aggregation of CypD+/+ and CypD−/− platelets in the presence of the indicated agonist(s). *, p < 0.05. n = 5–11 mice. C, adherence of CypD+/+ and CypD−/− platelets to type I collagen. Recalcified CTI/apyrase-treated whole blood was perfused over the collagen surface for 10 min at 1500/s shear rates. PE-CD61 was used to label platelets. JON/A was used at a 1:40 dilution. Scale bars = 20 μm. D, platelet coverage following perfusion was compared relative to CypD+/+. *, p < .01 relative to CypD+/+; #, p < 0.01 relative to CypD−/−. n = 3–6 mice.
Next, the role of mPTP-dependent events in regulating integrin αIIbβ3 function was assessed in a platelet aggregation assay (Fig. 2B and supplemental Fig. 3B). When platelets were stimulated with either thrombin or convulxin alone, aggregation progressed rapidly to a maximal amplitude. However, strongly stimulating conditions (Thr/Cvx) resulted in a significantly different pattern of aggregation. Initially, aggregation progressed smoothly. However, after 3–4 min, aggregation slowed, and a chaotic pattern consistent with intermittent disaggregation was observed. In CypD−/− platelets, this pattern of disaggregation was not observed (Fig. 2B, right panel). These results indicate that, in strongly stimulating conditions, a CypD-mediated event limits platelet aggregation.
The adherence and accumulation of CypD+/+ and CypD−/− platelets to type I collagen was evaluated in arterial conditions (1500 s−1) in recalcified whole blood (Figs. 2, C and D, and 5E). Relative to CypD+/+ blood, platelet accumulation was markedly increased when CypD−/− blood was perfused. Platelet accumulation was blocked by JON/A in both CypD+/+ and CypD−/− whole blood, demonstrating the dependence of the observed platelet accumulation on activated integrin αIIbβ3. Taken together, these results comparing the adhesive and aggregatory properties of CypD+/+ and CypD−/− platelets demonstrate that CypD-dependent mPTP formation limits platelet adhesion and aggregation in static and flow conditions. Furthermore, these results demonstrate the close association of integrin αIIbβ3 epitope modulation in procoagulant platelets with decreased integrin αIIbβ3 function or secondary integrin αIIbβ3 inactivation.
FIGURE 5.
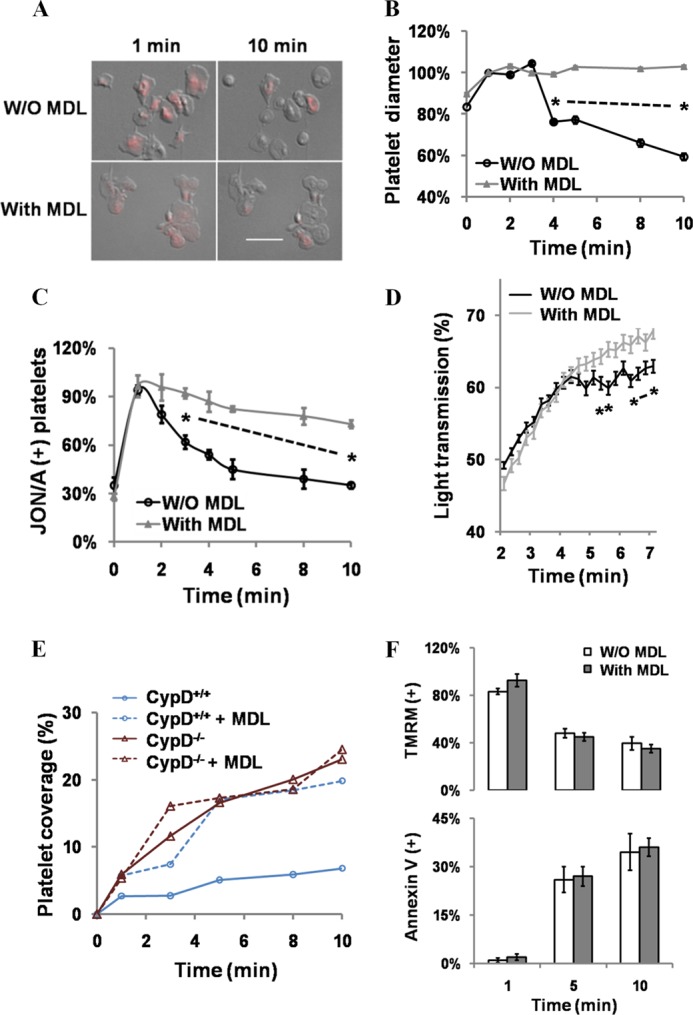
Calpain acts downstream of mPTP formation to mediate secondary integrin αIibβ3 inactivation. A–C, fibrinogen-adherent platelets that were untreated (W/O MDL) or MDL-treated were stimulated with Thr/Cvx. A, morphology and TMRM fluorescence of control and MDL-treated platelets 1 and 10 min after stimulation. Scale bar = 5 μm. B and C, platelet rounding (B) and JON/A binding (C) in control and MDL-treated fibrinogen-adherent platelets stimulated with Thr/Cvx. n = 3. D, platelet aggregation in control and MDL-treated platelets. *, p < 0.05, n = 6. E, adherence of control and MDL-treated CypD+/+ and CypD−/− platelets in flow conditions. CTI/apyrase-treated whole blood was perfused at 1500 s over type I collagen in a microfluidics assay. Representative of n = 3. F, effect of MDL treatment on mPTP formation and PS exposure in control and MDL-treated cells assessed using TMRM and annexin V, respectively. n = 3 in each condition.
Accelerated Thrombosis in Mice with Megakaryocyte/Platelet Deficiency of CypD
The physiologic role of CypD-mediated events in platelets, including integrin αIIbβ3 inactivation, was evaluated in vivo. CypD is a ubiquitously expressed protein (16). Because our previous in vivo experiments evaluating the role of CypD in thrombosis were performed in mice with germ line deletion of CypD (8), the absence of CypD in endothelial cells or other blood cell components associated with the forming thrombus may have contributed to the observed defect. As in the carotid artery (8), a prothrombotic phenotype was noted in CypD−/− mice relative to CypD+/+ mice in the smaller caliber mesenteric artery (supplemental Fig. 4). To directly evaluate the in vivo effects of mPTP dysfunction within the platelet lineage, mice with a megakaryocyte/platelet-specific deficiency of CypD (CypDplt−/−) were generated. Absent CypD expression and impaired procoagulant formation were confirmed in CypDplt−/− mice. As no difference was noted in procoagulant platelet formation or integrin inactivation between wild-type CypDplt+/+ and heterozygote CypDplt+/− mice3, CypDplt+/− and CypDplt−/− mice were compared in in vivo studies.
In comparison with CypDplt+/− mice, in which integrin inactivation is unimpaired, CypDplt−/− mice had a markedly shortened time to stable occlusion in a model of arterial thrombosis, and complete arterial occlusion was increased (Fig. 3A). There was no observed difference in the timing of initial platelet adherence.4 As was seen in CypD−/− mice (8), no significant difference was noted in the tail bleeding time of CypDplt+/− and CypDplt−/− mice (Fig. 3B). These in vivo results demonstrating the platelet-intrinsic effects of CypD deletion, together with the in vitro results demonstrating the importance of CypD in the negative regulation of integrin αIIbβ3 function, strongly point to mPTP-mediated integrin αIIbβ3 inactivation and procoagulant platelet formation as a critical negative determinant limiting platelet aggregation and thrombus formation.
FIGURE 3.
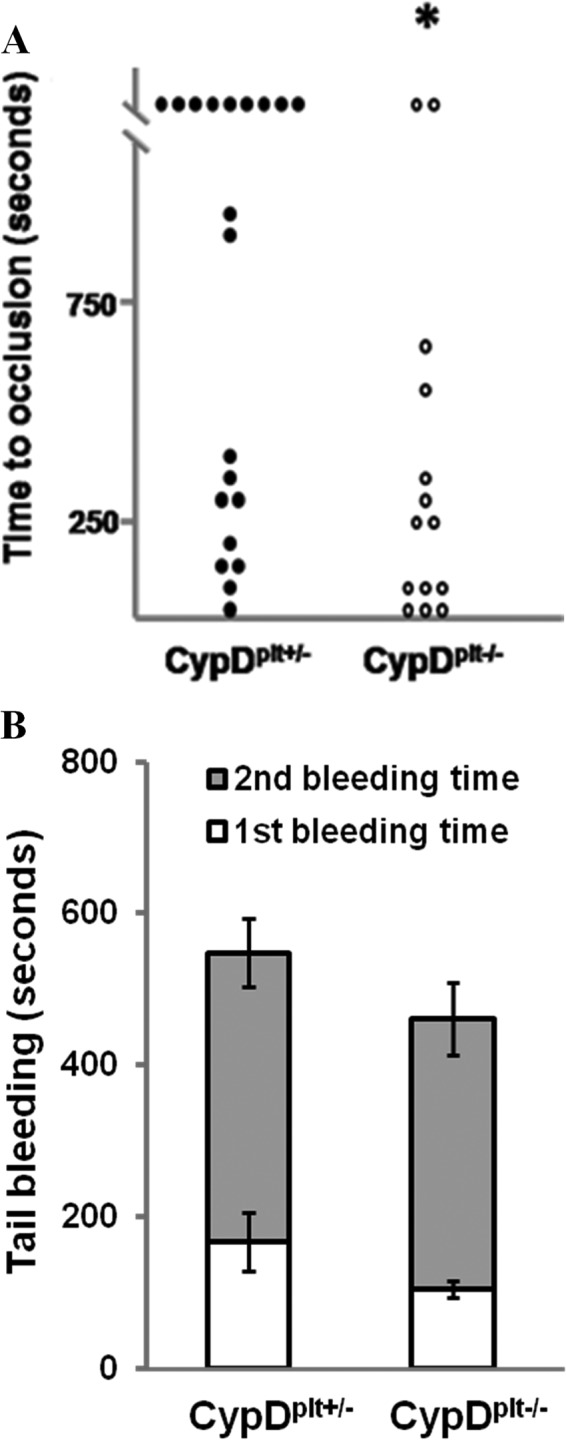
Thrombotic and hemostatic response in mice with megakaryocyte/platelet-specific deficiency of CypD. A, thrombosis was initiated within the mesenteric artery by photochemical injury, and time to stable occlusion was determined in littermate CypDplt+/− and CypDplt−/− mice. Stable occlusion occurred within 1200 s in 9 of 20 CypDplt+/− mice and 2 of 14 CypDplt−/− mice. *, p < 0.05 by Mann-Whitney U test. B, tail bleeding time. Shown is the time to first and final cessation of bleeding in CypDplt+/− and CypDplt−/− mice. p = 0.14, n = 17.
Calpain-mediated Cleavage of the Cytoplasmic Domain of Integrin β3 Is Dependent on CypD
Cleavage of the cytoplasmic tail of integrin β3 and associated cytoskeletal components occurs following platelet activation (23, 24). We examined whether these cleavage events could account for integrin αIIbβ3 inactivation in strongly stimulated platelets. Cleavage of integrin β3 and talin were assessed simultaneously by Western blot analysis using their respective antibodies. In strongly stimulated platelets (Thr/Cvx or Thr/ionomycin) but not in platelets stimulated with either thrombin or convulxin alone, ∼50% of both talin and integrin β3 were cleaved within 5 min (Fig. 4, A and B, and supplemental Fig. 5). In strongly stimulated platelets, a low molecular weight band consistent in size with the talin head domain appeared coincident with loss of full-length talin, and interrogation of integrin β3 using an antibody directed against the C-terminal cytoplasmic tail (C-20) demonstrated decreased binding. That this decreased binding was due to removal of the cytoplasmic tail from the full-length integrin β3 and not to loss of the integrin β3 protein was demonstrated by the persistent binding of an antibody directed against the integrin β3 extracellular domain (N-20) (Fig. 4C).
FIGURE 4.

Calpain-mediated cleavage of the cytoplasmic domain of integrin β3 is dependent on CypD. A and B, washed CypD+/+ or CypD−/− platelets were stimulated for 5 min with the indicated agonist(s) with or without MDL28170 (50 μm) or Q-VD-Oph (QVD) (50 μm), and cleavage of the integrin β3 cytoplasmic tail (C-20) and talin was assessed by Western blot analysis (0.5 units/ml Thr, 100 ng/ml Cvx, and 1 μm Iono). B, quantification of integrin β3 cleavage. Intensity of C-20 binding was assessed relative to unstimulated platelets. *, p < 0.05 compared with untreated platelets; #, p < 0.05 to relative to CypD+/+. n = 3. C, washed CypD+/+ or CypD−/− platelets were stimulated for 5 min with the indicated agonist(s) ± MDL and assessed by Western blot analysis using antibodies specific to the integrin β3 cytoplasmic tail (C-20) and integrin β3 extracellular domain (N-20). *, p < 0.05 relative to unstimulated, #, p < 0.05. D, kinetics of integrin β3 cleavage in CypD+/+ and CypD−/− platelets stimulated with thrombin and convulxin. *, p < 0.05. n = 3. Intensity of C-20 binding was assessed relative to unstimulated platelets.
The association of these cleavage events with integrin αIIbβ3 inactivation and their dependence on mPTP formation was examined. Both talin and integrin β3 cleavage were abrogated in CypD−/− platelets, demonstrating the mutual importance of mPTP formation in mediating both these cleavage events (Fig. 4 and supplemental Fig. 5) and integrin αIIbβ3 inactivation. Because CypD primarily acts to potentiate the calcium sensitivity of the mPTP (25), stimulation with high concentrations of the calcium ionophore ionomycin can initiate mPTP formation and subsequent PS exposure even in the absence of CypD (26). Similarly, ionomycin stimulation bypassed the inhibitory effect of the absence of CypD on talin and integrin β3 cleavage (Fig. 4, A and B, and supplemental Fig. 5). Procoagulant platelet formation recapitulates many events that occur in apoptotic and necrotic cells (27), where important mediators of proteolytic cleavage events include calpain and caspases. In strongly stimulated platelets, pharmacologic inhibition of calpain blocked integrin β3 and talin cleavage in both Thr/Cvx and Thr/ionomycin-stimulated platelets. In contrast, the pan-caspase inhibitor Q-VD-Oph (QVD) had no effect (Fig. 4, A and B, and supplemental Fig. 5). Finally, integrin β3 cleavage was temporally associated with integrin αIIbβ3 inactivation. Significant cleavage was first observed 3 min following agonist stimulation and was complete by 10 min (Fig. 4D).
Calpain Acts Downstream of mPTP Formation to Mediate Secondary Integrin αIIbβ3 Inactivation
The involvement of calpain in β3 C terminus cleavage led us to more closely examine its role in the regulation of platelet integrin αIIbβ3 function. Pharmacologic inhibition of calpain blocked platelet rounding and integrin αIIbβ3 epitope modulation (Fig. 5, A–C), a result consistent with the effects reported previously of calpain inhibition on sustained calcium-induced platelet morphology platelet formation (7). Inhibition of calpain similarly eliminated the pattern of attenuated platelet aggregation that occurred in strongly stimulating conditions (Fig. 5D). Furthermore, MDL treatment resulted in increased platelet accumulation in high shear conditions in CypD+/+ blood (Fig. 5E). No additive effect of MDL treatment was observed in CypD−/− platelets, consistent with inhibition of a common pathway.
To test whether calpain acts upstream or downstream of mPTP formation to regulate integrin αIIbβ3 inactivation, loss of Δψm and platelet shape change were observed simultaneously in MDL-treated platelets. MDL treatment had no effect on mPTP formation (Fig. 5, A and F), and large numbers of spread platelets demonstrated loss of Δψm, which, in untreated platelets, is almost invariably associated with a rounded platelet morphology. PS exposure was similarly unaffected by calpain inhibition (Fig. 5F). Together, these results demonstrate that calpain acts downstream of mPTP formation to mediate secondary integrin αIIbβ3 inactivation in platelets.
CypD-dependent Cytoplasmic Alkalinization Is Associated with Enhanced Calpain Activation and Integrin αIIbβ3 Inactivation
Ca2+ is a key regulator of calpain protease activation. However, as we demonstrated recently, Ca2+ transients in CypD+/+ and CypD−/− platelets are virtually indistinguishable (26) despite their markedly different levels of calpain activity as assessed by substrate cleavage. Because calpain activity can also be regulated by intracellular pH (pHi) (28), the effect of agonist stimulation on platelet pHi was investigated using the cell-permeable pH-sensitive dye SNARF-1. A rapid and pronounced increase in intracellular pHi was noted following Thr/Cvx stimulation (7.32 ± 0.07 to 7.92 ± 0.09). Stimulation with thrombin resulted in a slight increase in pHi (7.51 ± 0.10), as has been reported previously (Fig. 6A) (29). Thr/Cvx-mediated cytoplasmic alkalinization was not observed in CypD−/− platelets or in human platelets treated with the cyclophilin inhibitor cyclosporine (2 μm).4 Because the overlapping fluorescence of PE-JON/A and SNARF-1 AM precluded their simultaneous use, the association of cytoplasmic alkalinization with integrin inactivation was assessed using human platelets and the activation-dependent integrin αIIbβ3 antibody PAC-1. In Thr/Cvx-stimulated platelets, integrin inactivation (PAC-1-negative) and cytoplasmic alkalinization were closely associated (Fig. 6B). These results demonstrate that, in strongly stimulated platelets, mPTP-dependent cytoplasmic alkalinization occurs and that this CypD-mediated change in pHi is closely associated with enhanced calpain activation and integrin inactivation.
FIGURE 6.
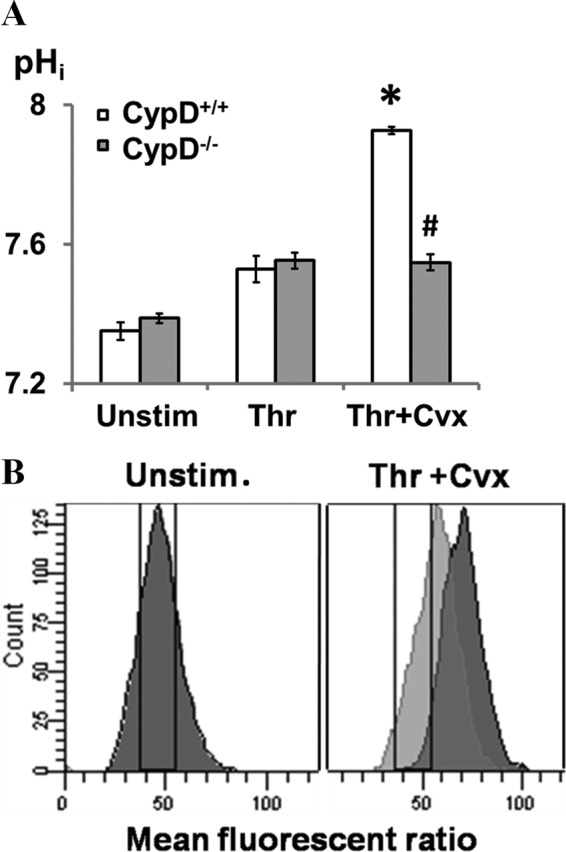
CypD-dependent cytoplasmic alkalinization is associated with enhanced calpain activation and integrin αIibβ3 inactivation. A and B, intracellular pH was measured by flow cytometry assay using SNARF-1 AM as a pH indicator. pHi was determined by the ratio of fluorescent emission at 640 nm and 580 nm. A, pHi was assessed in CypD+/+ and CypD−/− platelets, respectively, after platelets were treated with the indicated agonists. n = 4. *, p < 0.05 compared with unstimulated (Unstim.) platelets; #, p < 0.05 to relative to CypD+/+ platelets. B, pHi and integrin αIIbβ3 activation in strongly stimulated platelets. PAC-1 was used to evaluate integrin αIIbβ3 in human platelets. The dark peak indicates PAC-1(-) platelets and the light gray peak PAC-1(+) platelets. A pHi increase using the SNARF-1 AM indicator is associated with an increase in the 640/580 nm fluorescence ratio. The drawn gate encompasses the fluorescent ratio associated with a pHi of 7.2 (left panel) to 7.4 (right panel). n = 3.
DISCUSSION
Here we demonstrate a novel mitochondrial mechanism that initiates inactivation of integrin αIIbβ3 in strongly stimulated platelets and limits thrombus formation in vivo. In strongly stimulated platelets, CypD and mitochondrially mediated events initiate cytosolic alkalinization and enhanced calpain activation that results in proteolytic cleavage of the integrin β3 cytoplasmic domain, together with other integrin-regulatory proteins, and integrin αIIbβ3 inactivation. Abrogation of mitochondrially mediated calpain activation, both in CypD−/− platelets or using calpain inhibition, prevented secondary integrin αIIbβ3 inactivation and accentuated platelet aggregation. Finally, using platelet-specific CypD−/− mice, we directly demonstrate the physiologic importance of platelet-intrinsic mitochondrial events in limiting thrombus formation.
Previous studies have suggested that integrin αIIbβ3 epitope modulation might be a relevant functional characteristic of the procoagulant platelet (7, 8, 14, 24), but various aspects of these studies have limited their applicability. Pharmacologic induction of a procoagulant platelet monolayer using ionomycin has been shown to result in a decreased ability to stably recruit platelets in flow conditions (7). Impaired clot retraction has been demonstrated in conditions that favor procoagulant platelet formation (8, 24). Furthermore, strongly stimulating conditions have been shown to result in blunted and slowed aggregation (14). Limitations of these studies have included their correlative approach (7, 14), the utilization of the pharmacologic agonist ionomycin (7, 24), or, as in the case of clot retraction (8, 24), the plausibility of other explanations apart from integrin inactivation. Because clot retraction is an energy-dependent event (30), a decrease in mitochondrial ATP production caused by mPTP formation (16) could also account for the inhibition of clot retraction. Here, by utilizing CypD−/− platelets and direct assessment of integrin inactivation, we firmly establish that integrin αIIbβ3 epitope modulation in procoagulant platelets is indicative of inactivation of integrin αIIbβ3. In strongly stimulated CypD+/+ platelets, rounding and lamellipodal retraction occurred subsequent to initial platelet spreading. Platelet aggregation was disrupted, and platelet accumulation in flow conditions was blunted. These limiting responses, all consistent with functional inactivation of integrin αIIbβ3, were impaired in CypD−/− platelets. Rounding and integrin αIIbβ3 inactivation were delayed, aggregation proceeded without disruption, and platelet accumulation and adherence were enhanced markedly.
These epitope and functional changes led us to more closely examine integrin αIIbβ3 in strongly stimulated platelets. Calpain- and CypD-dependent cleavage of both talin and the integrin β3 cytoplasmic tail were found to occur in strongly stimulated platelets within minutes of activation. Proteolytic cleavage of integrin β3 and talin were closely correlated with integrin αIIbβ3 inactivation, and these events were mutually abrogated in CypD−/− and calpain-inhibited platelets. However, the precise calpain-mediated event(s) resulting in integrin αIIbβ3 inactivation remains to be established. Besides integrin β3 and talin, kindlin 3 is cleaved in ionomycin-stimulated platelets (31). Additionally, within integrin β3, there are multiple potential calpain cleavage sites (19). Truncation of integrin β3 at the more membrane distal site Tyr759 selectively effects outside-in integrin signaling, whereas more membrane-proximal truncation disrupts integrin activation (19). However, because calpain cleavage of integrin β3 both eliminates the ability of the talin head domain to activate αIIbβ3 in a purified system (20) and removes the kindlin 3 binding site (32), proteolysis of integrin β3 is likely to contribute in some way to integrin αIIbβ3 inactivation in strongly stimulated platelets.
mPTP formation is an important upstream mediator of both calpain-mediated secondary integrin αIIbβ3 inactivation and PS exposure (26). In this as in other studies (33, 34), PS exposure was unaffected by calpain inhibition. These results are consistent with two distinct pathways regulating events downstream of mPTP formation, one that initiates PS exposure and a second calpain-dependent pathway that initiates integrin αIIbβ3 inactivation and platelet rounding. The dependence of calpain activation on calcium is firmly established (35). However, because cytoplasmic calcium fluxes do not differ in CypD+/+ and CypD−/− platelets (26), differences in calcium flux alone cannot explain the activation of calpain subsequent to mPTP formation. One potential mechanism is that mitochondrial matrix contents released as a consequence of mPTP formation might further up-regulate calpain activity analogous to the effects of cytochrome c as a mediator of apoptosis (36). pHi elevation has been demonstrated to enhance calpain activity (28). Here we demonstrate the association of mPTP-mediated cytoplasmic alkalinization and integrin inactivation. A potential mechanism that could account for this alkalinization is equilibration of the alkalotic mitochondrial matrix (37) with the cytoplasm. Alternatively, activation of the Na+/H+ exchanger could facilitate cytosolic alkalinization. Interestingly, multiple studies have suggested the involvement of Na+/H+ exchanger activity in mediating procoagulant activity and, presumably, procoagulant platelet formation (38, 39).
The prothrombotic effects of the absence of CypD on in vivo thrombosis have been demonstrated previously (8). Whether this prothrombotic effect of the absence of CypD was intrinsic to the platelet could not be determined in those initial experiments because of the ubiquitous expression of CypD. Using mice with a platelet-specific deficiency of CypD, we conclusively demonstrate that CypD-mediated events intrinsic to the platelet limit thrombus formation in vivo. We propose that integrin αIIbβ3 inactivation is the primary CypD-mediated mechanism down-regulating thrombus formation in vivo. However, an alternative hypothesis is suggested by an in vivo study that demonstrated the formation of a fibrin cap on the formed thrombus in a laser-induced model (40). Impaired PS exposure in CypD−/− platelets, by preventing formation of the limiting fibrin cap (41), might allow thrombus growth to proceed unabated.
Interestingly, the phenotype of CypD−/− and CypDplt−/− mice differs markedly from that of organisms with Scott syndrome in which a bleeding, not a prothrombotic phenotype, is observed (42–44). This is despite their mutual abrogation of PS exposure in vitro. Loss of Δψm is unaffected in Scott syndrome, indicating that the effects of TMEM16F are mediated downstream of mPTP formation (45, 46). Because different mPTP-dependent pathways mediate PS exposure and integrin αIIbβ3 inactivation, it is likely that preservation of integrin αIIbβ3 inactivation in Scott syndrome is responsible for the observed bleeding defect. Future studies examining the in vivo effects of specific inhibition of each of the pathways downstream of CypD, namely PS exposure and integrin αIIbβ3 inactivation, would be expected to provide exciting new mechanistic insights.
Together, the results presented here indicate the central role of platelet mPTP formation in regulating both integrin αIIbβ3 function and arterial thrombosis. Strongly stimulating conditions in which multiple soluble or adherent agonists are present promote mPTP formation and secondary integrin αIIbβ3 inactivation. Together, this dependence on strongly stimulating conditions and the prothrombotic effect of the absence of CypD suggest that CypD-dependent integrin αIIbβ3 inactivation could act as a negative feedback mechanism limiting thrombus growth in vivo. Interestingly, use of the cyclophilin D inhibitor, cyclosporine, is associated with thrombotic complications (47). Modulation of procoagulant platelet formation and integrin αIIbβ3 function using agents that favor mPTP formation may offer a novel alternative for the prevention of thrombosis.
Acknowledgments
We thank Keith Neeves (Colorado School of Mines) and Jorge Di Paola (University of Colorado, Denver) for the microfluidic system, David R. Archer (Emory University) for providing β-actin-GFP transgenic mice, and the Emory and Children's Pediatric Research Flow Cytometry Core.
This work was supported, in whole or in part, by National Institutes of Health grant HL095858. This work was also supported by American Heart Association Fellow-to-Faculty Transition Award 2006-2011.

This article contains supplemental Figs. 1–6.
S. M. Jobe, unpublished observations.
F. Liu, G. Gamez, D. R. Myers, W. Clemmons, W. A. Lam, and S. M. Jobe, unpublished observations.
- PS
- phosphatidylserine
- mPTP
- mitochondrial permeability transition pore
- CypD
- cyclophilin D
- TMRM
- tetramethylrhodamine methyl ester
- TRITC
- tetramethylrhodamine isothiocyanate
- SNARF-1
- seminaphtharhodafluor
- Thr
- thrombin
- Cvx
- convulxin
- MDL
- MDL28170
- CTI
- corn trypsin inhibitor.
REFERENCES
- 1. Dale G. L., Friese P., Batar P., Hamilton S. F., Reed G. L., Jackson K. W., Clemetson K. J., Alberio L. (2002) Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 415, 175–179 [DOI] [PubMed] [Google Scholar]
- 2. Munnix I. C., Cosemans J. M., Auger J. M., Heemskerk J. W. (2009) Platelet response heterogeneity in thrombus formation. Thromb. Haemost. 102, 1149–1156 [DOI] [PubMed] [Google Scholar]
- 3. Alberio L., Safa O., Clemetson K. J., Esmon C. T., Dale G. L. (2000) Surface expression and functional characterization of α-granule factor V in human platelets. Effects of ionophore A23187, thrombin, collagen, and convulxin. Blood 95, 1694–1702 [PubMed] [Google Scholar]
- 4. Dale G. L. (2005) Coated-platelets. An emerging component of the procoagulant response. J. Thromb. Haemost 3, 2185–2192 [DOI] [PubMed] [Google Scholar]
- 5. Heemskerk J. W., Vuist W. M., Feijge M. A., Reutelingsperger C. P., Lindhout T. (1997) Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets. Evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood 90, 2615–2625 [PubMed] [Google Scholar]
- 6. Hess M. W., Siljander P. (2001) Procoagulant platelet balloons. Evidence from cryopreparation and electron microscopy. Histochem. Cell Biol. 115, 439–443 [DOI] [PubMed] [Google Scholar]
- 7. Kulkarni S., Jackson S. P. (2004) Platelet factor XIII and calpain negatively regulate integrin αIIbβ3 adhesive function and thrombus growth. J. Biol. Chem. 279, 30697–30706 [DOI] [PubMed] [Google Scholar]
- 8. Jobe S. M., Wilson K. M., Leo L., Raimondi A., Molkentin J. D., Lentz S. R., Di Paola J. (2008) Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 111, 1257–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Briedé J. J., Wielders S. J., Heemskerk J. W., Baruch D., Hemker H. C., Lindhout T. (2003) Von Willebrand factor stimulates thrombin-induced exposure of procoagulant phospholipids on the surface of fibrin-adherent platelets. J. Thromb. Haemost. 1, 559–565 [DOI] [PubMed] [Google Scholar]
- 10. Cosemans J. M., Schols S. E., Stefanini L., de Witt S., Feijge M. A., Hamulyák K., Deckmyn H., Bergmeier W., Heemskerk J. W. (2011) Key role of glycoprotein Ib/V/IX and von Willebrand factor in platelet activation-dependent fibrin formation at low shear flow. Blood 117, 651–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leytin V., Allen D. J., Mykhaylov S., Mis L., Lyubimov E. V., Garvey B., Freedman J. (2004) Pathologic high shear stress induces apoptosis events in human platelets. Biochem. Biophys. Res. Commun. 320, 303–310 [DOI] [PubMed] [Google Scholar]
- 12. Munnix I. C., Kuijpers M. J., Auger J., Thomassen C. M., Panizzi P., van Zandvoort M. A., Rosing J., Bock P. E., Watson S. P., Heemskerk J. W. (2007) Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation. Regulation by transient integrin activation. Arterioscler. Thromb. Vasc. Biol. 27, 2484–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jobe S. M., Leo L., Eastvold J. S., Dickneite G., Ratliff T. L., Lentz S. R., Di Paola J. (2005) Role of FcRγ and factor XIIIA in coated platelet formation. Blood 106, 4146–4151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yakimenko A. O., Verholomova F. Y., Kotova Y. N., Ataullakhanov F. I., Panteleev M. A. (2012) Identification of different proaggregatory abilities of activated platelet subpopulations. Biophys. J. 102, 2261–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Remenyi G., Szasz R., Friese P., Dale G. L. (2005) Role of mitochondrial permeability transition pore in coated-platelet formation. Arterioscler. Thromb. Vasc. Biol. 25, 467–471 [DOI] [PubMed] [Google Scholar]
- 16. Halestrap A. P. (2009) What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 46, 821–831 [DOI] [PubMed] [Google Scholar]
- 17. Giorgio V., Soriano M. E., Basso E., Bisetto E., Lippe G., Forte M. A., Bernardi P. (2010) Cyclophilin D in mitochondrial pathophysiology. Biochim. Biophys. Acta 1797, 1113–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yan B., Calderwood D. A., Yaspan B., Ginsberg M. H. (2001) Calpain cleavage promotes talin binding to the β 3 integrin cytoplasmic domain. J. Biol. Chem. 276, 28164–28170 [DOI] [PubMed] [Google Scholar]
- 19. Xi X., Bodnar R. J., Li Z., Lam S. C., Du X. (2003) Critical roles for the COOH-terminal NITY and RGT sequences of the integrin β3 cytoplasmic domain in inside-out and outside-in signaling. J. Cell Biol. 162, 329–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ye F., Hu G., Taylor D., Ratnikov B., Bobkov A. A., McLean M. A., Sligar S. G., Taylor K. A., Ginsberg M. H. (2010) Recreation of the terminal events in physiological integrin activation. J. Cell Biol. 188, 157–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Leo L., Di Paola J., Judd B. A., Koretzky G. A., Lentz S. R. (2002) Role of the adapter protein SLP-76 in GPVI-dependent platelet procoagulant responses to collagen. Blood 100, 2839–2844 [DOI] [PubMed] [Google Scholar]
- 22. Neeves K. B., Maloney S. F., Fong K. P., Schmaier A. A., Kahn M. L., Brass L. F., Diamond S. L. (2008) Microfluidic focal thrombosis model for measuring murine platelet deposition and stability. PAR4 signaling enhances shear-resistance of platelet aggregates. J. Thromb. Haemost. 6, 2193–2201 [DOI] [PubMed] [Google Scholar]
- 23. Du X., Saido T. C., Tsubuki S., Indig F. E., Williams M. J., Ginsberg M. H. (1995) Calpain cleavage of the cytoplasmic domain of the integrin β 3 subunit. J. Biol. Chem. 270, 26146–26151 [DOI] [PubMed] [Google Scholar]
- 24. Schoenwaelder S. M., Yuan Y., Cooray P., Salem H. H., Jackson S. P. (1997) Calpain cleavage of focal adhesion proteins regulates the cytoskeletal attachment of integrin αIIbβ3 (platelet glycoprotein IIb/IIIa) and the cellular retraction of fibrin clots. J. Biol. Chem. 272, 1694–1702 [DOI] [PubMed] [Google Scholar]
- 25. Basso E., Fante L., Fowlkes J., Petronilli V., Forte M. A., Bernardi P. (2005) Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J. Biol. Chem. 280, 18558–18561 [DOI] [PubMed] [Google Scholar]
- 26. Choo H. J., Saafir T. B., Mkumba L., Wagner M. B., Jobe S. M. (2012) Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler. Thromb. Vasc. Biol. 32, 2946–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schoenwaelder S. M., Yuan Y., Josefsson E. C., White M. J., Yao Y., Mason K. D., O'Reilly L. A., Henley K. J., Ono A., Hsiao S., Willcox A., Roberts A. W., Huang D. C., Salem H. H., Kile B. T., Jackson S. P. (2009) Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood 114, 663–666 [DOI] [PubMed] [Google Scholar]
- 28. Zhao X., Newcomb J. K., Posmantur R. M., Wang K. K., Pike B. R., Hayes R. L. (1998) pH dependency of μ-calpain and m-calpain activity assayed by casein zymography following traumatic brain injury in the rat. Neurosci Lett. 247, 53–57 [DOI] [PubMed] [Google Scholar]
- 29. Horne W. C., Norman N. E., Schwartz D. B., Simons E. R. (1981) Changes in cytoplasmic pH and in membrane potential in thrombin-stimulated human platelets. Eur. J. Biochem. 120, 295–302 [DOI] [PubMed] [Google Scholar]
- 30. Mürer E. H. (1969) Clot retraction and energy metabolism of platelets. Effect and mechanism of inhibitors. Biochim. Biophys. Acta 172, 266–276 [DOI] [PubMed] [Google Scholar]
- 31. Zhao Y., Malinin N. L., Meller J., Ma Y., West X. Z., Bledzka K., Qin J., Podrez E. A., Byzova T. V. (2012) Regulation of cell adhesion and migration by Kindlin-3 cleavage by calpain. J. Biol. Chem. 287, 40012–40020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moser M., Nieswandt B., Ussar S., Pozgajova M., Fässler R. (2008) Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14, 325–330 [DOI] [PubMed] [Google Scholar]
- 33. Gaffet P., Bettache N., Bienvenüe A. (1995) Phosphatidylserine exposure on the platelet plasma membrane during A23187-induced activation is independent of cytoskeleton reorganization. Eur. J. Cell Biol. 67, 336–345 [PubMed] [Google Scholar]
- 34. Wolf B. B., Goldstein J. C., Stennicke H. R., Beere H., Amarante-Mendes G. P., Salvesen G. S., Green D. R. (1999) Calpain functions in a caspase-independent manner to promote apoptosis-like events during platelet activation. Blood 94, 1683–1692 [PubMed] [Google Scholar]
- 35. Perrin B. J., Huttenlocher A. (2002) Calpain. Int. J. Biochem. Cell Biol. 34, 722–725 [DOI] [PubMed] [Google Scholar]
- 36. Oberst A., Bender C., Green D. R. (2008) Living with death. The evolution of the mitochondrial pathway of apoptosis in animals. Cell Death Differ. 15, 1139–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Llopis J., McCaffery J. M., Miyawaki A., Farquhar M. G., Tsien R. Y. (1998) Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. U.S.A. 95, 6803–6808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bucki R., Pastore J. J., Giraud F., Janmey P. A., Sulpice J. C. (2006) Involvement of the Na+/H+ exchanger in membrane phosphatidylserine exposure during human platelet activation. Biochim. Biophys. Acta 1761, 195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Samson J., Stelmach H., Tomasiak M. (2001) The importance of Na+/H+ exchanger for the generation of procoagulant activity by porcine blood platelets. Platelets 12, 436–442 [DOI] [PubMed] [Google Scholar]
- 40. Kamocka M. M., Mu J., Liu X., Chen N., Zollman A., Sturonas-Brown B., Dunn K., Xu Z., Chen D. Z., Alber M. S., Rosen E. D. (2010) Two-photon intravital imaging of thrombus development. J. Biomed Opt. 15, 016020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lishko V. K., Yermolenko I. S., Ugarova T. P. (2010) Plasminogen on the surfaces of fibrin clots prevents adhesion of leukocytes and platelets. J. Thromb. Haemost 8, 799–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Brooks M. B., Catalfamo J. L., Brown H. A., Ivanova P., Lovaglio J. (2002) A hereditary bleeding disorder of dogs caused by a lack of platelet procoagulant activity. Blood 99, 2434–2441 [DOI] [PubMed] [Google Scholar]
- 43. Yang H., Kim A., David T., Palmer D., Jin T., Tien J., Huang F., Cheng T., Coughlin S. R., Jan Y. N., Jan L. Y. (2012) TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 151, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zwaal R. F., Comfurius P., Bevers E. M. (2004) Scott syndrome, a bleeding disorder caused by defective scrambling of membrane phospholipids. Biochim. Biophys. Acta 1636, 119–128 [DOI] [PubMed] [Google Scholar]
- 45. Brooks M. B., Catalfamo J. L., Friese P., Dale G. L. (2007) Scott syndrome dogs have impaired coated-platelet formation and calcein-release but normal mitochondrial depolarization. J. Thromb. Haemost. 5, 1972–1974 [DOI] [PubMed] [Google Scholar]
- 46. van Kruchten R., Mattheij N. J., Saunders C., Feijge M. A., Swieringa F., Wolfs J. L., Collins P. W., Heemskerk J. W., Bevers E. M. (2013) Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood 121, 1850–1857 [DOI] [PubMed] [Google Scholar]
- 47. Lopez E., Rosado J. A., Redondo P. C. (2011) Immunophilins and thrombotic disorders. Curr. Med. Chem. 18, 5414–5423 [DOI] [PubMed] [Google Scholar]