

Background: HSC-derived CTGF is an important mediator of liver fibrogenesis.
Results: Stat3 is an essential factor for TGF-β mediated CTGF expression in activated HSCs.
Conclusion: TGF-β is using a complex signaling network including Stat3 to trigger CTGF production in HSCs.
Significance: Stat3 is defined for the first time as crucial TGF-β downstream signaling mediator to regulate extracellular matrix production in liver.
Keywords: ERK, Jak Kinase, Jun N-terminal Kinase (JNK), MAP Kinases (MAPKs), PI 3-kinase (PI3K), SMAD Transcription Factor, Liver Fibrosis
Abstract
In fibrotic liver, connective tissue growth factor (CTGF) is constantly expressed in activated hepatic stellate cells (HSCs) and acts downstream of TGF-β to modulate extracellular matrix production. Distinct from other cell types in which Smad signaling plays major role in regulating CTGF production, TGF-β stimulated CTGF expression in activated HSCs is only in part dependent on Smad3. Other signaling molecules like MAPKs and PI3Ks may also participate in this process, and the underlying mechanisms have yet to be clarified. In this study, we report involvement of Stat3 activation in modulating CTGF production upon TGF-β challenge in activated HSCs. Stat3 is phosphorylated via JAK1 and acts as a critical ALK5 (activin receptor-like kinase 5) downstream signaling molecule to mediate CTGF expression. This process requires de novo gene transcription and is additionally modulated by MEK1/2, JNK, and PI3K pathways. Cell-specific knockdown of Smad3 partially decreases CTGF production, whereas it has no significant influence on Stat3 activation. The total CTGF production induced by TGF-β in activated HSCs is therefore, to a large extent, dependent on the balance and integration of the canonical Smad3 and Stat3 signaling pathways.
Introduction
Connective tissue growth factor (CTGF)3 is a secreted matricellular protein and participates in regulation of various important cell functions such as proliferation, differentiation, cell adhesion, migration, and extracellular matrix production. It interacts with a variety of molecules, including cytokines and growth factors, and modulates signaling pathways, which leads to changes in cellular responses. Among these, most importantly, CTGF acts downstream and synergizes with action of the fibrogenic master cytokine transforming growth factor-β (TGF-β) and is a central mediator of tissue remodeling and fibrosis (1–3). Up-regulated expression of CTGF is observed in numerous fibrotic tissues, e.g. kidney, lung, heart, liver, pancreas, bowel, and skin (3). CTGF production in healthy liver is usually very low, whereas elevated levels of CTGF are common in patients with liver fibrosis/cirrhosis of various etiologies as well as in experimental animal models of liver fibrosis (4–13). Accordingly, inhibition of CTGF expression not only prevents but also can regress established hepatic fibrosis in experiment models (14–16).
The cellular distribution of CTGF in liver fibrosis is largely dependent on etiology and disease progression. Diverse cellular sources for CTGF have been reported in fibrotic liver including hepatocytes (7, 12), activated hepatic stellate cells (HSCs), myofibroblasts (5–8, 10, 16), endothelial cells (5, 12), proliferating bile duct epithelial cells (5, 10, 12), and inflammatory cells (12). Its sustained expression in HSCs is of special importance, as these cells assume an activated phenotype in liver fibrogenesis and play a central role in excessive fibrillar collagen production and extracellular matrix remodeling. Indeed, activated HSCs are a biologically significant source of CTGF, as supported by intensive cellular localization of CTGF in α-smooth muscle actin (SMA) positive sinusoidal cells (6). Importantly, clinical studies and animal models revealed that CTGF is intensely detectable in activated HSCs or myofibroblasts in fibrotic liver but not in quiescent HSCs in healthy liver, indicating that CTGF is induced in HSCs as a specific response to liver damage (5, 8, 17).
Expression of CTGF in HSCs is up-regulated by numerous profibrotic factors, including TGF-β (4, 18, 19), endothelin-1 (20), PDGF-BB, acetaldehyde (21), ethanol (22), and the Th2 cytokine IL-13 (23, 24), with TGF-β being the most important trigger as elevated TGF-β levels are observed in almost all fibrotic liver diseases, irrespective of etiology. Although in vitro experiments have shown that TGF-β plays only a minor role in CTGF production in quiescent HSCs (20, 23, 24), activated HSCs as well as established HSC cell lines secrete high levels of CTGF in response to TGF-β treatment (22, 23).
TGF-β exerts its cellular function through binding to its cognate serine/threonine kinase receptors type II (TβRII) and type I (TβRI, ALK5, or ALK1), which is subsequently followed by transactivation of TβRI by TβRII, leading to R-Smad (Smad1/2/3) activation. Phosphorylated R-Smads then heteromerize with Smad4 and shuttle into the nucleus to regulate gene expression. CTGF is an immediate early gene upon TGF-β challenge, and its up-regulation is at least in part dependent on intracellular Smad signaling (22). In addition to the canonical Smad pathway, TGF-β can also activate other signaling molecules such as Erk1/2, JNK, p38 MAPK, PKC, and PI3K/Akt, which is largely cell type-dependent (25). Previous studies have demonstrated that selective inhibition of those signaling pathways resulted in a potent reduction of CTGF mRNA expression in activated HSCs (26, 27). The underlying mechanisms, however, have not been clarified yet.
In this study, we show for the first time that signal transducer and activator of transcription 3 (Stat3) is involved in modulating CTGF production upon TGF-β treatment in activated HSCs (CFSC-2G and hTERT HSC cell lines). Stat3 is phosphorylated via JAK1 and acts as an essential downstream signaling molecule to mediate CTGF expression. This process is independent from Smad2/3 phosphorylation but is additionally modulated by MEK1/2, JNK, and PI3K pathways. The total CTGF production induced by TGF-β in activated HSCs is thus, to a large extent, dependent on the balance and integration of the canonical Smad3 and Stat3 signaling pathways.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Human recombinant TGF-β1 was purchased from Peprotech; DMEM, penicillin/streptomycin, and l-glutamine were obtained from Cambrex. Non-essential amino acids and fetal bovine serum were from Invitrogen. siRNAs for Stat3 (SI02040752), JAK1 (SI01526616), JAK2 (SI01526623), Smad2 (SI03108140), and Smad3 (SI03056515) were delivered from Qiagen, siRNA Tyk2 (SASI_Rn01_00149727) was purchased from Sigma-Aldrich and siRNA ALK5 (M-098121 siGenome SMARTpool) was from Dharmacon. Transfection reagent (RNAiMAX) was obtained from Invitrogen. The following inhibitors were used: Stattic (p-Stat3 inhibitor) was purchased from Merck/Calbiochem; actinomycin D (transcription inhibitor), cycloheximide (translation inhibitor), SB431542 (ALK5 inhibitor), and SP600125 (JNK inhibitor) were obtained from Sigma-Aldrich. U0126 (Erk1/2 inhibitor) and LY294002 (PI3K/Akt inhibitor) were from Tocris. Antibodies were as follows: p-Smad3 (Epitomics/Biomol); β-actin (Sigma-Aldrich); α-SMA (Dako); CTGF, TGF-β1, p-Erk1/2, and GAPDH (Santa Cruz Biotechnology); and p-Stat3, Stat3, p-Smad2, Smad2, Smad3, p-c-Jun, and p-Akt (Cell Signaling). Horseradish peroxidase-linked secondary anti-mouse, anti-rabbit, and anti-goat antibodies were from Santa Cruz Biotechnology.
Liver Tissues
Surgical liver samples were obtained from nine patients at the Department of General, Visceral Surgery, and Transplantation (University Hospital Tübingen, Germany). Eight had hepatocellular carcinoma on non-fibrosis (n = 1), fibrosis (n = 1), or cirrhosis (n = 6) background, and one had focal nodular hyperplasia with otherwise normal liver structure. Partial hepatectomy (n = 6) or liver transplantation (n = 3) was performed. The collected tissues are all tumor-free (at least >5 cm away from the tumor area). The etiology for liver fibrosis/cirrhosis was chronic hepatitis C (n = 1), chronic hepatitis B (n = 3), alcoholic liver disease (n = 2), and unknown reasons (n = 1). The study was approved by the local ethics committee, and written informed consent was obtained from all patients. The study protocol conformed to the ethical guidelines of the Declaration of Helsinki (1975).
Cell Culture
The immortalized rat HSC cell line CFSC-2G, generated from carbon tetrachloride (CCl4)-induced cirrhotic rat liver (28), was cultured in DMEM containing 10% FCS, 4 mm l-glutamine, penicillin/streptomycin, and non-essential amino acids. The activated human HSC cell line hTERT HSC (29) was a kind gift from Dr. Claus Hellerbrand (University Hospital of Regensburg, Regensburg, Germany) and cultured in DMEM, supplemented with 10% FCS, 4 mm l-glutamine, penicillin/streptomycin, and 400 ng/liter Geneticin (G418, Invitrogen). Cells were grown and maintained at 37 °C, 5% CO2 in a humidified atmosphere. FCS was reduced to 0.2% for overnight starvation prior to chemical and cytokine treatment.
siRNA Transfection
siRNA transfection was performed in CFSC-2G cells using RNAiMAX. Transfection was maintained overnight without medium change, and TGF-β challenge was performed 48 h post transfection. Knockdown of specific genes was verified at RNA or protein level.
RNA Isolation and PCR for Knockdown Verification
Total RNA was isolated using the RNeasy mini kit (Qiagen). For each sample, 1 μg RNA was reverse transcribed into cDNA with the Quantitech reverse transcription kit (Qiagen). Semiquantitative PCR was performed to proof knockdown efficiency of JAK1, JAK2, and Tyk2 siRNA oligonucleotides. The following primers were used: JAK1, 5′-TGACATTGGCCCGTTCATCA-3′ (forward) and 5′-CGCTCTATGCACTCTTGCCT-3′ (reverse); JAK2, 5′-AGTGGAGGAGACAAGCCTCT-3′ (forward) and 5′-AGTTACCCTTGCCAAGTTGC-3′ (reverse); Tyk2, 5′-ACAAGTGCTTGTTGCTGTGC-3′ (forward) and 5′-GCTACGGTGAGGATCAGTCG-3′ (reverse).
Immunohistochemical Staining
Liver tissues were fixed in 4% formaldehyde and embedded in paraffin. 3-μm sections were prepared for immunohistochemical staining using the DAKO EnVision system. Briefly, slides were deparaffinized in xylene and rehydrated in a dilution series of graded ethanol to distilled water. Antigen retrieval was performed by microwave treatment in 1 mm EDTA buffer (pH 8.0). The slides were blocked with DAKO Dual Endogenous Enzyme Block reagent for 30 min at room temperature and then incubated with 3% H2O2 for 15 min. After rinsing with PBS, the slides were incubated with primary antibodies overnight at 4 °C. The next day, after extensive washing with PBS, the slides were incubated with EnVision peroxidase-labeled anti-rabbit or anti-goat antibodies (Dako) for 1 h at room temperature. Peroxidase activity was detected with diaminobenzidine. The slides were counterstained with hematoxylin. Immunoreactivity was visualized under light microscopy.
Immunofluorescent Staining
CFSC-2G cells were cultured on glass coverslips at a density of 7 × 105 cells per well in a 12-well plate and stimulated with 5 ng/ml TGF-β1 after overnight starvation. For immunostaining, cells were fixed with 4% (w/v) paraformaldehyde in PBS for 15 min at room temperature. After fixation, cells were rinsed with PBS and then permeabilized with ice-cold methanol for 10 min at −20 °C. Unspecific binding sites were blocked with 0.1% (w/v) BSA for 1 h at room temperature. Cells were then incubated with primary antibody overnight at 4 °C. After extensive washing, cells were incubated with secondary antibody (Alexa Fluor 488 goat anti-rabbit immunoglobulin G, Invitrogen) for 45 min at room temperature, and nuclei were stained with draq5 (Dako). Confocal microscopy and image acquisition were carried out using the confocal microscope Leica TCS SP2.
For liver tissue staining, sections were deparaffinized in xylene, followed by serial ethanol dilutions. After washing with PBS, sections were transferred into 1 mm EDTA buffer (pH 8.0), and antigen unmasking was performed in a microwave. After cooling down, sections were incubated in peroxidase blocking reagent (Dako) for 1 h and with primary antibody overnight at 4 °C. Secondary antibodies were Alexa Fluor 488 donkey anti-goat immunoglobulin G and Alexa Fluor 633 goat anti-mouse immunoglobulin G (Invitrogen). Nuclei were stained with draq5. Samples were mounted using Dako Cytomation fluorescent mounting medium.
Western Blot Analysis
Cells were lysed on ice with radioimmune precipitation assay lysis buffer (50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, and 0.1% SDS) in the presence of complete protease inhibitor mixture (Roche Applied Science) and phosphatase inhibitors (Sigma-Aldrich). Protein concentration was determined using Bradford method with a Bio-Rad protein assay. 20 μg protein per sample were separated by SDS-polyacrylamide gel (10%) electrophoresis and subsequently transferred to a nitrocellulose membrane (Pierce). Nonspecific binding was blocked with 5% milk in TBST (Tris-buffered saline plus 1% Tween-20), and the membrane was allowed to react with primary antibody overnight at 4 °C. Horseradish peroxidase-linked anti-mouse, anti-rabbit, and anti-goat antibodies were used as secondary antibodies. The membranes were developed using Chemi-Smart (Vilber Lourmat).
Densitometry and Statistical Analysis
Western blot data were quantitatively analyzed using the software program Aida Image Analyzer (version 2.11). Results were expressed as means ± S.E. and calculated from at least three independent experiments. Data were analyzed using two-tailed unpaired Student's t test. p < 0.05 was considered statistically significant.
RESULTS
Elevated p-Stat3, TGF-β/p-Smad3, and CTGF Levels in Liver Samples of Fibrosis/cirrhosis Patients
As reported previously (3, 6), both TGF-β and CTGF expressions are elevated in the sinusoids of fibrotic/cirrhotic livers (Fig. 1A). To define the cell type expressing CTGF, we performed immunofluorescent co-staining with α-SMA. CTGF expression was found localized in α-SMA-positive cells (activated HSCs or myofibroblasts) (Fig. 1A). Accordingly, immunoblot analyses of liver lysates from fibrosis/cirrhosis patients with different etiologies showed up-regulated p-Smad3 and CTGF levels in the majority of investigated cases, which was accompanied by significantly elevated α-SMA expression (Fig. 1B). Furthermore, a marked increase of Stat3 phosphorylation (Tyr-705) was observed in fibrotic/cirrhotic livers, and to a large extent, this was associated with elevated levels in p-Smad3 and CTGF (Fig. 1B, red boxes). These findings prompted us to investigate whether there is a relationship between TGF-β signaling and Stat3 activation and to study the functional importance of this toward CTGF production in activated HSCs.
FIGURE 1.

Elevated p-Stat3, TGF-β/p-Smad3, and CTGF levels in liver samples of fibrosis/cirrhosis patients. A, immunohistochemical stainings of TGF-β and CTGF expression were performed in healthy liver tissues from a patient with gallstones and in cirrhotic liver tissues from a patient with hepatitis B (HBV) infection (upper panel). Elevated TGF-β and CTGF levels were only detected in cirrhotic liver but not in the liver with normal architecture. Both TGF-β and CTGF displayed positive staining in sinusoidal areas in this patient. Immunofluorescent co-staining demonstrated CTGF overexpression in activated HSCs (α-SMA-positive cells, lower panel). B, liver lysates of fibrosis/cirrhosis patients from different etiologies were analyzed for profibrotic signaling activation and CTGF production. A marked increase of p-Stat3 was detected in association with Smad3 activation and CTGF production in the majority of liver cirrhosis cases (red boxes).
TGF-β Induces CTGF Expression and Stat3 Activation in Activated HSCs
Although TGF-β only marginally stimulates CTGF expression in quiescent HSCs (24), activated HSCs (CFSC-2G cells and hTERT HSCs) respond to TGF-β treatment by expressing high levels of CTGF (Fig. 2A). Additionally, we observed activation of Stat3 (phosphorylation on Tyr-705) upon TGF-β challenge prior to significant CTGF induction (Fig. 2A). To further consolidate this result, p-Stat3 subcellular distribution with TGF-β challenge was measured by immunofluorescent staining in CFSC-2G cells. Here, a time-dependent nuclear accumulation of p-Stat3 was detected, being pronounced 2 h after TGF-β incubation (Fig. 2B). These results imply that the Stat3-mediated signaling pathway is activated by TGF-β in activated HSCs and may participate in stimulation of CTGF expression.
FIGURE 2.

TGF-β induces Stat3 activation and CTGF expression in activated HSCs. A, CFSC-2G (CFSC) cells and hTERT HSCs were treated with 5 ng/ml TGF-β1 for the indicated time periods. Western blot analysis unraveled tyrosine Stat3 phosphorylation prior to significant CTGF expression upon TGF-β challenge. CTGF and GAPDH were quantified by densitometric analysis and expressed as a ratio of CTGF to GAPDH. Data are means ± S.E. p < 0.01, p < 0.001, and p < 0.0001 indicate the statistical significance. B, CFSC-2G cells were stimulated with TGF-β1 for 0.5–2 h. Time-dependent p-Stat3 and p-Smad2/3 nuclear translocation was shown with immunofluorescence.
Stat3 Activation Is Required for TGF-β-mediated CTGF Expression in Activated HSCs
To clarify involvement of Stat3 in TGF-β induced CTGF expression, CFSC-2G cells were transfected with siRNA that selectively depletes Stat3 expression. Silencing Stat3 resulted in a significant decrease of TGF-β-mediated CTGF expression without affecting the canonical TGF-β/Smad signaling (Fig. 3A). Moreover, addition of a chemical inhibitor, Stattic, which was shown to selectively inhibit activation of Stat3 by blocking tyrosine phosphorylation and Stat3 dimerization (31), attenuated TGF-β induced CTGF expression in a dose-dependent manner in both CFSC-2G cells and hTERT HSCs (Fig. 3B). These findings further underscore a role for Stat3 activation in TGF-β-dependent CTGF expression in activated HSCs.
FIGURE 3.

Stat3 activation is required for TGF-β-mediated CTGF production. A, CFSC-2G (CFSC) cells were transfected with Stat3 or control siRNA for 48 h, followed by exposure to 5 ng/ml TGF-β1 as indicated. Lack of Stat3 resulted in reduced CTGF production. Knockdown was verified at protein level by measurement of total Stat3 and p-Stat3. B, Stattic, a chemical inhibitor, was added to CFSC-2G cells and hTERT HSCs 30 min prior to TGF-β1 treatment to blunt Stat3 phosphorylation. CTGF expression was analyzed by Western blot. Stattic interfered with TGF-β-induced CTGF in a dosage-dependent manner in both cell lines. Data are expressed as a ratio of CTGF to GAPDH and are means ± S.E., p < 0.01 and p < 0.001 indicate the statistical significance. DMSO, dimethyl sulfoxide. ut, untreated; siCo, siControl.
TGF-β-induced Stat3 Activation Requires ALK5, but Not Smad2/3
TGF-β can utilize both ALK1 and ALK5 to exert its fibrogenic function in HSCs with ALK5/Smad3 being the predominant signaling pathway (32). We thus examined whether Stat3 activation by TGF-β is downstream of ALK5/Smad2/3 signaling. The ALK5 kinase inhibitor SB431542 was applied to CFSC-2G cells and hTERT HSCs. SB431542 blunted both TGF-β-induced Stat3 phosphorylation and CTGF expression (Fig. 4A). Moreover, siRNA-mediated knockdown of ALK5 in CFSC-2G cells strongly mitigated TGF-β-mediated Stat3 activation and CTGF induction (Fig. 4A). Together, these findings indicate that TGF-β induces Stat3 phosphorylation and CTGF production in activated HSCs in an ALK5 receptor kinase-dependent manner.
FIGURE 4.

Stat3 activation is downstream of ALK5 and independent of Smad2/3. A, ALK5 is inactivated in CFSC-2G cells and hTERT HSCs by pre-incubation with 5 μm SB431542 for 30 min or by transfection of siRNA for ALK5 in CFSC-2G cells prior to TGF-β1 (5 ng/ml) treatment. Western blot showed repressed Stat3 activation and CTGF expression upon loss of ALK5 activity. Successful inactivation of ALK5 was indicated by reduced p-Smad2/3. B, CFSC-2G cells were transfected with siRNA for Smad2, Smad3, or both, followed by stimulation with 5 ng/ml TGF-β1 as indicated. Knockdown of Smad2 and Smad3 did not interfere with initiation of Stat3 activation. CTGF expression is Smad2-independent but at least in part associated with Smad3 signaling. Results are expressed as a ratio of CTGF to GAPDH and are means ± S.E.; p < 0.05, p < 0.01, and p < 0.001 indicate the statistical significance. DMSO, dimethyl sulfoxide; ut, untreated; siCo, siControl.
Smad2 and Smad3 are pivotal downstream effectors of TGF-β/ALK5 signaling (33). In particular, Smad3 plays a critical role in mediating TGF-β-induced fibrogenic response (34). Interestingly, siRNA-mediated Smad2 and/or Smad3 knockdown in CFSC-2G cells did not significantly affect TGF-β-induced Stat3 phosphorylation (Fig. 4B). Depletion of Smad3 partially attenuated TGF-β-mediated CTGF expression, suggesting that the transcription factors Smad3 and Stat3 act together for CTGF induction, and the total amount of CTGF production upon TGF-β challenge in activated HSCs may be a result from balance and integration of the two signaling pathways.
TGF-β-stimulated Stat3 Activation Requires de Novo Protein Synthesis
Considering that both type I and type II TGF-β receptors possess mainly Ser/Thr kinase function, and the tyrosine Stat3 phosphorylation by TGF-β occurs with delayed kinetics as compared with R-Smad phosphorylation (Fig. 2A), we hypothesized that the effect is not direct and requires new protein synthesis. We therefore subjected both cell lines to the transcriptional and translational inhibitors actinomycin D and cycloheximide. Both inhibitors blunted TGF-β-induced Stat3 phosphorylation without affecting total Stat3 levels and Smad signaling (Fig. 5A), implicating that tyrosine phosphorylation of Stat3 in activated HSCs may be a secondary response to TGF-β and requires de novo protein synthesis.
FIGURE 5.

TGF-β stimulated Stat3 activation requires de novo protein synthesis. A, CFSC-2G cells and hTERT HSCs were pre-incubated with 1 μg/ml actinomycin D (ActD) or 10 μg/ml cycloheximide (CHX) for 30 min prior to TGF-β1 (5 ng/ml) treatment. TGF-β-induced Stat3 phosphorylation was blocked by both inhibitors. B, CFSC-2G cells were treated with or without TGF-β1 (5 ng/ml) for 1 h, and conditioned media were collected and added into new cell cultures (after overnight starvation). Western blot analysis did not detect earlier Stat3 activation and CTGF expression in conditioned (cond.) media-treated cells. DMSO, dimethyl sulfoxide.
We subsequently tested whether Stat3 activation is the result of a fast secreted cellular product of an immediate early TGF-β target. To estimate this, we incubated CFSC-2G cells with or without TGF-β for 1 h and collected the conditioned media to treat new cells for different time periods (Fig. 5B). If Stat3 phosphorylation was induced by such a secretory product in responsive to TGF-β, an earlier p-Stat3 signal (<1 h) would be expected. However, an accelerated Stat3 activation was not observed upon incubation of CFSC-2G cells with the conditioned media (Fig. 5B). The p-Stat3 detected in this setting was still resulting from the remaining TGF-β in the conditioned media and occurred 1 h after incubation started. This finding indicates that instead of producing/secreting new proteins, an intracellular level of regulatory mechanism is suggested as initiator of Stat3 activation upon TGF-β treatment.
JAK1 Is the Dominant Upstream Kinase to Activate Stat3 upon TGF-β Challenge
Next, our studies aimed to get further insight into the molecular mechanism mediating Stat3 activation upon TGF-β treatment. As the Stat family of transcription factors is canonically activated by members of the JAK family, and JAK2/Stat3 signaling has been shown previously to play a critical role in the early stages of HSC transdifferentiation (35), we examined whether JAKs participate in initiation of Stat3 activation and CTGF production upon TGF-β stimulation in activated HSCs. To achieve this, we silenced all three ubiquitously expressed JAK family members (JAK1, JAK2, and Tyk2) in CFSC-2G cells, respectively. Knockdown of JAK1, but not JAK2 and Tyk2, attenuated TGF-β-mediated Stat3 activation as well as CTGF production (Fig. 6). These results suggest a JAK1-dependent mechanism being dominant for TGF-β induced Stat3 activation and CTGF production in activated HSCs.
FIGURE 6.

TGF-β-mediated Stat3 phosphorylation requires JAK1. CFSC-2G (CFSC) cells were transfected with JAK1, JAK2, or Tyk2 siRNA and subsequently exposed to 5 ng/ml TGF-β1 for 2 h. Knockdown of JAK1 blunted Stat3 phosphorylation and resulted in reduced CTGF expression. Silencing JAK2 and Tyk2 had no influence on this process. CTGF and GAPDH expression was quantified, and data are means ± S.E.; p < 0.01 indicates the statistical significance. Knockdown of JAKs was verified at the RNA level by semiquantitative PCR. ut, untreated; siCo, siControl; n.s., not significant.
TGF-β-mediated Stat3 Activation and CTGF Expression Is Regulated by MAPKs and PI3K
Because TGF-β-stimulated tyrosine Stat3 phosphorylation plays an important role in regulation of total CTGF production in activated HSCs, we wanted to identify in more detail how TGF-β signaling integrates into initiation of Stat3 activation. This may be of special relevance to determine the ultimate cellular effect of Stat3 in HSCs, as its tyrosine phosphorylation often shows cell type and stimuli-dependent functional variance. Src, MAPKs, and PI3K have been described as modulators of Stat3 activation by promoting either tyrosine or serine phosphorylation (36–38). Considering that all of these signaling components can also be activated by TGF-β (25), we investigated their potential participations in regulation of Stat3 activation in activated HSCs. A time course experiment in CFSC-2G cells was performed to identify TGF-β non-canonical signaling activation in HSCs with impact on initiating Stat3 phosphorylation. A significant activation of different branches of MAPK signaling pathways (Erk1/2 and JNK) as well as transient PI3K/Akt signaling was observed prior to Stat3 phosphorylation upon TGF-β treatment (Fig. 7A). Significant p-p38 and p-Src elevation, however, were not observed in the same time course in response to TGF-β (data not shown), thus excluding a crucial role of these pathways in affecting Stat3 activation. Further analyses suggested that activation of Erk, JNK, and PI3K/Akt signaling cascades were also mediated by ALK5 (Fig. 7A). Subsequently, we investigated whether these signaling pathways were involved in TGF-β-mediated Stat3 activation and CTGF expression. To achieve this, MEK1/2, JNK, and PI3K were blunted with specific inhibitors (U0126 for MEK/Erk1/2, SP600125 for JNK, and LY294002 for PI3K/Akt). Interference with Erk1/2, JNK, and Akt pathways all mitigated TGF-β-induced Stat3 phosphorylation (Fig. 7, B–D), suggesting that TGF-β-stimulated Stat3 signaling initiation in activated HSCs is a very complex process, integrating several intracellular signaling branches. When further investigating CTGF expression, we observed that PI3K/Akt pathway inhibition had no significant influence on TGF-β-induced CTGF production (Fig. 7B). Analysis of p-Smad3 demonstrated a strong cell type specific modulation of PI3K/Akt on the TGF-β canonical signaling pathway. Blunting PI3K in CFSC-2G cells enhanced Smad3 activation, whereas Akt pathway inhibition in hTERT HSCs decreased p-Smad3 levels (Fig. 7B). This observation suggests that in addition to Smad3 and Stat3, other signaling molecules that are negatively regulated by PI3K/Akt pathway may also participate to modulate total CTGF expression in the absence of PI3K activity. In contrast to PI3K/Akt, Erk1/2, and JNK pathways inhibition strongly interfered with the TGF-β/Stat3 downstream effect and ultimately resulted in decreased CTGF expression (Fig. 7, C and D). Together, these data suggest that MEK1/2, JNK and PI3K participate in regulation of TGF-β stimulated Stat3 activation and subsequent CTGF production in activated HSCs.
FIGURE 7.

MAPKs and PI3K are required for TGF-β-induced Stat3 activation in activated HSCs. A, Western blot revealed ALK5 (SB431542, 5 μm) mediated activation of Erk1/2, JNK, and PI3K/Akt pathways prior to Stat3 phosphorylation upon TGF-β treatment. B–D, PI3K/Akt (LY294002, 10 μm), Erk1/2 (U0126, 10 μm), and JNK (SP600125, 5 μm) inhibitors were used to block specific signaling pathways. CFSC-2G cells and hTERT HSCs were then treated with TGF-β1 (5 ng/ml) for indicated times to detect Stat3 activation and CTGF expression. All inhibitor treatments attenuated early Stat3 phosphorylation. CTGF expression was reduced by Erk1/2 and JNK inhibition but remained unchanged in the absence of PI3K. In contrast to the enhanced p-Smad3 level in LY294002 treated CFSC-2G cells (TGF-β1, 2 h), a reduced p-Smad3 level was observed in hTERT HSCs after PI3K inhibition (TGF-β1, 4 h), indicating a third signaling pathway participating in modulation of total CTGF production in this setting (see “Discussion”). CTGF and GAPDH expression was densitometrically quantified, and results are means ± S.E., p < 0.01 and p < 0.001 indicate the statistical significance. ut, untreated; DMSO, dimethyl sulfoxide.
DISCUSSION
CTGF is often highly expressed in fibrotic/cirrhotic liver tissues and is considered a reliable marker for the severity of fibrosis (39). Sustained expression of CTGF by activated HSCs is of particular interest due to its direct contribution to the function of this cell type in response to liver injury. Following a fibrotic stimulus, HSC undergoes a phenotype change from a quiescent to an activated form, which is associated with increased proliferation and production of large amounts of fibrillar collagens. The master fibrotic cytokine, TGF-β1 is suggested a major trigger for CTGF overproduction in liver disorders and animal experiments (17). Culture activated primary HSCs and HSC cell lines show enhanced CTGF expression when exposed to TGF-β. The molecular details how TGF-β induces CTGF production in HSCs are still largely unsolved. Current available data suggest an ALK5-dependent mechanism as dominant for this induction (26). Other signaling pathways such as the TGF-β non-canonical MAPK, PI3K/Akt, and PKC pathways may also participate in this process; the underlying mechanisms, however, are yet to be clarified (26).
The present study aims at dissecting molecular details downstream of TGF-β/ALK5 signaling leading to CTGF expression in activated HSCs. We show for the first time that Stat3 activation is involved in the process of TGF-β-induced CTGF expression. Because culture-activated primary HSCs in general have low sensitivity to exogenous TGF-β stimulation in terms of Smad activation (40), we chose the CFSC-2G cell line, generated from cirrhotic rat liver, as major cell material. CFSC-2G cells are able to remain genetically stable during culture passages, show high sensitivity to TGF-β, and exhibit expression profiles of fibrotic factors and their cognate receptors, which resemble the character of fully activated HSCs or myofibroblasts in vivo (41). Critical results were also consolidated in a second cell type, hTERT HSC, to exclude cell type specificity of findings.
Clinical observations of tyrosine Stat3 phosphorylation accompanied by enhanced Smad3 activation and up-regulated CTGF/α-SMA expression in fibrotic/cirrhotic livers prompted us to consider the involvement of Stat3 in TGF-β cellular effects during fibrogenesis. In our studies, both CFSC-2G and hTERT HSC cell lines are capable of overexpressing CTGF in response to exogenous TGF-β. Stat3 activation was observed in both cell lines upon TGF-β treatment, and inactivation of Stat3 by either siRNA or specific chemical inhibitors significantly reduced CTGF expression. Thus, our results support a crucial role of p-Stat3 in TGF-β-mediated CTGF overproduction in activated HSCs. This finding also suggests an ambivalent function of Stat3 in modulating TGF-β cellular effects, as IL-6 type cytokines induced Stat3 activation generally counteracts TGF-β/Smad3-mediated CTGF expression in different cell types (42, 43). Our further studies could show that TGF-β-induced p-Stat3 was not significantly affected by silencing Smad3. Therefore, it is very likely that Smad3 and Stat3 independently modulate CTGF gene expression. This is further supported by the observation that either knockdown of Smad3 or Stat3 showed only partial inhibition on TGF-β-induced CTGF expression. Thus, the total CTGF production may to a large extent depend on the balance of both signaling pathways in presence of TGF-β.
Initiation of Stat3 activation upon TGF-β requires JAK1 and de novo gene transcription. However, conditioned media from TGF-β treated CFSC-2G cells failed to induce early Stat3 phosphorylation, suggesting that an intracellular regulation, rather than a simple secondary effect induced by early secretory products, plays a role in activating Stat3. This finding is supported by a follow-up study, in which we found that TGF-β stimulation was not able to induce Stat1 tyrosine phosphorylation in CFSC-2G cells (data not shown), implicating that a unique mechanism distinct from general cytokine receptor-dependent JAK/Stat activation is involved in the TGF-β-mediated Stat3 phosphorylation process, as cytokine-driven JAK1 activation is usually followed by both Stat1 and Stat3 phosphorylation. It is possible that a yet to be identified adaptor protein(s) participate(s) in this process. Another explanation is that TGF-β could rapidly induce negative regulators of Stat1 and thus selectively enable activation of Stat3. Current knowledge could not explain the underlying details arising from this study. Further investigations are needed to define the mediator leading to JAK/Stat3 activation and ultimately to CTGF production.
We also observed elevated MAPKs and PI3K/Akt signaling by TGF-β in CFSC-2G cells shortly before Stat3 phosphorylation and thus hypothesized involvement of these kinases in initiation of activating Stat3. Further investigations with MAPK and PI3K inhibitors confirmed the participation of Erk1/2, JNK, and Akt signaling events in modulating TGF-β-induced Stat3 activation as blocking any of these pathways nearly completely abolished phosphorylation of Stat3 in both CFSC-2G cells and hTERT HSCs. Thus, we confirmed that a complex signaling network orchestrates initiation of Stat3 phosphorylation that is required for CTGF expression. This further indicates that fine modulation of Stat3-mediated CTGF gene expression in HSCs is tightly regulated and largely depends on the combination of available upstream initiating kinases.
Another interesting finding is that MEK/Erk1/2 and JNK pathways are capable of regulating tyrosine phosphorylation of Stat3 upon TGF-β treatment. Previous studies have shown that MEK and JNK pathways mainly induce Stat3 serine phosphorylation, which is required for transcriptional activities of Stat3 (36). Other studies also reported that Erk1/2 is indirectly involved in tyrosine Stat3 phosphorylation in cardiomyocytes (37, 38). Interestingly, the current investigation observed many similarities, including delayed kinetics of Stat3 activation and involvement of de novo protein synthesis. Thus, it is possible that TGF-β utilizes the same mechanism to activate and modulate Stat3 in activated HSCs.
The PI3K/Akt pathway was also previously shown to regulate tyrosine phosphorylation of Stat3 (30, 44). Similarly, PI3K is required for TGF-β-induced Stat3 activation in activated HSCs. Interestingly, PI3K/Akt pathway inhibition had no significant influence on total CTGF production induced by TGF-β. Analysis of Smad3 activation further showed that PI3K modulated TGF-β canonical signaling pathway in a cell type-dependent manner. Although these observations seem contradictory, it may indicate that in addition to Smad3 and Stat3, another CTGF-inducing pathway is switched on in the absence of PI3K/Akt signaling. A potential candidate might be GSK3-transduced signaling as it is repressed with active Akt. Future studies will shed light on how PI3K/Akt integrates and modulates TGF-β fibrogenic signaling pathways in HSCs.
Taken together, our study demonstrated that Stat3 activation is required for TGF-β mediated CTGF induction in activated HSCs. JAK1 is identified as the dominant tyrosine kinase for Stat3 phosphorylation, and the latter is further modulated by TGF-β non-canonical signaling pathways, namely MEK, JNK, and PI3K. The total amount of CTGF expressed in activated HSCs can therefore be regulated at different levels and depend on a balance of Stat3 and Smad3 signaling pathways (Fig. 8). These data provide novel and advanced information for a better understanding of molecular and cellular mechanisms related to fibrogenic TGF-β function in chronic liver diseases.
FIGURE 8.
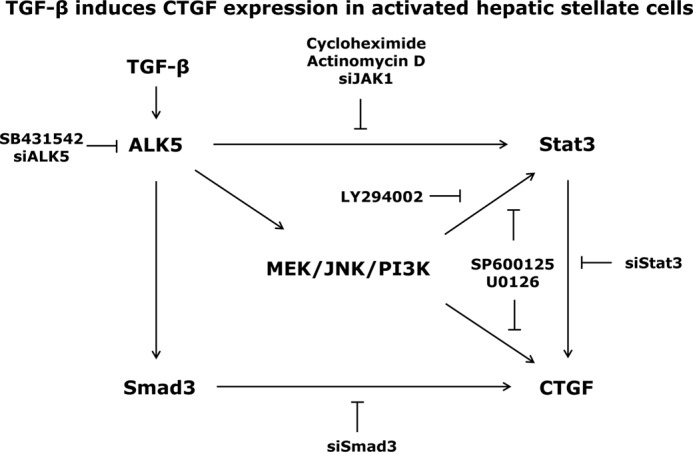
Possible mechanism for TGF-β-mediated CTGF expression in activated HSCs. TGF-β binding to ALK5 receptor induces direct Smad phosphorylation and indirect JAK/Stat3 activation, resulting in enhanced CTGF expression in activated HSCs. This process is additionally modulated by ALK5 mediated activation of MEK1/2, JNK, and PI3K pathways. The total amount of CTGF production upon TGF-β stimulation thus results from integration of both Smad3 and Stat3 signaling pathways.
Acknowledgment
We are grateful for excellent technical assistance from Alexandra Müller.
This work was supported by a Marie Curie Initial Training Network (ITN) IT-Liver grant; the Netherlands Institute for Regenerative Medicine and Cancer Genomics Centre Netherlands, Centre for Biomedical Genetics; German Research Foundation Programs “SFB TRR77 Liver Cancer” and “Do373/8-1”; and Federal Ministry of Education and Research grants “The Virtual Liver” and “Cell Therapy in Liver Regeneration.”
- CTGF
- connective tissue growth factor
- ALK
- activin receptor-like kinase
- α-SMA
- α-smooth muscle actin
- CFSC
- cirrhotic fat storing cell
- ERK
- extracellular signal-regulated kinase
- HSC
- hepatic stellate cell.
REFERENCES
- 1. Leask A., Abraham D. J. (2003) The role of connective tissue growth factor, a multifunctional matricellular protein, in fibroblast biology. Biochem. Cell Biol. 81, 355–363 [DOI] [PubMed] [Google Scholar]
- 2. Lipson K. E., Wong C., Teng Y., Spong S. (2012) CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 5, Suppl. 1, S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gressner O. A., Gressner A. M. (2008) Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int. 28, 1065–1079 [DOI] [PubMed] [Google Scholar]
- 4. Williams E. J., Gaça M. D., Brigstock D. R., Arthur M. J., Benyon R. C. (2000) Increased expression of connective tissue growth factor in fibrotic human liver and in activated hepatic stellate cells. J. Hepatol. 32, 754–761 [DOI] [PubMed] [Google Scholar]
- 5. Abou-Shady M., Friess H., Zimmermann A., di Mola F. F., Guo X. Z., Baer H. U., Büchler M. W. (2000) Connective tissue growth factor in human liver cirrhosis. Liver 20, 296–304 [DOI] [PubMed] [Google Scholar]
- 6. Paradis V., Dargere D., Vidaud M., De Gouville A. C., Huet S., Martinez V., Gauthier J. M., Ba N., Sobesky R., Ratziu V., Bedossa P. (1999) Expression of connective tissue growth factor in experimental rat and human liver fibrosis. Hepatology 30, 968–976 [DOI] [PubMed] [Google Scholar]
- 7. Kobayashi H., Hayashi N., Hayashi K., Yamataka A., Lane G. J., Miyano T. (2005) Connective tissue growth factor and progressive fibrosis in biliary atresia. Pediatr. Surg. Int. 21, 12–16 [DOI] [PubMed] [Google Scholar]
- 8. Hayashi N., Kakimuma T., Soma Y., Grotendorst G. R., Tamaki K., Harada M., Igarashi A. (2002) Connective tissue growth factor is directly related to liver fibrosis. Hepato-gastroenterology 49, 133–135 [PubMed] [Google Scholar]
- 9. Hora C., Negro F., Leandro G., Oneta C. M., Rubbia-Brandt L., Muellhaupt B., Helbling B., Malinverni R., Gonvers J. J., Dufour J. F. (2008) Connective tissue growth factor, steatosis and fibrosis in patients with chronic hepatitis C. Liver Int. 28, 370–376 [DOI] [PubMed] [Google Scholar]
- 10. Sedlaczek N., Jia J. D., Bauer M., Herbst H., Ruehl M., Hahn E. G., Schuppan D. (2001) Proliferating bile duct epithelial cells are a major source of connective tissue growth factor in rat biliary fibrosis. Am. J. Pathol. 158, 1239–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paradis V., Perlemuter G., Bonvoust F., Dargere D., Parfait B., Vidaud M., Conti M., Huet S., Ba N., Buffet C., Bedossa P. (2001) High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 34, 738–744 [DOI] [PubMed] [Google Scholar]
- 12. Tache D., Bogdan F., Pisoschi C., Banită M., Stănciulescu C., Fusaru A. M., Comănescu V. (2011) Evidence for the involvement of TGF-β1-CTGF axis in liver fibrogenesis secondary to hepatic viral infection. Rom. J. Morphol. Embryol. 52, 409–412 [PubMed] [Google Scholar]
- 13. Kamada Y., Tamura S., Kiso S., Matsumoto H., Saji Y., Yoshida Y., Fukui K., Maeda N., Nishizawa H., Nagaretani H., Okamoto Y., Kihara S., Miyagawa J., Shinomura Y., Funahashi T., Matsuzawa Y. (2003) Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 125, 1796–1807 [DOI] [PubMed] [Google Scholar]
- 14. Li G., Xie Q., Shi Y., Li D., Zhang M., Jiang S., Zhou H., Lu H., Jin Y. (2006) Inhibition of connective tissue growth factor by siRNA prevents liver fibrosis in rats. J. Gene Med. 8, 889–900 [DOI] [PubMed] [Google Scholar]
- 15. Brigstock D. R. (2009) Strategies for blocking the fibrogenic actions of connective tissue growth factor (CCN2): From pharmacological inhibition in vitro to targeted siRNA therapy in vivo. J. Cell Commun. Signal. 3, 5–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. George J., Tsutsumi M. (2007) siRNA-mediated knockdown of connective tissue growth factor prevents N-nitrosodimethylamine-induced hepatic fibrosis in rats. Gene Ther. 14, 790–803 [DOI] [PubMed] [Google Scholar]
- 17. Huang G., Brigstock D. R. (2012) Regulation of hepatic stellate cells by connective tissue growth factor. Front. Biosci. 17, 2495–2507 [DOI] [PubMed] [Google Scholar]
- 18. Sun K., Wang Q., Huang X. H. (2006) PPARγ inhibits growth of rat hepatic stellate cells and TGFβ-induced connective tissue growth factor expression. Acta Pharmacol. Sin. 27, 715–723 [DOI] [PubMed] [Google Scholar]
- 19. Gao R., Brigstock D. R. (2003) Low density lipoprotein receptor-related protein (LRP) is a heparin-dependent adhesion receptor for connective tissue growth factor (CTGF) in rat activated hepatic stellate cells. Hepatol. Res. 27, 214–220 [DOI] [PubMed] [Google Scholar]
- 20. Gressner O. A., Lahme B., Demirci I., Gressner A. M., Weiskirchen R. (2007) Differential effects of TGF-β on connective tissue growth factor (CTGF/CCN2) expression in hepatic stellate cells and hepatocytes. J. Hepatol. 47, 699–710 [DOI] [PubMed] [Google Scholar]
- 21. Paradis V., Dargere D., Bonvoust F., Vidaud M., Segarini P., Bedossa P. (2002) Effects and regulation of connective tissue growth factor on hepatic stellate cells. Lab. Invest. 82, 767–774 [DOI] [PubMed] [Google Scholar]
- 22. Chen L., Charrier A. L., Leask A., French S. W., Brigstock D. R. (2011) Ethanol-stimulated differentiated functions of human or mouse hepatic stellate cells are mediated by connective tissue growth factor. J. Hepatol. 55, 399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weng H. L., Liu Y., Chen J. L., Huang T., Xu L. J., Godoy P., Hu J. H., Zhou C., Stickel F., Marx A., Bohle R. M., Zimmer V., Lammert F., Mueller S., Gigou M., Samuel D., Mertens P. R., Singer M. V., Seitz H. K., Dooley S. (2009) The etiology of liver damage imparts cytokines transforming growth factor β1 or interleukin-13 as driving forces in fibrogenesis. Hepatology 50, 230–243 [DOI] [PubMed] [Google Scholar]
- 24. Liu Y., Meyer C., Müller A., Herweck F., Li Q., Müllenbach R., Mertens P. R., Dooley S., Weng H. L. (2011) IL-13 induces connective tissue growth factor in rat hepatic stellate cells via TGF-beta-independent Smad signaling. J. Immunol. 187, 2814–2823 [DOI] [PubMed] [Google Scholar]
- 25. Mu Y., Gudey S. K., Landström M. (2012) Non-Smad signaling pathways. Cell Tissue Res. 347, 11–20 [DOI] [PubMed] [Google Scholar]
- 26. Leask A., Chen S., Pala D., Brigstock D. R. (2008) Regulation of CCN2 mRNA expression and promoter activity in activated hepatic stellate cells. J. Cell Commun. Signal. 2, 49–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Son G., Hines I. N., Lindquist J., Schrum L. W., Rippe R. A. (2009) Inhibition of phosphatidylinositol 3-kinase signaling in hepatic stellate cells blocks the progression of hepatic fibrosis. Hepatology 50, 1512–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Greenwel P., Schwartz M., Rosas M., Peyrol S., Grimaud J. A., Rojkind M. (1991) Characterization of fat storing cell lines derived from normal and CCl4-cirrhotic livers. Lab. Invest. 65, 644–653 [PubMed] [Google Scholar]
- 29. Schnabl B., Choi Y. H., Olsen J. C., Hagedorn C. H., Brenner D. A. (2002) Immortal activated human hepatic stellate cells generated by ectopic telomerase expression. Lab. Invest. 82, 323–333 [DOI] [PubMed] [Google Scholar]
- 30. Vogt P. K., Hart J. R. (2011) PI3K and STAT3: a new alliance. Cancer Discov. 1, 481–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schust J., Sperl B., Hollis A., Mayer T. U., Berg T. (2006) Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 13, 1235–1242 [DOI] [PubMed] [Google Scholar]
- 32. Wiercinska E., Wickert L., Denecke B., Said H. M., Hamzavi J., Gressner A. M., Thorikay M., ten Dijke P., Mertens P. R., Breitkopf K., Dooley S. (2006) Id1 is a critical mediator in TGF-β-induced transdifferentiation of rat hepatic stellate cells. Hepatology 43, 1032–1041 [DOI] [PubMed] [Google Scholar]
- 33. Itoh S., Itoh F., Goumans M. J., Ten Dijke P. (2000) Signaling of transforming growth factor-β family members through Smad proteins. Eur. J. Biochem. 267, 6954–6967 [DOI] [PubMed] [Google Scholar]
- 34. Flanders K. C. (2004) Smad3 as a mediator of the fibrotic response. Int. J. Exp. Pathol. 85, 47–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lakner A. M., Moore C. C., Gulledge A. A., Schrum L. W. (2010) Daily genetic profiling indicates JAK/STAT signaling promotes early hepatic stellate cell transdifferentiation. World J. Gastroenterol. 16, 5047–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Decker T., Kovarik P. (2000) Serine phosphorylation of STATs. Oncogene 19, 2628–2637 [DOI] [PubMed] [Google Scholar]
- 37. Frias M. A., Rebsamen M. C., Gerber-Wicht C., Lang U. (2007) Prostaglandin E2 activates Stat3 in neonatal rat ventricular cardiomyocytes: A role in cardiac hypertrophy. Cardiovasc. Res. 73, 57–65 [DOI] [PubMed] [Google Scholar]
- 38. Ng D. C., Long C. S., Bogoyevitch M. A. (2001) A role for the extracellular signal-regulated kinase and p38 mitogen-activated protein kinases in interleukin-1 β-stimulated delayed signal tranducer and activator of transcription 3 activation, atrial natriuretic factor expression, and cardiac myocyte morphology. J. Biol. Chem. 276, 29490–29498 [DOI] [PubMed] [Google Scholar]
- 39. Leask A., Abraham D. J. (2006) All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J. Cell Sci. 119, 4803–4810 [DOI] [PubMed] [Google Scholar]
- 40. Dooley S., Hamzavi J., Breitkopf K., Wiercinska E., Said H. M., Lorenzen J., Ten Dijke P., Gressner A. M. (2003) Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 125, 178–191 [DOI] [PubMed] [Google Scholar]
- 41. Herrmann J., Gressner A. M., Weiskirchen R. (2007) Immortal hepatic stellate cell lines: useful tools to study hepatic stellate cell biology and function? J. Cell Mol. Med. 11, 704–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gressner O. A., Peredniene I., Gressner A. M. (2011) Connective tissue growth factor reacts as an IL-6/STAT3-regulated hepatic negative acute phase protein. World J. Gastroenterol. 17, 151–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sarközi R., Flucher K., Haller V. M., Pirklbauer M., Mayer G., Schramek H. (2012) Oncostatin M inhibits TGF-β1-induced CTGF expression via STAT3 in human proximal tubular cells. Biochem. Biophys. Res. Commun. 424, 801–806 [DOI] [PubMed] [Google Scholar]
- 44. Cho M. L., Kang J. W., Moon Y. M., Nam H. J., Jhun J. Y., Heo S. B., Jin H. T., Min S. Y., Ju J. H., Park K. S., Cho Y. G., Yoon C. H., Park S. H., Sung Y. C., Kim H. Y. (2006) STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J. Immunol. 176, 5652–5661 [DOI] [PubMed] [Google Scholar]