

Background: p40 is a Lactobacillus rhamnosus GG-derived protein.
Results: p40 stimulates ADAM17 activation and HB-EGF release, which is required for EGF receptor transactivation, prevention of apoptosis, and preservation of barrier function in intestinal epithelial cells.
Conclusion: p40 transactivates the EGF receptor through ADAM17-mediated HB-EGF release in intestinal epithelial cells.
Significance: These results define a mechanism of p40 in modulating intestinal epithelial cell homeostasis.
Keywords: Apoptosis, Epidermal Growth Factor (EGF), Intestinal Epithelium, Probiotics, Tight Junctions
Abstract
p40, a Lactobacillus rhamnosus GG (LGG)-derived soluble protein, ameliorates intestinal injury and colitis, reduces apoptosis, and preserves barrier function by transactivation of the EGF receptor (EGFR) in intestinal epithelial cells. The aim of this study is to determine the mechanisms by which p40 transactivates the EGFR in intestinal epithelial cells. Here we show that p40-conditioned medium activates EGFR in young adult mouse colon epithelial cells and human colonic epithelial cell line, T84 cells. p40 up-regulates a disintegrin and metalloproteinase domain-containing protein 17 (ADAM17) catalytic activity, and broad spectrum metalloproteinase inhibitors block EGFR transactivation by p40 in these two cell lines. In ADAM17-deficient mouse colonic epithelial (ADAM17−/− MCE) cells, p40 transactivation of EGFR is blocked, but can be rescued by re-expression with WT ADAM17. Furthermore, p40 stimulates release of heparin binding (HB)-EGF, but not transforming growth factor (TGF)α or amphiregulin, in young adult mouse colon cells and ADAM17−/− MCE cells overexpressing WT ADAM17. Knockdown of HB-EGF expression by siRNA suppresses p40 effects on transactivating EGFR and Akt, preventing apoptosis, and preserving tight junction function. The effects of p40 on HB-EGF release and ADAM17 activation in vivo are examined after administration of p40-containing pectin/zein hydrogel beads to mice. p40 stimulates ADAM17 activity and EGFR activation in colonic epithelial cells and increases HB-EGF levels in blood from WT mice, but not from mice with intestinal epithelial cell-specific ADAM17 deletion. Thus, these data define a mechanism of a probiotic-derived soluble protein in modulating intestinal epithelial cell homeostasis through ADAM17-mediated HB-EGF release, leading to transactivation of EGFR.
Introduction
The symbiotic relationship between intestinal microbiota and the host plays an important role in maintaining human health, including regulating host metabolism and biosynthetic pathways, promoting homeostasis, and eliminating toxic substances (1). Interruption of this relationship has been found in several pathological conditions, including inflammatory bowel disease (2). Probiotics are live microorganisms which, when consumed in adequate amounts as part of food, confer a health benefit on the host and may serve as a potential alternative therapy for disease prevention and treatment. Currently, both clinical and basic research has revealed several distinct cellular and molecular mechanisms underlying the beneficial effects of probiotics, including blocking pathogenic bacterial effects, regulating immune responses, and modulating intestinal epithelial homeostasis (3–5).
However, there are several concerns regarding the use of viable probiotics. First, it has been a challenge to determine the bioavailability and efficacy of probiotic bacteria in the gastrointestinal tract. In addition, the use of viable probiotic bacteria raises concerns about biosafety, with several cases of bacterium-associated infections in very young (6) and immuno-compromised patients (7), and the increased risk of mortality in patients with severe acute pancreatitis (8). In light of these concerns, an alternative approach of probiotic-derived products has been used to develop novel therapeutic reagents with improved clinical efficacy.
Lactobacillus rhamnosus GG (LGG)2 is a naturally occurring Gram-positive bacterium originally isolated from the healthy human intestine (9). LGG is one of the probiotic bacteria used in clinical trials for treating and/or preventing several diseases, including ulcerative colitis (10), diarrhea (11, 12), and atopic dermatitis (13). Our group has purified and cloned a LGG-derived soluble protein, p40, which contains 412 amino acid residues with a calculated molecular mass of 42 kDa (14). Sequence analysis of p40 revealed that the N terminus of p40 contains an uncharacterized protein domain conserved in bacteria, whereas the C-terminal region of p40 exhibits high homology to a cysteine, histidine-dependent amidohydrolase/peptidase domain (14). Further characterization of p40 showed that p40 in Lactobacilllus casei BL23 is located at the bacterial cell surface, is secreted to the culture media, and is able to hydrolyze the muropeptides from L. casei cell walls (15), whereas another study reported that p40 has d-glutamyl-l-lysyl endopeptidase activity (16, 17).
We have shown that p40 prevents cytokine-induced epithelial damage and apoptosis (14, 18) and hydrogen peroxide disruption of epithelial barrier function (19) through activation of EGF receptor (EGFR) and its downstream target, Akt, in intestinal epithelial cells. Furthermore, specific delivery of p40 to the colon using special hydrogel beads to protect p40 from degradation prevents and treats colonic epithelial cell injury and inflammation in mouse models of colitis in an EGFR-dependent manner (20).
EGFR, a member of ErbB family, has an extracellular ligand-binding domain and an intracellular portion that contains a tyrosine kinase domain (21, 22). Ligation of EGFR by its soluble ligands, EGF, heparin-binding (HB)-EGF, transforming growth factor (TGF)α, or amphiregulin, triggers formation of homo- and heterodimers with other ErbB family members and autophosphorylation of cytoplasmic tyrosine residues (21, 22). These phosphorylated amino acids provide docking sites for a variety of signaling molecules that regulate intracellular signaling networks, such as Akt. Activation of EGFR promotes cell proliferation, differentiation, migration, and survival (21, 22). Although EGFR is considered a tumor promoter, EGF has shown therapeutic potential in human ulcerative colitis (23), and EGFR activation plays a role in ameliorating chronic inflammation, thus limiting colitis-associated tumorigenesis (24).
EGFR ligands are synthesized as transmembrane precursors, which are released by a highly regulated process to produce soluble active ligands. Membrane-bound or uncleavable ligands impede EGFR activation by preventing dimerization (25). A wide variety of pharmacological and physiological stimuli, such as ligand-mediated stimulation of G protein-coupled receptors (26), can trigger EGFR ligand shedding and release. A disintegrin and metalloproteinases (ADAMs) are membrane-anchored proteases that are able to cleave the extracellular domains of membrane-bound proteins in a process known as “ectodomain shedding.” Regulation of ADAM proteolytic activity is still poorly defined, although recent studies have demonstrated that intracellular trafficking of ADAM17 (also termed tumor necrosis factor-α-converting enzyme, TACE) is critical for its function (27, 28).
Although we have identified the ability of p40 to activate EGFR (20), the mechanisms underlying this process are unknown. We show here that p40-stimulated HB-EGF release is mediated by ADAM17, leading to EGFR activation in intestinal epithelial cells. Our data define a mechanism of probiotic-derived soluble proteins in modulating intestinal homeostasis.
EXPERIMENTAL PROCEDURES
Cell Culture
T84 cells (ATCC, CCL-248), a human colonic adenocarcinoma cell line, were cultured in 1:1 mixture of Ham's F12 medium and DMEM with 2.5 mm glutamine, 5% fetal bovine serum (FBS), and 100 units/ml penicillin and streptomycin at 37 °C with 5% CO2.
A young adult mouse colonic epithelium (YAMC) cell line was generated using a mouse harboring thermolabile mutation (tsA58) under the control of an interferon (IFN)-γ-inducible H-2kb promoter and a temperature-sensitive simian virus 40 large T antigen (Immortomouse) (29). The functional expression of the SV40 large T antigen is induced by culturing the cells in vitro in RPMI 1640 medium containing IFNγ at a temperature permissive (33 °C) for function of the tsA58 mutation. Expression of this gene is required for YAMC cell proliferation. YAMC cells die when the temperature is raised to the nonpermissive temperature (37 °C) or when the cells are cultured without IFNγ for three passages.
An EGFR−/− mouse colonic epithelial (MCE) cell line was generated from the colonic epithelium of EGFR-null mice crossed to the Immortomouse (30).
YAMC and EGFR−/− MCE cells were maintained in RPMI 1640 medium supplemented with 5% FBS, 5 units/ml murine IFN-γ, 100 units/ml penicillin and streptomycin, 5 μg/ml insulin, 5 μg/ml transferrin, and 5 ng/ml selenous acid at 33 °C (permissive condition) with 5% CO2.
An ADAM17-deficient MCE (termed ADAM17−/− MCE) cell line was generated from the colonic epithelium of Adam17ΔZn/ΔZn-null mice crossed to the Immortomouse (31). ADAM17−/− MCE cells reconstituted to stably express HA-tagged WT or proteolytically inactive (E>A) ADAM17 mutant mouse ADAM17 were prepared using the pBM–ires-PURO retroviral vector, as described before (31). Empty vector was used as a control. Transduced cells were cultured in the same medium as that used for YAMC cells plus 5 μg/ml puromycin at 33 °C with 5% CO2.
Prior to treatment, T84 and YAMC, EGFR−/− MCE and ADAM17−/− MCE cells were maintained in serum-starved medium (F12 medium and DMEM for T84, RPMI 1640 medium (no IFN-γ) for YAMC, a EGFR−/− MCE and ADAM17−/− MCE cells) containing 0.5% FBS and 100 units/ml penicillin and streptomycin at 37 °C for 18 h.
Mice and Treatments
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committees at University of Vanderbilt and the University of Michigan. Villin-Cre and Adam17Fl/Fl mouse strains on a C57BL/6J background have been described previously (32, 33). Intestinal epithelial cell (IEC)-specific Adam17-deficient mice (Villin-Cre; Adam17Fl/Fl, referred to as IEC-Adam17KO) and genotype controls (Adam17Fl/Fl) were used in this study. IEC-Adam17KO mice show no overt intestinal phenotype and are a suitable model to study the loss of ADAM17 signaling within IECs without altering either intestinal function or baseline inflammatory cytokine responses.3
8–10-week old C57BL/6J WT, IEC-Adam17KO, and Adam17Fl/Fl mice were gavaged with pectin/zein beads containing p40 or pectin/zein control beads. Preparation of p40-containing pectin/zein beads was described in our previous publication (20). Blood was collected before and after p40 treatment. Colonic epithelial cells were isolated from mice at sacrifice.
Isolation of Colonic Epithelial Cells from Mice
Mouse colonic epithelial cells were isolated using a modified protocol (34). The colon was opened and incubated with 0.5 mm dithiothreitol and 3 mm EDTA at room temperature for 1.5 h without shaking. After gently removing the solution, PBS was added to the colon. Crypts released from the colon by shaking the tube were washed with PBS and then solubilized in cell lysis buffer, containing 50 mm Tris-HCl (pH 7.4), 120 mm NaCl, 1% Nonidet P-40, with protease and phosphatase 1 and 2 inhibitor cocktails (Sigma-Aldrich). The protein concentration was determined using a BCA protein assay kit (Pierce Thermo Scientific).
Transient Transfection of HB-EGF siRNA
YAMC cells were transiently transfected with either 20 nm nontargeting siRNA or 20 nm mouse HB-EGF siRNA (Santa Cruz Biotechnology) at 80% confluence using Lipofectamine 2000 (Invitrogen) for 6 h, according to the manufacturer's instructions. Cells were cultured for 24 h after transfection at 33 °C and then were cultured under the nonpermissive condition for 18 h before treatment.
Immunocytochemistry
For detecting p40 effects on H2O2-induced disruption of ZO-1 localization, cells were treated with H2O2 in the presence or absence of p40. Then, cells were fixed and permeabilized with 2% Triton X-100 in PBS for 5 min at room temperature followed by treatment with 5% bovine serum albumin in PBS containing 1% Triton X-100 for 1 h at room temperature. Slides were then incubated with a rabbit anti-mouse ZO-1 (Invitrogen) antibody overnight at 4 °C and a Cy3-labeled goat anti-rabbit IgG (Jackson ImmunoResearch) antibody at room temperature for 1 h. Slides were mounted using Vectashield® mounting medium and observed under fluorescence microscopy.
Real-time PCR Analysis
Total RNA was isolated from YAMC cells using an RNA isolation kit (Qiagen) and was treated with RNase-free DNase. Reverse transcription was performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). For Real-time PCRs, 25-μl reactions were set up by the addition of 1.25 μl of the HB-EGF primer mix (containing 5 μm reverse and forward primers) (Mm 00439306, Applied Biosystems), 5 μl of diluted cDNA template, and 12.5 μl of Taqman Gene Expression Master Mix. Real-time PCR was performed using the 7300 Real-time PCR System (Applied Biosystems). The data were analyzed using the Sequence Detection System V1.4.0 software. The relative abundance of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was used to normalize levels of the mRNAs of HB-EGF. All cDNA samples were analyzed in triplicate.
ELISA
Cell culture media and mouse sera were collected to determine EGFR ligand levels, including HB-EGF, TGFα, and amphiregulin, using the corresponding kits from DuoSet® ELISA Development System (R&D Systems), according to the manufacturer's instructions. The protein concentration of the cellular lysate was determined using a BCA protein assay kit (Pierce Thermo Scientific). The indicated ligand concentration in the cell culture medium was calculated as pg of ligand/mg of cellular protein. The indicated ligand concentration in serum was calculated as pg of ligand/ml of serum.
ADAM17 Activity Assay
Cultured human and mouse cell lines and isolated MCE cells were solubilized in cell lysis buffer containing 50 mm Tris-HCl (pH 7.4), 120 mm NaCl, and 1% Nonidet P-40 with protease and phosphatase 1 and 2 inhibitor mixtures. The protein concentration was determined using a BCA protein assay kit. ADAM17 activity was detected using a InnoZymeTM TACE Activity Kit (Calbiochem) according to the manufacturer's instructions using recombinant human ADAM17 as the standard. Although this assay was designed to detect human ADAM17 activity, our analysis of ADAM17−/− MCE cells transduced with WT mouse ADAM17, catalytically inactive ADAM17 E>A, or vector alone indicated that mouse ADAM17 could be detected. Importantly, p40 stimulated ADAM17 activity in ADAM17−/− MCE cells expressing WT mouse ADAM17 but not ADAM17 E>A mutant or vector alone (see Fig. 2C). Both human and mouse ADAM17 activities were calculated as ng of recombinant human ADAM17 equivalents/mg of cellular protein.
FIGURE 2.

p40 activates ADAM17 in intestinal epithelial cells. YAMC (A), T84 (B), and ADAM17−/− (C) MCE cells transduced with WT ADAM, E>A mutant ADAM17, or vector control were treated with p40 (10 ng/ml) for the indicated times. Cellular lysates were collected for analysis of ADAM17 proteolytic activity for peptide substrates and protein concentration. Data are presented as ng of active ADAM17 per mg of cellular protein. *, p < 0.05 compared with the control group for each cell line. Error bars, S.D. Data are quantified from three to five separate experiments.
Cellular Lysate Preparation and Western Blot Analysis
p40 was purified from LGG culture broth, as described in Ref. 14. p40 or recombinant murine HB-EGF (Pepro Tech) was used to treat cells in the presence or absence of the broad spectrum metalloproteinase inhibitors GM6001 (Millipore) and TAPI-1 (Enzo Life Sciences, Farmingdale, NY), or recombinant murine TNF (Pepro Tech) and cycloheximide (Sigma-Aldrich). For preparing total cellular proteins, cell monolayers were rinsed twice with cold PBS and then scraped into cell lysis buffer. The cellular suspensions were centrifuged (14,000 × g for 10 min) at 4 °C, and the supernatants were saved. The protein concentration was determined using a BCA protein assay kit. Cellular proteins isolated from cultured cells and colonic epithelial cells were mixed with Laemmli sample buffer and separated by SDS-polyacrylamide gel electrophoresis for Western blot analysis using antibodies against total EGFR (Millipore), phospho-EGFR (Tyr-1068) (Cell Signaling Technology), phospho-Akt (Ser-473) (Cell Signaling Technology), total Akt (Cell Signaling Technology), PARP (which identifies both full-length and cleaved PARP) (Cell Signaling Technology), Caspase-3 (which identifies both full-length and cleaved Caspase-3) (Cell Signaling Technology), ADAM17 (Cell Signaling Technology, 3976), and β-actin (Sigma-Aldrich).
Statistical Analysis
Statistical significance was determined by one-way analysis of variance followed by Newman-Keuls analysis using Prism 5.0 (GraphPad Software, San Diego, CA) for multiple comparisons and t test for paired samples. A p value < 0.05 was defined as statistically significant. Data are presented as mean ± S.D.
RESULTS
p40-stimulated ADAM17 Activation Mediates EGFR Transactivation in Intestinal Epithelial Cells
Our previous studies have demonstrated that p40 activates EGFR in intestinal epithelial cells, which is required for decreasing apoptosis and preserving epithelial barrier function in intestinal epithelial cells in mouse models of colitis (20). However, how EGFR is activated by p40 is unknown. Stimulation of EGFR ligand release serves as one of the mechanisms involved in transactivation of EGFR. Thus, this work was focused on determining whether p40 stimulated intestinal epithelial cells to release EGFR ligands. We first determined whether p40 stimulates release of any factors from YAMC and T84 cells to activate EGFR. p40-treated conditioned medium was prepared by collecting cell culture medium from YAMC and T84 cells treated with p40 for 60 and 120 min. Then the p40-treated conditioned medium was used to treat cells for 10 min. Because p40 was present in the conditioned medium, we treated YAMC and T84 cells with p40 for 10 min as a control. As reported before (14, 18), p40 treatment for 60 and 120 min, but not 10 min, activated EGFR and Akt. By contrast, EGFR and Akt were activated in YAMC and T84 cells treated with p40-conditioned medium for 10 min (Fig. 1A). These data suggest that p40-conditioned medium contains soluble factors required for activation of EGFR in YAMC and T84 cells.
FIGURE 1.

p40 stimulates ligand release from intestinal epithelial cells to activate EGFR. A, conditioned medium (CM) was collected from YAMC and T84 cells treated with p40 (10 ng/ml) for 60 and 120 min. YAMC and T84 cells were treated with p40 (10 ng/ml) for the indicated times or conditioned medium for 10 min. B, YAMC and T84 cells were treated with p40 (10 ng/ml) for 60 min or EGF (10 ng/ml for YAMC and 30 ng/ml for T84 cells) for 5 min in the presence or absence of 1-h pretreatment with metalloproteinase inhibitors, GM6001 (2.5 μm) and TAPI-1 (10 μm), or an antibody blocking human EGF receptor ligand binding (C225, 5 nm). Cellular lysates were collected for Western blot analysis of total EGFR and Akt levels and EGFR (Tyr-1068) and Akt (Ser-473) phosphorylation. β-Actin protein levels were used as loading controls. Data are representative of at least three separate experiments.
To test whether EGFR ligand release might mediate p40-induced signaling in intestinal epithelial cells, we used a monoclonal antibody, C225, which interacts with the ectodomain of human EGFR to block ligand binding (35). C225 inhibited p40 activation of EGFR and Akt in T84 cells (Fig. 1B), indicating that p40 stimulation of EGFR and Akt phosphorylation requires ligand binding to EGFR. Because the proteolytic processing of EGF family members is a regulated event and disintegrin-metalloproteinases play a critical role in the release of EGFR ligands (36, 37), we tested whether broad spectrum metalloproteinase inhibitors, GM6001 and TAPI-1, could suppress the p40 effect on EGFR activation. p40-stimulated EGFR and Akt activation in T84 and YAMC cells was blocked by treatment of cells with the combination of these two metalloproteinase inhibitors (Fig. 1B), indicating that metalloproteinase activity is required for p40-induced signaling. Importantly, EGF-stimulated Akt and EGFR activation was blocked by the EGFR ligand-binding antibody, but not by metalloproteinase inhibitors, confirming that metalloproteinase inhibition had not perturbed functional EGFR signaling (Fig. 1B). These data suggest that p40 requires metalloproteinase activity to release ligands for transactivation of EGFR in IECs.
ADAM17 plays an important role in the shedding of HB-EGF, TGFα, and amphiregulin for activation of EGFR (36, 37). Thus, we focused on determining whether ADAM17 mediated p40-regulated EGFR transactivation in intestinal epithelial cells. p40 stimulated ADAM17 activity in YAMC cells (Fig. 2A) and T84 cells (Fig. 2B) in a time-dependent manner. To further validate these findings, we examined the effects of p40 treatment on ADAM17 activity in ADAM17−/− MCE cells expressing either WT ADAM17, catalytically inactive ADAM17 E>A mutant, or vector alone. p40 stimulated ADAM17 activity only in ADAM17−/− MCE cells expressing WT ADAM17 (Fig. 2C). Next, we tested the requirement of ADAM17 for p40 effects on EGFR and Akt signaling using the ADAM17−/− MCE cells. Whereas activation of EGFR and Akt by p40 was lost in ADAM17−/− MCE cells transduced with control vector, overexpression of WT ADAM17 in ADAM17−/− MCE cells rescued p40 effects on EGFR and Akt activation (Fig. 3A). As a positive control, EGF activated EGFR and Akt in ADAM17−/− MCE transduced with control vector (Fig. 3A).
FIGURE 3.
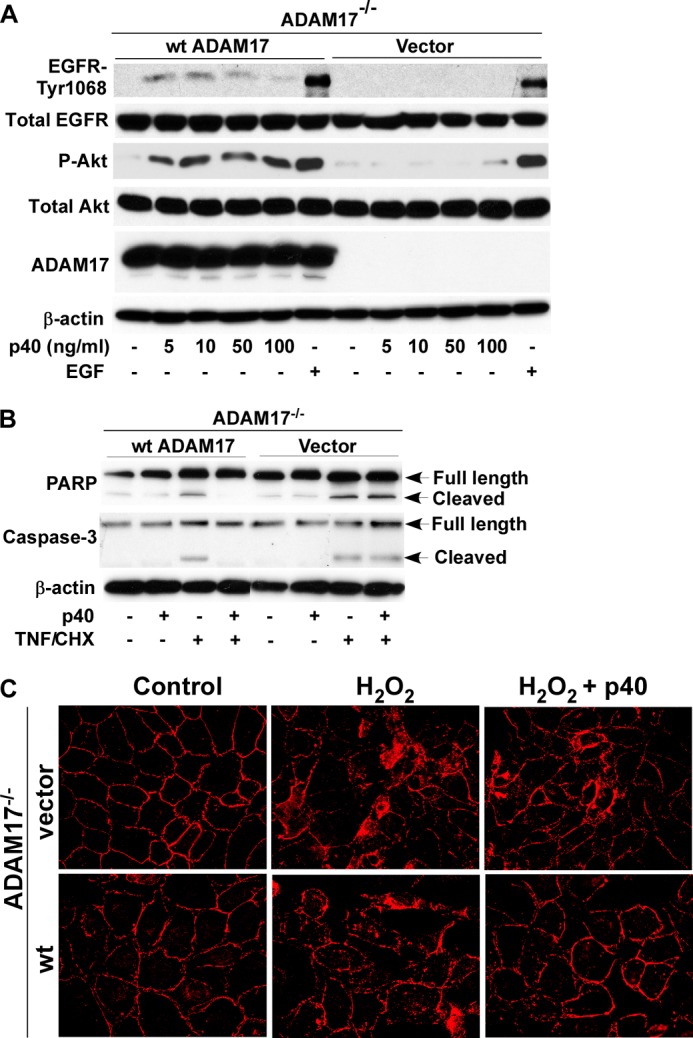
ADAM17 mediates p40 effects on intestinal epithelial cells. A, ADAM17−/− MCE cells transduced with WT ADAM17 or control vector were treated with p40 (10 ng/ml) for the indicated times or EGF (10 ng/ml) for 5 min. Cellular lysates were collected for Western blot analysis of total and phosphorylation levels of EGFR and Akt, and ADAM17 expression. β-Actin levels were used to control for protein loading. B, ADAM17−/− MCE cells transduced with WT ADAM17 or control vector were treated with TNF (100 ng/ml) and cycloheximide (CHX, 1 μg/ml) for 6 h in the presence or absence of 1-h p40 (10 ng/ml) pretreatment. Cellular lysates were collected for Western blot analysis of full-length and cleaved PARP and Caspase-3. C, cells were treated with H2O2 (40 μm) for 4 h in the presence or absence of 1-h pretreatment with p40 (10 ng/ml). ZO-1 distribution was detected by immunostaining using a ZO-1 antibody and a Cy3-conjugated secondary antibody and monitored by fluorescence microscopy. Images shown are representative of three separate experiments.
We have reported that p40 activation of EGFR is required for inhibition of apoptosis and preservation of epithelial barrier function in intestinal epithelial cells (20). Thus, we determined the role of ADAM17 in p40 regulation of these two cellular responses. We found that p40 prevented TNF and cycloheximide-induced apoptosis detected by Western blot analysis of cleaved PARP and Caspase-3 in ADAM17−/− MCE cells expressing WT ADAM17, but not with control vector (Fig. 3B). Tight junctions were detected by immunostaining for the tight junctional protein, ZO-1. H2O2-induced redistribution of this protein from apical tight junctional complexes to the cytoplasmic compartment of epithelial cells was prevented by p40 treatment in ADAM17−/− MCE cell with WT ADAM17, but not vector transfection (Fig. 3C). These data suggest that ADAM17 mediates p40 regulatory effects on intestinal epithelial cells.
HB-EGF Release by p40 Mediates Activation of EGFR in Intestinal Epithelial Cells
We next examined which ErbB ligand released by p40 from YAMC cells is responsible for transactivation of EGFR. Because we have found that YAMC cells express endogenous HB-EGF, TGFα, and amphiregulin, but not EGF,4 we focused on detecting these three ligands in the cell culture medium isolated from p40 treated YAMC cells by ELISA. p40 treatment increased HB-EGF levels in cell culture medium of YAMC cells (Fig. 4A). To gain additional insights into p40-mediated HB-EGF release, we examined the effects of p40 treatment on EGFR−/− MCE cells in which released HB-EGF will accumulate in the conditioned medium due to the lack of ligand binding to and consumption by EGFRs. We found that the basal level of HB-EGF and p40-stimulated HB-EGF release were higher in EGFR−/− MCE cells (Fig. 4A). The peak level of HB-EGF was found in the medium of 1-h p40-treated YAMC cells, but the HB-EGF level returned to base line by 4 h after p40 treatment, which is consistent with the finding that p40-stimulated EGFR activation occurs 1 h after p40 treatment. However, in p40-treated EGFR−/− MCE cells, maximal levels of HB-EGF in conditioned medium were observed 2 h after p40 treatment, which persisted over time, due presumably to the lack of ligand consumption. Importantly, p40 did not affect TGFα and amphiregulin shedding in the conditioned medium of EGFR−/− MCE cells, suggesting that the effects of p40 were specific for HB-EGF release (Fig. 4A).
FIGURE 4.

p40 stimulates release of HB-EGF, but not TGFα or amphiregulin, in intestinal epithelial cells. A,YAMC and EGFR−/− MCE cells were treated with p40 (10 ng/ml) for the indicated times, and cell culture supernatants were collected for ELISA analysis of HB-EGF, TGFα, and amphiregulin. Data are presented as pg of ligand in the culture medium per mg of cellular protein. B, mRNA was isolated from YAMC cells treated with p40 (10 ng/ml) for the indicated times. Levels of HB-EGF were quantified using real-time PCR, and its expression level in untreated cells was set as 100% for comparison with p40-treated cells. C, ADAM17−/− MCE cells transfected with WT ADAM17 or vector control were treated with p40 (10 ng/ml) for the indicated times. The HB-EGF level in the cell culture supernatant was determined as described in A. D, phosphorylation of EGFR and Akt was evaluated after p40 (10 ng/ml) or HB-EGF (0.25 and 2.5 ng/ml) treatment of YAMC cells for the indicated times. Cellular lysates were collected for Western blot analysis of total and phosphorylation levels of EGFR and Akt. β-Actin blotting was used as a control for protein loading. E, the relative densities of protein bands on Western blots shown in D were determined by comparing densities of EGFR-Tyr-1068 and phosphorylated Akt to total EGFR and Akt bands from the same sample, respectively. The relative density of bands from the untreated group was set as 1, and the relative densities of bands from treatment groups were compared with those in the untreated group to obtain the -fold changes. In A, C, and E, *, p < 0.05 compared with the untreated group of the same cell line. #, p < 0.05 compared with the same time point and treatment group of YAMC cells. Data in A, B, C, and E are quantified from at least three separate experiments. Error bars, S.D.
To test whether p40 up-regulates HB-EGF gene expression, real-time PCR analysis was performed to measure HB-EGF mRNA levels in YAMC cells treated with p40. p40 did not affect HB-EGF gene expression in YAMC cells (Fig. 4B). Thus, gene expression does not contribute to the shedding events observed. Importantly, we confirmed that p40 stimulated significant HB-EGF release in ADAM17−/− cells transduced with WT ADAM17, but not in vector controls (Fig. 4C). These data suggest that p40 up-regulates HB-EGF release from intestinal epithelial cells, which is mediated by ADAM17 activation.
We next treated YAMC cells with exogenous HB-EGF to compare EGFR and Akt activation by HB-EGF released in response to p40. YAMC cells treated with HB-EGF at 0.25 ng/ml, which is the same concentration of HB-EGF that was detected in 1-h p40-treated cell culture medium (Fig. 4A), produced levels of EGFR and Akt activation similar to that observed in p40-treated cells (Fig. 4D). However, EGFR activation by HB-EGF treatment occurred (15 min) earlier than that by p40 (30 min) (Fig. 4, D and E), indicating that EGFR and Akt activation was significantly delayed in p40-treated cells.
To determine the requirement of HB-EGF for p40's action, the siRNA method was used to knock down HB-EGF expression in YAMC cells. HB-EGF expression, detected as a protein band with molecular mass of about 20 kDa by Western blotting, was suppressed in cells transduced with HB-EGF siRNA, but not in cells treated with nontargeting siRNA (Fig. 5, A and B). p40 failed to activate EGFR in cells with knockdown of HB-EGF expression (Fig. 5, A and B). Next, p40 effects on apoptosis and preservation of intestinal integrity were examined (20). p40 inhibited TNF and cycloheximide-induced apoptosis, as shown by the reduction of cleaved PARP and Caspase-3 (Fig. , 5, C and D). In addition, the presence of p40 prevented H2O2-induced disruption of the tight junction complex protein, ZO-1 (Fig. 5E), in YAMC cells transduced with nontargeting siRNA but not when HB-EGF siRNA was used. These data indicate a requirement of HB-EGF for p40 effects on EGFR signaling and cellular responses in intestinal epithelial cells.
FIGURE 5.

p40 regulation of intestinal epithelial cell responses requires HB-EGF. A and C, YAMC cells transduced with HB-EGF or nontargeting siRNA were treated with p40 (10 ng/ml) for 60 min (A), or TNF (100 ng/ml) and cycloheximide (CHX, 1 μg/ml) (C) for 6 h in the presence or absence of 1-h p40 (10 ng/ml) pretreatment. Cellular lysates were collected for Western blot analysis of HB-EGF and total and phosphorylation levels of EGFR (A), and full-length and cleaved PARP and Caspase-3 levels (C). β-Actin blotting was used as the protein loading control. B, the -fold changes of relative densities of EGFR-Tyr-1068 bands shown in A were determined as described in Fig. 4E. D, the relative densities of cleaved PARP and cleaved Caspase-3 bands shown in C were determined by comparing densities of cleaved PARP and Caspase-3 with that of total PARP and Caspase-3, respectively. The relative density of bands from the untreated group with nontargeting siRNA transfection was set as 1, and the relative densities of bands from treatment groups were compared with those in the untreated group with nontargeting siRNA transfection to obtain the -fold changes. E, cells were treated with H2O2 (40 μm) for 4 h in the presence or absence of 1-h pretreatment of p40 (10 ng/ml). Cells were immunostained using an anti-ZO-1 antibody and a Cy3-conjugated secondary antibody to detect ZO-1 distribution. Then cells were mounted and observed under fluorescence microscopy. Images shown are representative of three separate experiments. In B and D, *, p < 0.05 compared with the untreated group with nontargeting RNA transfection. #, p < 0.05 compared with the TNF/cycloheximide-treated group with nontargeting RNA transfection. Data are quantified from three separate experiments.
p40 Stimulates HB-EGF Release and Activates ADAM17 in Intestinal Epithelial Cells in Mice
Our in vitro data indicate that p40 stimulates HB-EGF release, which is required for p40 activation of EGFR and regulation of intestinal epithelial cellular responses. To determine whether p40 exerts the same function in vivo, WT mice were gavaged with p40 containing pectin/zein beads, a hydrogel formulation that protects associated drugs from abundant intestinal enzymes (38, 39). Colonic epithelial cells were isolated from mice for detecting ADAM17 activity. p40 increased ADAM17 activity in colonic epithelial cells at 6 h after treatment, but this had returned to base line at 24 h after treatment (Fig. 6A). Although it was not possible to directly measure soluble levels of HB-EGF in the intestine after p40 treatment, we examined HB-EGF levels in serum. p40 treatment increased the HB-EGF level in sera, with maximal levels at 6 h, which is coincident with increased ADAM17 activity at this time point (Fig. 6B). Consistent with the specificity of p40 for HB-EGF shedding in vitro (Fig. 4A), p40 did not increase serum levels of TGFα (Fig. 6C) or amphiregulin (Fig. 6D). To determine whether ADAM17 mediates p40 regulation of HB-EGF release in vivo, we used mice with ADAM17 specifically deleted in the intestinal epithelial cells (IEC-Adam17KO). Their littermates, Adam17Fl/Fl mice, were used as controls. First, we confirmed that p40 activated EGFR in colonic epithelial cells of Adam17Fl/Fl but not in IEC-Adam17KO mice (Fig. 7, A and B). In addition, p40 stimulated ADAM17 activity (Fig. 7C) in the colonic epithelial cells and up-regulated the HB-EGF level in sera (Fig. 7D) of Adam17Fl/Fl mice but not IEC-Adam17KO mice. Thus, these data suggested that p40 activation of ADAM17 in the intestinal epithelial cell plays a role in release of HB-EGF in vivo.
FIGURE 6.
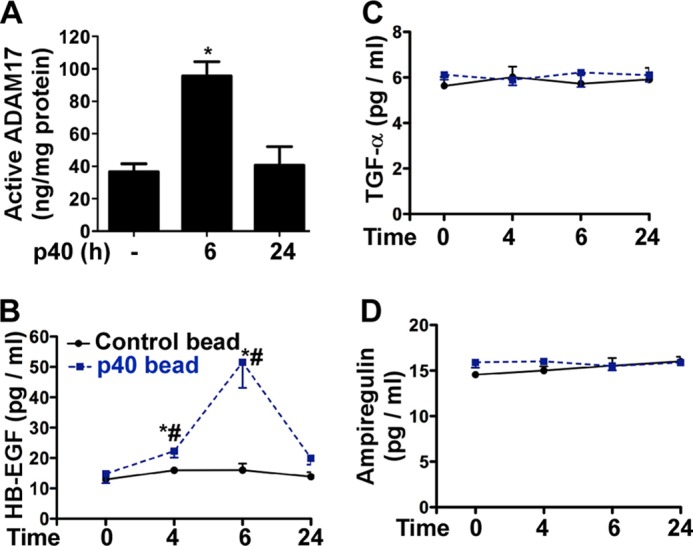
p40 stimulates the ADAM17 activity in the colonic epithelial cells and the HB-EGF level in serum in WT mice. C57BL/6J mice were gavaged with 10 μg of p40 in two pectin/zein beads (each bead contains 5 μg of p40) or two pectin/zein beads without p40 (control) beads. A, ADAM17 enzyme activity was evaluated from cell lysates of colonic epithelial cells collected from mice with no p40 treatment and 6 h and 24 h after p40-containing bead gavage. B–D, ELISAs of sera collected at different times after gavage were used to evaluate serum levels of HB-EGF (B), TGFα (C), and amphiregulin (D). In A, *, p < 0.05 compared with the untreated group (n = 5/group). In B, *, p < 0.05 compared with the control bead treatment group at the same corresponding time point. #, p < 0.05 compared with the level before p40 treatment in the same mouse, n = 5/group. Error bars, S.D.
FIGURE 7.

Intestinal epithelial expression of ADAM17 is required for p40 activity in mice. A, IEC-Adam17KO and Adam17Fl/Fl mice were gavaged with 10 μg of p40 in two pectin/zein beads or two pectin/zein beads without p40 (control) beads. Colonic epithelial cells were collected from mice without any treatment and from mice at 6 h after gavage. Cell lysates were prepared for Western blot analysis of total and phosphorylated levels of EGFR and ADAM17 expression. B, The -fold change of EGFR-Tyr-1068 relative density was determined by normalizing protein load to the total EGFR band of each sample. The average of relative densities of the control bead treated Adam17Fl/Fl mice was set at 1. C, the same cell lysates evaluated in A were also assayed with peptide substrates for ADAM17 proteolytic activity. D, sera were collected at the indicated times after gavage for ELISA analysis of HB-EGF. In B, *, p < 0.05 compared with the control bead-treated Adam17Fl/Fl mice. In C, *, p < 0.05 compared with the untreated and the control bead-treated Adam17Fl/Fl groups. In D, *, p < 0.05 compared with the control bead treatment group at the corresponding time point. #, p < 0.05 compared with the level in mice before p40 treatment. n = 5/group. Error bars, S.D.
DISCUSSION
The discovery of probiotic-derived proteins that regulate host homeostasis represents an area of recent progress in probiotic research. A LGG-derived-soluble protein, p40, has been shown to transactivate EGFR signaling to ameliorate cytokine-induced apoptosis and disruption of epithelial barrier in intestinal epithelial cells in vitro and in mice (20). Results from the present study indicate that p40 regulation of ADAM17 and its release of HB-EGF may serve as the mechanism for transactivation of EGFR in intestinal epithelial cells.
Our in vivo studies show that p40 increases HB-EGF levels in mouse sera. The source of HB-EGF may be intestinal epithelial cells, but it is also possible that p40 may regulate other cell types through direct or indirect effects. However, the evidence that p40 fails to up-regulate HB-EGF in sera in IEC-Adam17KO mice suggests that ADAM17 activity in intestinal epithelial cells plays a significant role in p40 stimulation of HB-EGF production.
We have previously reported that the full-length amino acid sequence of p40 shows no sequence homology to any EGFR ligands (14). Consistent with the idea that p40 cannot directly bind to and activate EGFR, we show that exogenous HB-EGF can stimulate rapid EGFR activation in cultured cells whereas p40-induced EGFR activation is markedly delayed. This delayed EGFR activation may reflect the time needed for p40 to stimulate ADAM17 activity, a process which may require trafficking of both ADAM17 and HB-EGF to the appropriate cellular compartment to achieve functional substrate processing. If p40 does increase ADAM17 proteolytic activity, it would be expected to enhance the shedding of other ADAM17 substrates such as TNF and other EGFR ligands including TGFα and amphiregulin. Interestingly, although we found that p40 can stimulate TNF release in YAMC cells (data not shown), we did not detect increased release of TGFα or amphiregulin by p40 treatment in YAMC or EGFR−/− MCE cells. The ability to detect p40-induced HB-EGF and TNF cleavage, but not TGFα or amphiregulin processing, could reflect lower expression levels of these ligands as seen in EGFR−/− MCE cells or alternatively differences in the consumption or fate of specific soluble EGFR ligands. It also remains possible that p40 can selectively enhance processing of specific ADAM17 substrates. In addition, because TNF can transactivate the EGFR in YAMC cells (40), we cannot rule out the possibility that released TNF can contribute to p40-induced EGFR activation. Further investigations are clearly needed to distinguish between these different possibilities.
The exact mechanism(s) by which p40 regulates the catalytic activity of ADAM17 is still unclear. Given the variety of stimuli that activate ADAM17 and the wide range of ADAM17 substrates, there must be tight control over its enzymatic activity. At least in the human leukemic cell line THP-1 and a SV40-transformed kidney cell line COS-7 from African green monkey, the majority of endogenous ADAM17 precursor is localized in the endoplasmic reticulum and Golgi apparatus, and little mature protein is observed at the plasma membrane (41).
However, it is still unresolved whether the majority of substrate processing by catalytically active ADAM17 occurs at the cell surface or mainly within an intracellular compartment. Several reports have shown that phosphorylation at T735 in the cytoplasmic tail of ADAM17 by either p38 or ERK/MAPK is important for trafficking and regulation of ADAM17 activity (42–44). However, we have reported that p40 does not activate ERK/MAPK and p38 in intestinal epithelial cells (14), suggesting that this is less likely in intestinal epithelial cells. Recent studies showed that iRHOM2 is critical for ADAM17 function and that iRHOM2 is required for the export and maturation of ADAM17 from the endoplasmic reticulum to the plasma membrane (27, 28). Importantly, the binding of iRHOM2 to ADAM17 also contributes to innate immunity and pathogen defense (27). In future studies, we will focus on investigations into whether p40 directly or indirectly regulates ADAM17 activity and whether these actions involve modulation of accessory binding partners of ADAM17 in intestinal epithelial cells. In addition, we observe that a high concentration of p40 (100 ng/ml) with an extended incubation time (4 h) results in residual HB-EGF release and EGFR activation in ADAM17−/− MCE cells. This result suggests that ADAM17 may not be the only enzyme mediating p40 stimulation of EGFR ligand release. It will be important therefore to determine whether p40 actions are specific for ADAM17 or can regulate the activity of other ADAMs.
Although ADAM17 is ubiquitously present in the human gastrointestinal tract, its expression and catalytic activity are increased in ulcerative colitis, and it regulates TNF levels in rodent models of colitis (45). In mice with trinitrobenzenesulfonic acid-induced colitis, ADAM17 activity was elevated, resulting in concomitant increases in soluble TNF. Treatment with a broad based hydroxamate inhibitor of metalloproteinases ameliorated trinitrobenzenesulfonic acid-induced colonic damage and inflammation (46). However, whereas ADAM17 and other metalloproteinases are targets of drug development for inflammatory conditions, ADAM17 deficiency in humans leads to increased intestinal inflammation (47), indicating that this metalloproteinase may also play important roles in gut homeostasis. Importantly, the increased susceptibility of hypomorphic ADAM17 mice to DSS-induced colitis has been directly associated with a loss of ErbB ligand processing and functional EGFR signaling (48, 49). Our results show that p40 is involved in activation of ADAM17 in intestinal epithelial cells in vitro and in mice, thus supporting the notion that p40 may play a role in protecting the gastrointestinal tract from acute injury-induced inflammation through ADAM17-mediated production of soluble EGFR ligands. Therefore, a better understanding of the function of ADAM17 in HB-EGF release by p40 may provide insights into its potential role in intestinal inflammation.
In summary, this is the first report to our knowledge showing transactivation of EGFR by a probiotic-derived protein through stimulation of EGFR ligand release in intestinal epithelial cells. Results from our previous studies show that p40 exerts protective effects in an experimental colitis mouse model through transactivation of the EGFR (20). We now demonstrate that p40 regulates overlapping signaling and biological cytoprotective pathways in human and mouse colon cells, suggesting possibly similar functions in humans and mice.
This work was supported, in whole or in part, by National Institutes of Health Grants R01DK081134 (to F. Y.), R01DK93697 (to P. J. D), R01HL067267 and P01HL018645 (to E. W. R.), and R01DK56008, R01DK54993, and R01DK066176 (to D. B. P.). This work was also supported by Tianjin Research Program of Application Foundation and Advanced Technology for Young Scientists of China Grant 13JCQNJC10600 (to H. C.) and core services performed through Vanderbilt University Medical Center Digestive Disease Research Center supported by National Institutes of Health Grant P30DK058404.
Y. Feng et al., unpublished data.
F. Yan, L. Liu, P. J. Dempsey, Y.-H. Tsai, E. W. Raines, C. L. Wilson, H. Cao, Z. Cao, L. Liu, and D. B. Polk, unpublished data.
- LGG
- Lactobacillus rhamnosus GG
- ADAM17
- a disintegrin and metalloproteinase domain-containing protein 17
- EGFR
- epidermal growth factor receptor
- HB-EGF
- heparin-binding epidermal growth factor
- IEC
- intestinal epithelial cell
- MCE
- mouse colonic epithelial
- PARP
- poly(ADP-ribose) polymerase
- YAMC
- young adult mouse colon.
REFERENCES
- 1. Gill S. R., Pop M., Deboy R. T., Eckburg P. B., Turnbaugh P. J., Samuel B. S., Gordon J. I., Relman D. A., Fraser-Liggett C. M., Nelson K. E. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Elson C. O., Cong Y. (2012) Host-microbiota interactions in inflammatory bowel disease. Gut Microbes 3, 332–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vanderpool C., Yan F., Polk D. B. (2008) Mechanisms of probiotic action: implications for therapeutic applications in inflammatory bowel diseases. Inflamm. Bowel Dis. 14, 1585–1596 [DOI] [PubMed] [Google Scholar]
- 4. Yan F., Polk D. B. (2010) Probiotics: progress toward novel therapies for intestinal diseases. Curr. Opin. Gastroenterol. 26, 95–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yan F., Polk D. B. (2011) Probiotics and immune health. Curr. Opin. Gastroenterol. 27, 496–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Land M. H., Rouster-Stevens K., Woods C. R., Cannon M. L., Cnota J., Shetty A. K. (2005) Lactobacillus sepsis associated with probiotic therapy. Pediatrics 115, 178–181 [DOI] [PubMed] [Google Scholar]
- 7. Vahabnezhad E., Mochon A. B., Wozniak L. J., Ziring D. A. (2013) Lactobacillus bacteremia associated with probiotic use in a pediatric patient with ulcerative colitis. J. Clin. Gastroenterol. 47, 437–439 [DOI] [PubMed] [Google Scholar]
- 8. Besselink M. G., van Santvoort H. C., Buskens E., Boermeester M. A., van Goor H., Timmerman H. M., Nieuwenhuijs V. B., Bollen T. L., van Ramshorst B., Witteman B. J., Rosman C., Ploeg R. J., Brink M. A., Schaapherder A. F., Dejong C. H., Wahab P. J., van Laarhoven C. J., van der Harst E., van Eijck C. H., Cuesta M. A., Akkermans L. M., Gooszen H. G. (2008) Probiotic prophylaxis in predicted severe acute pancreatitis: a randomised, double-blind, placebo-controlled trial. Lancet 371, 651–659 [DOI] [PubMed] [Google Scholar]
- 9. Gorbach S. L. (1996) The discovery of Lactobacillus GG. Nutrition Today 31, 2S–4S [Google Scholar]
- 10. Zocco M. A., dal Verme L. Z., Cremonini F., Piscaglia A. C., Nista E. C., Candelli M., Novi M., Rigante D., Cazzato I. A., Ojetti V., Armuzzi A., Gasbarrini G., Gasbarrini A. (2006) Efficacy of Lactobacillus GG in maintaining remission of ulcerative colitis. Aliment. Pharmacol. Ther. 23, 1567–1574 [DOI] [PubMed] [Google Scholar]
- 11. Basu S., Paul D. K., Ganguly S., Chatterjee M., Chandra P. K. (2009) Efficacy of high-dose Lactobacillus rhamnosus GG in controlling acute watery diarrhea in Indian children: a randomized controlled trial. J. Clin. Gastroenterol. 43, 208–213 [DOI] [PubMed] [Google Scholar]
- 12. Szajewska H., Wanke M., Patro B. (2011) Meta-analysis: the effects of Lactobacillus rhamnosus GG supplementation for the prevention of healthcare-associated diarrhoea in children. Aliment. Pharmacol. Ther. 34, 1079–1087 [DOI] [PubMed] [Google Scholar]
- 13. Doron S., Snydman D. R., Gorbach S. L. (2005) Lactobacillus GG: bacteriology and clinical applications. Gastroenterol. Clin. North Am. 34, 483–498 [DOI] [PubMed] [Google Scholar]
- 14. Yan F., Cao H., Cover T. L., Whitehead R., Washington M. K., Polk D. B. (2007) Soluble proteins produced by probiotic bacteria regulate intestinal epithelial cell survival and growth. Gastroenterology 132, 562–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bäuerl C., Pérez-Martínez G., Yan F., Polk D. B., Monedero V. (2010) Functional analysis of the p40 and p75 proteins from Lactobacillus casei BL23. J. Mol. Microbiol. Biotechnol. 19, 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Claes I. J., Schoofs G., Regulski K., Courtin P., Chapot-Chartier M. P., Rolain T., Hols P., von Ossowski I., Reunanen J., de Vos W. M., Palva A., Vanderleyden J., De Keersmaecker S. C., Lebeer S. (2012) Genetic and biochemical characterization of the cell wall hydrolase activity of the major secreted protein of Lactobacillus rhamnosus GG. PloS One 7, e31588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Regulski K., Courtin P., Meyrand M., Claes I. J., Lebeer S., Vanderleyden J., Hols P., Guillot A., Chapot-Chartier M. P. (2012) Analysis of the peptidoglycan hydrolase complement of Lactobacillus casei and characterization of the major γ-d-glutamyl-l-lysyl-endopeptidase. PloS One 7, e32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yan F., Polk D. B. (2002) Probiotic bacterium prevents cytokine-induced apoptosis in intestinal epithelial cells. J. Biol. Chem. 277, 50959–50965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seth A., Yan F., Polk D. B., Rao R. K. (2008) Probiotics ameliorate the hydrogen peroxide-induced epithelial barrier disruption by a PKC- and MAP kinase-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1060–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan F., Cao H., Cover T. L., Washington M. K., Shi Y., Liu L., Chaturvedi R., Peek R. M., Jr., Wilson K. T., Polk D. B. (2011) Colon-specific delivery of a probiotic-derived soluble protein ameliorates intestinal inflammation in mice through an EGFR-dependent mechanism. J. Clin. Invest. 121, 2242–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yarden Y. (2001) The EGFR family and its ligands in human cancer: signalling mechanisms and therapeutic opportunities. Eur. J. Cancer 37, S3–8 [DOI] [PubMed] [Google Scholar]
- 22. Yarden Y., Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137 [DOI] [PubMed] [Google Scholar]
- 23. Sinha A., Nightingale J., West K. P., Berlanga-Acosta J., Playford R. J. (2003) Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N. Engl. J. Med. 349, 350–357 [DOI] [PubMed] [Google Scholar]
- 24. Dubé P. E., Yan F., Punit S., Girish N., McElroy S. J., Washington M. K., Polk D. B. (2012) Epidermal growth factor receptor inhibits colitis-associated cancer in mice. J. Clin. Invest. 122, 2780–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arribas J., Merlos-Suárez A. (2003) Shedding of plasma membrane proteins. Curr. Top. Dev. Biol. 54, 125–144 [DOI] [PubMed] [Google Scholar]
- 26. Ohtsu H., Dempsey P. J., Eguchi S. (2006) ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am. J. Physiol. Cell Physiol. 291, C1–10 [DOI] [PubMed] [Google Scholar]
- 27. McIlwain D. R., Lang P. A., Maretzky T., Hamada K., Ohishi K., Maney S. K., Berger T., Murthy A., Duncan G., Xu H. C., Lang K. S., Häussinger D., Wakeham A., Itie-Youten A., Khokha R., Ohashi P. S., Blobel C. P., Mak T. W. (2012) iRhom2 regulation of TACE controls TNF-mediated protection against Listeria and responses to LPS. Science 335, 229–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adrain C., Zettl M., Christova Y., Taylor N., Freeman M. (2012) Tumor necrosis factor signaling requires iRhom2 to promote trafficking and activation of TACE. Science 335, 225–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Whitehead R. H., VanEeden P. E., Noble M. D., Ataliotis P., Jat P. S. (1993) Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 90, 587–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dise R. S., Frey M. R., Whitehead R. H., Polk D. B. (2008) Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G276–285 [DOI] [PubMed] [Google Scholar]
- 31. Garton K. J., Gough P. J., Philalay J., Wille P. T., Blobel C. P., Whitehead R. H., Dempsey P. J., Raines E. W. (2003) Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) Is mediated by tumor necrosis factor-α-converting enzyme (ADAM 17). J. Biol. Chem. 278, 37459–37464 [DOI] [PubMed] [Google Scholar]
- 32. el Marjou F., Janssen K. P., Chang B. H., Li M., Hindie V., Chan L., Louvard D., Chambon P., Metzger D., Robine S. (2004) Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186–193 [DOI] [PubMed] [Google Scholar]
- 33. Wilson C. L., Gough P. J., Chang C. A., Chan C. K., Frey J. M., Liu Y., Braun K. R., Chin M. T., Wight T. N., Raines E. W. (2013) Endothelial deletion of ADAM17 in mice results in defective remodeling of the semilunar valves and cardiac dysfunction in adults. Mech. Dev. 130, 272–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Whitehead R. H., Demmler K., Rockman S. P., Watson N. K. (1999) Clonogenic growth of epithelial cells from normal colonic mucosa from both mice and humans. Gastroenterology 117, 858–865 [DOI] [PubMed] [Google Scholar]
- 35. Fan S., el-Deiry W. S., Bae I., Freeman J., Jondle D., Bhatia K., Fornace A. J., Jr., Magrath I., Kohn K. W., O'Connor P. M. (1994) p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA damaging agents. Cancer Res. 54, 5824–5830 [PubMed] [Google Scholar]
- 36. Blobel C. P. (2005) ADAMs: key components in EGFR signalling and development. Nat. Rev. Mol. Cell Biol. 6, 32–43 [DOI] [PubMed] [Google Scholar]
- 37. Blobel C. P., Carpenter G., Freeman M. (2009) The role of protease activity in ErbB biology. Exp. Cell Res. 315, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu L., Fishman M. L., Hicks K. B., Kende M., Ruthel G. (2006) Pectin/zein beads for potential colon-specific drug delivery: synthesis and in vitro evaluation. Drug Deliv. 13, 417–423 [DOI] [PubMed] [Google Scholar]
- 39. Liu L., Fishman M. L., Kost J., Hicks K. B. (2003) Pectin-based systems for colon-specific drug delivery via oral route. Biomaterials 24, 3333–3343 [DOI] [PubMed] [Google Scholar]
- 40. Yamaoka T., Yan F., Cao H., Hobbs S. S., Dise R. S., Tong W., Polk D. B. (2008) Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc. Natl. Acad. Sci. U.S.A. 105, 11772–11777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schlöndorff J., Becherer J. D., Blobel C. P. (2000) Intracellular maturation and localization of the tumour necrosis factor-α convertase (TACE). Biochem. J. 347, 131–138 [PMC free article] [PubMed] [Google Scholar]
- 42. Díaz-Rodríguez E., Montero J. C., Esparís-Ogando A., Yuste L., Pandiella A. (2002) Extracellular signal-regulated kinase phosphorylates tumor necrosis factor α-converting enzyme at threonine 735: a potential role in regulated shedding. Mol. Biol. Cell 13, 2031–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soond S. M., Everson B., Riches D. W., Murphy G. (2005) ERK-mediated phosphorylation of Thr-735 in TNFα-converting enzyme and its potential role in TACE protein trafficking. J. Cell Sci. 118, 2371–2380 [DOI] [PubMed] [Google Scholar]
- 44. Xu P., Derynck R. (2010) Direct activation of TACE-mediated ectodomain shedding by p38 MAP kinase regulates EGF receptor-dependent cell proliferation. Mol. Cell 37, 551–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Brynskov J., Foegh P., Pedersen G., Ellervik C., Kirkegaard T., Bingham A., Saermark T. (2002) Tumour necrosis factor α-converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease. Gut 51, 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Colón A. L., Menchén L. A., Hurtado O., De Cristóbal J., Lizasoain I., Leza J. C., Lorenzo P., Moro M. A. (2001) Implication of TNF-α convertase (TACE/ADAM17) in inducible nitric oxide synthase expression and inflammation in an experimental model of colitis. Cytokine 16, 220–226 [DOI] [PubMed] [Google Scholar]
- 47. Blaydon D. C., Biancheri P., Di W. L., Plagnol V., Cabral R. M., Brooke M. A., van Heel D. A., Ruschendorf F., Toynbee M., Walne A., O'Toole E. A., Martin J. E., Lindley K., Vulliamy T., Abrams D. J., MacDonald T. T., Harper J. I., Kelsell D. P. (2011) Inflammatory skin and bowel disease linked to ADAM17 deletion. N. Engl. J. Med. 365, 1502–1508 [DOI] [PubMed] [Google Scholar]
- 48. Brandl K., Sun L., Neppl C., Siggs O. M., Le Gall S. M., Tomisato W., Li X., Du X., Maennel D. N., Blobel C. P., Beutler B. (2010) MyD88 signaling in nonhematopoietic cells protects mice against induced colitis by regulating specific EGF receptor ligands. Proc. Natl. Acad. Sci. U.S.A. 107, 19967–19972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chalaris A., Adam N., Sina C., Rosenstiel P., Lehmann-Koch J., Schirmacher P., Hartmann D., Cichy J., Gavrilova O., Schreiber S., Jostock T., Matthews V., Häsler R., Becker C., Neurath M. F., Reiss K., Saftig P., Scheller J., Rose-John S. (2010) Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J. Exp. Med. 207, 1617–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]