

Abstract
The epithelial sodium channel (ENaC) plays an important role in homeostasis of blood pressure and of the airway surface liquid, and excess function of ENaC results in refractory hypertension (in Liddle's syndrome) and impaired mucociliary clearance (in cystic fibrosis). The regulation of ENaC by molecular chaperones, such as the 70-kDa heat shock protein Hsc70, is not completely understood. Our previously published data suggest that Hsc70 negatively affects ENaC activity and surface expression in Xenopus oocytes; here we investigate the mechanism by which Hsc70 acts on ENaC in epithelial cells. In Madin-Darby canine kidney cells stably expressing epitope-tagged αβγ-ENaC and with tetracycline-inducible overexpression of Hsc70, treatment with 5 μg/ml doxycycline increased total Hsc70 expression 20%. This increase in Hsc70 expression led to a decrease in ENaC activity and surface expression that corresponded to an increased rate of functional ENaC retrieval from the cell surface. In addition, Hsc70 overexpression decreased the association of newly synthesized ENaC subunits. These data support the hypothesis that Hsc70 inhibits ENaC functional expression at the apical surface of epithelia by regulating ENaC biogenesis and ENaC trafficking at the cell surface.
Keywords: ENaC, chaperone, heat shock protein, trafficking, epithelia
the critical role of the epithelial sodium channel (ENaC) in maintaining salt and water homeostasis in a number of organ systems is evidenced by the severe conditions caused by ENaC dysregulation (18, 51, 67). In the absence of ENaC in pseudohypoaldosteronism type 1, the kidney wastes salt, which leads to hypotension (20, 48, 52). In contrast, ENaC functional overexpression in the distal nephron results in salt-sensitive hypertension, such as in Liddle's syndrome (10, 27, 47, 50, 52, 55).
ENaC also plays an important role in the airway epithelia. In patients with cystic fibrosis (CF), ENaC activity appears relatively increased, which is hypothesized to cause airway surface liquid volume depletion, decreased mucociliary clearance, and increased bacterial colonization of the airway (39). It is therefore critical to understand the mechanisms underlying ENaC regulation to better understand disease pathophysiology in these organs.
ENaC is likely a heterotrimer of three homologous subunits, α, β, and γ. This hypothesized structure is based on the structure of a related ENaC/degenerin family member, the acid-sensing ion channel (30), as well as recent atomic force microscopy data (62). Each subunit of ENaC contains NH2- and COOH-terminal intracellular domains, as well as a large extracellular domain with many glycosylation sites (8, 9). As with many secreted and membrane proteins, ENaC subunits are most likely assembled in the endoplasmic reticulum (ER). Either the initial assembly of ENaC subunits or their exit from the ER appears to be inefficient in model systems, and a significant fraction of the newly synthesized channel is targeted for degradation by an ER-associated degradation (ERAD) pathway (35, 60, 64). The COOH terminus of α-ENaC appears to control ENaC's exit from the ER and contains a consensus sequence for interaction with coat complex II (COP II) machinery (44). In fact, our group's recent data present evidence that ENaC does, in fact, interact with COP II and that promotion of this interaction by overexpression of Hsp70, the stress-induced 70-kDa heat shock protein, is associated with increased ENaC activity (11).
As it transits the Golgi, ENaC can undergo modification of its N-linked glycans, as well as proteolytic cleavage of the α- and γ-subunit extracellular domains by furin, a Golgi-resident protein convertase (24). This proteolysis is associated with an increase in the open probability (Po) of ENaC once it reaches the apical epithelial surface. ENaC can also, by an unknown mechanism, reach the cell surface without undergoing glycosyl and proteolytic processing in the Golgi, arriving at the membrane in an uncleaved, low-Po form (25).
Regulation of ENaC at the cell surface can occur by modification of the total number of channels present (N) or by cleavage of the uncleaved channels that were delivered to the cell surface, thus modifying ENaC Po. Conversion of uncleaved, low-Po ENaC to cleaved, higher-Po ENaC at the cell surface can result from the action of prostasin or other endogenous channel-activating proteases or from the action of exogenous proteases such as trypsin and elastase (1, 4, 6, 7, 16, 24, 31). The number of active ENaCs at the cell surface can also be regulated by the rate at which channels are retrieved, a process that is regulated by the E-3 ubiquitin ligase Nedd4-2 and disrupted in Liddle's syndrome (33, 34, 36, 59).
Our group's work previously demonstrated that sodium 4-phenylbutyrate (4PBA) corrects the aberrant intracellular trafficking of the most common disease-causing mutant of the CFTR, F508del. Our data (53, 54, 63) and the data of others (13, 66) have implicated 4PBA's differential regulation of the 70-kDa heat shock proteins Hsc70 and Hsp70 as a potential mechanism by which such correction of F508del trafficking may occur. As the work of others also suggested that 4PBA may similarly promote ENaC trafficking and activity in epithelia (28, 49), we became interested in testing whether the 4PBA-induced small changes in the abundance of Hsc70 [∼30–40% decrease (54, 63)] and Hsp70 (transient ∼50% increase) might influence ENaC trafficking and activity.
We initially demonstrated that Hsc70, the ubiquitously and constitutively expressed 70-kDa heat shock cognate protein, inhibits surface expression and activity of ENaC when overexpressed in Xenopus oocytes (19); this effect of Hsc70 was opposite and antagonistic to that of Hsp70, even though Hsc70 and Hsp70 are highly homologous (19). On the basis of these oocyte data (19) and our recently published data detailing the regulation of ENaC trafficking by Hsp70 in epithelia (11), using Madin-Darby canine kidney (MDCK) cells as a model system, we investigated the mechanisms by which ENaC activity and surface expression are regulated by Hsc70 in mammalian epithelia. In contrast to the effects of nearly twofold increased expression of Hsp70 to increase ENaC activity (11) and consistent with our oocyte data (19), we found that Hsc70 acts to decrease ENaC activity and surface expression in two ways: 1) Hsc70 promotes the internalization of active ENaC from the apical cell membrane and decreases its association with recycling endosomes, and 2) Hsc70 impairs association of newly synthesized ENaC subunits, therefore likely decreasing the amount of ENaC that reaches the cell surface. These data support the hypothesis that Hsc70 regulates ENaC trafficking by actions at multiple locations in the cell.
MATERIALS AND METHODS
The primary aim of this study is to determine the role of the molecular chaperone protein Hsc70 in ENaC trafficking and activity. We employ a cell-based system of inducible Hsc70 overexpression to examine changes in ENaC whole cell expression, cell surface expression, and activity.
Cell culture.
Type 1 MDCK epithelial cells that stably express COOH-terminal epitope-tagged ENaC subunits (α-HA, β-V5, and γ-Myc; gift of Dr. Thomas Kleyman, University of Pittsburgh) were used as a representative model system. Similar epitope-tagged ENaC subunits have been shown to traffic and function in the same manner as the native subunits in model systems (21, 26). These cells were selected to have doxycycline (Dox)-inducible expression of Hsc70 or ATPase-deficient Hsc70 (K71M), which are also epitope-tagged (COOH-terminal Myc/His).
Cells were cultured in 50:50 Ham's F-12 (Cellgro)-DMEM (GIBCO) containing 10% fetal bovine serum (Gemini) and 1% penicillin-streptomycin (Pen/Strep, Invitrogen). Cells were maintained under antibiotic selective pressure by addition of puromycin (Sigma), G418 sulfate (Cellgro), blasticidin S HCl (Invitrogen), hygromycin B (Roche), and Zeocin (Invitrogen) to the medium.
Cells were grown in polarized monolayers on Transwell plates (Costar, Corning Life Sciences) for surface expression assays or on Snapwell plates (Costar) for ion transport assays. Once cells had achieved resistance >500 Ω·cm2, they were treated with 1 μg/ml dexamethasone (Dex; Sigma-Aldrich) for 48 h prior to experiment. Unless otherwise indicated, cells were treated with Dox (Sigma-Aldrich) for the final 24 h of the Dex treatment.
Antibodies.
Primary antibodies were as follows: rabbit anti-Hsc70 (StressGen), mouse anti-Hsp70 (StressMarq), and rat anti-HA (Roche) for α-ENaC detection; mouse anti-V5 (Invitrogen) and rabbit anti-V5 (Sigma) for β-ENaC detection; rabbit anti-γ-ENaC (Abcam); mouse anti-Rab7 (Abcam); rabbit anti-Rab11 (Sigma); and mouse anti-GAPDH (Calbiochem).
Immunoblot.
Cells were lysed for 30 min in RIPA buffer (150 mM NaCl, 50 mM Tris·HCl, pH 8, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) containing protease inhibitor cocktail (Sigma-Aldrich), which was used according to the manufacturer's instructions. Lysates were collected, passed through a 21-gauge needle, and centrifuged (14,000 g for 15 min at 4°C) to remove cellular debris.
Protein content was determined using DC Protein Assay reagents (Bio-Rad) and BSA as a standard. Equal amounts of protein (25 μg, unless otherwise indicated) were resolved using SDS-PAGE and transferred to nitrocellulose using a semidry technique (Bio-Rad). Nonspecific protein binding was diminished by incubation of the membrane in 5% BSA or 5% nonfat milk in Tris-buffered saline (10 mM Tris·HCl, pH 8, and 150 mM NaCl) with 0.05% Tween 20. Primary antibodies and horseradish peroxidase-conjugated secondary antibodies (from Millipore or Amersham) were applied in Tris-buffered saline with 0.05% Tween 20 with 1% nonfat milk or 1% BSA. Immunoreactivity was detected by chemiluminescence (SuperSignal, ThermoFisher Scientific) and fluorography. Densitometry was performed using an Alpha Imager 2200 system (Alpha Innotech), as described below.
Coimmunoprecipitation.
Cell lysates were prepared as described above in RIPA buffer lacking SDS. Sepharose beads with conjugated protein A (for rabbit primary antibodies; Invitrogen) or protein G (for rat and mouse primary antibodies; Invitrogen) were washed with PBS and combined with primary antibody for 1 h at room temperature. Beads were washed again with PBS and incubated with 250 μg of cell lysates overnight at 4°C. Beads were then sequentially washed in RIPA buffer lacking SDS and PBS, and precipitated proteins were released from the beads by heating in SDS-PAGE sample buffer (125 mM Tris, pH 6.8, 4% SDS, 10% glycerol, 0.006% bromophenol blue, and 1.8% 2-mercaptoethanol). The released proteins were resolved by SDS-PAGE, and specific associated proteins were detected by immunoblot.
Short-circuit current measurement.
Cells were grown on Snapwell plates, and incubations prior to experimentation were initiated after transepithelial resistance was ≥500 Ω·cm2. After the Dex and Dox incubations described above (48 h total), cells were mounted in a vertical Ussing chamber setup. Bath solution (115 mM NaCl, 25 mM NaHCO3, 2.4 mM KH2PO4, 1.2 mM K2HPO4, 1.2 mM MgCl2, 1.2 mM CaCl2, and 10 mM glucose, pH 7.4) was maintained at 37°C. The short-circuit current (Isc) was determined under continuously voltage-clamped conditions using Acquire & Analyze data acquisition software (Physiologic Instruments), as we previously described (11); transepithelial resistance during these assays was 1,000–3,000 Ω·cm2. Amiloride, cycloheximide, and trypsin (all from Sigma-Aldrich) were dissolved in bath solution. Apical application of 10 μM amiloride and 10 μg/ml trypsin (final concentration) was used where indicated. Apical and basolateral application of 100 μg/ml cycloheximide (final concentration) was used in ENaC retrieval assays.
Cell surface expression assay.
Determination of ENaC surface expression by surface biotinylation was performed as previously described (11). Briefly, cells were grown on Transwell plates until transepithelial resistance was ≥500 Ω·cm2 and then incubated as described above. After these incubations (48 h total), cells and buffers were placed on ice for 20 min and washed with cold PBS containing 2.5 mM (final concentration) excess Ca2+ and Mg2+, pH 7.4. The apical surface of the cells was then treated with 1 mg/ml Sulfo-NHS-SS-biotin (200 mg/ml stock in DMSO; ThermoScientific) diluted in biotinylation buffer (10 mM H3BO4, 137 mM NaCl, and 1 mM CaCl2, pH 8.0) for 25 min on ice. This step was repeated one time for an additional 25 min. Cells were then washed three times with quenching buffer (192 mM glycine and 25 mM Tris·HCl, pH 8.3) and incubated on ice in quenching buffer for 20 min. Cells were washed in PBS and lysed in RIPA buffer as described above. Biotinylated proteins were precipitated using NeutrAvidin beads (Invitrogen) and analyzed by SDS-PAGE as described above.
Pulse-chase assay.
Cells were grown in six-well plates (Corning), and pulse-chase assays were performed using modifications of a published protocol (44). After Dex and Dox treatments as described above, cells were starved in DMEM lacking Met and Cys (GIBCO) for 1 h. After starvation, medium was replaced with 0.5 ml of the same medium containing 50–100 μCi of [35S]Met/Cys for 20 min. Cells were washed in PBS containing excess Met and Cys (2.5 mM each) and chased for indicated times in the 50:50 Ham's F-12-DMEM (see Cell culture) with 10% fetal bovine serum, also containing 2.5 mM Met and Cys. Cells were washed with PBS and lysed in RIPA without SDS as described above. Proteins were recovered by immunoprecipitation and resolved by SDS-PAGE as described above.
Where indicated, two sequential immunoprecipitation steps were conducted. After the initial immunoprecipitation, recovered proteins were released from beads by heating for 30 min at 65°C in sample buffer. After heating, 10% of the sample was removed for analysis, while the remaining fraction was diluted with RIPA buffer lacking SDS to reduce the concentration of SDS to <0.2%. This sample was combined with beads conjugated to a second primary antibody and incubated overnight at 4°C. After the beads were washed, the samples were released from the beads by heating for 3.5 min at 95°C and resolved by SDS-PAGE.
To reveal the precipitated and resolved proteins, gels were fixed in 30% methanol-10% acetic acid for 20 min and dried using a gel dryer (model FB GD45, Fisher). Dried gels were exposed to phosphor screens and quantified using a Typhoon PhosphorImager (GE), also as described below.
Immunofluorescence.
MDCK cells expressing αβγ-ENaC and Dox-inducible Hsc70 were grown on glass chamber slides (Falcon). Cells were treated with Dex (48 h) in the presence and absence of Dox (24 h). The cells were fixed with 50% methanol-50% acetone for 5 min at −20°C, and nonspecific binding was blocked with a solution of 10% goat serum (GIBCO-BRL)-1% BSA in PBS (1 h at room temperature). α-ENaC was detected using rabbit anti-HA (Abcam; 1:200 dilution) in PBS containing 1% BSA and revealed with anti-rabbit Alexa 488 (Invitrogen), which was used according to the manufacturer's directions. Rab7 and Rab11 were detected with mouse anti-Rab7 (Abcam) and mouse anti-Rab11 (Abcam), respectively, in PBS-1% BSA and revealed with Alexa 594-conjugated antibodies (Invitrogen). The slides were also counterstained with 4,6-diaminido-2-phenylindole nuclear stain (final concentration 300 nM in PBS) after the last wash.
Fluorescence was viewed on an Olympus IX81 inverted microscope with appropriate filters. Images were captured using Slidebook software (Intelligent Imaging Innovations, Denver, CO). Digital images were elaborated using Adobe Photoshop (version CS2).
Densitometric analyses.
As we previously described (11), images of immunoblots were digitized using an AlphaImager 2200 digital analysis system (AlphaInnotech, San Leandro, CA). These images were analyzed using AlphaImager analysis software (version 5.5, AlphaInnotech) with two-dimensional integration of the selected band. For pulse-chase analyses, densitometric signal was determined using ImageQuant TL software on a Typhoon PhosphorImager. For comparison within an individual experiment, the density of untreated control was arbitrarily set to 1.0, with the remaining densities expressed relative to this reference density.
Statistical analyses.
As we previously described for similar types of experiments (11), statistical significance between two groups was determined by a two-tailed Student's t-test, as all data were normally distributed. One-way ANOVA was utilized for multiple comparisons. Nonlinear regressions for determination of apparent first-order rate constants of decay in Fig. 5 were performed using the Regression Wizard function of SigmaPlot (version 8.0, Aspire, Ashburn, VA). Statistical analysis was performed using SigmaStat software (version 2.03). P ≤ 0.05 was considered significant.
Fig. 5.

Influence of Hsc70 on rate of ENaC retrieval from the apical cell surface. MDCK cells were treated without or with Dox for 24 h, and a baseline Isc was established in Ussing chambers. Cycloheximide (100 μg/ml) was added, and amiloride-sensitive Isc was determined at the indicated times. Values are means ± SE for n repeats. Pseudo-1st-order rate constants (kapp) for decay of amiloride-sensitive Isc were determined by fitting these data to the following equation: (It/I0) = e−kt, where (It/I0) is the fraction of ENaC current remaining at time t and k (kapp) is the apparent 1st-order rate constant, by a nonlinear regression using the Regression Wizard function of Sigma Plot (version 8.0). This analysis yields kapp ± SE, which were then compared by t-test as a function of ± Dox. A: cells expressing Hsc70. B: cells expressing ATPase-deficient Hsc70.
RESULTS
Increased Hsc70 expression in MDCK epithelial cells decreases ENaC activity.
MDCK epithelial cells were used as a representative model for all experiments. These cells are commonly used as an epithelial model to study ENaC trafficking and activity (21, 37, 43), because they form polarized monolayers when grown on permeable supports. This allows measurement of ion transport in Ussing chambers, as well as correlation of these ion transport measurements with direct biochemical assessment of surface expression. In addition, these cells do not endogenously express high levels of ENaC under normal conditions (29, 43) and were selected to stably express epitope-tagged αβγ-ENaC (α-HA, β-V5, and γ-Myc). Similar COOH-terminal epitope-tagged ENaC subunits have previously been shown to traffic and function in a manner consistent with the native subunits in model systems (21, 26) while allowing increased sensitivity for detection of ENaC by antibody-based techniques. We further selected MDCK cell clones that stably express αβγ-ENaC and had a Dox-inducible promoter driving the expression of Hsc70 or an ATPase-deficient form of Hsc70 (K71M), which were also epitope-tagged (COOH-terminal Myc/His).
On the basis of our previous data obtained from Xenopus oocytes (19), we hypothesized that Hsc70 overexpression would negatively impact ENaC function in mammalian cells. To determine whether Hsc70 affects the activity of ENaC, using a range of Dox concentrations, we examined the amiloride-sensitive (ENaC-specific) Isc in Ussing chamber assays. As shown in Fig. 1A, increased Hsc70 expression induced by 5 μg/ml Dox led to a significant decrease in amiloride-sensitive Isc, consistent with Hsc70 decreasing the ENaC-mediated current in these cells. In contrast, when expression of an ATPase-deficient Hsc70 construct was induced with 5 μg/ml Dox, amiloride-sensitive Isc was similar to that of cells not treated with Dox (Fig. 1B). These control data suggest that Hsc70's negative impact on ENaC activity is not merely an artifact of our overexpression system.
Fig. 1.

Hsc70 decreases epithelial sodium channel (ENaC) functional expression in Madin-Darby canine kidney (MDCK) cells. MDCK cells were grown as epithelial monolayers on semipermeable supports and incubated with doxycycline (Dox, 0–5 μg/ml) for 24 h, and amiloride-sensitive short-circuit current (Isc) was determined in Ussing chambers. Hsc70-expressing (A) or ATPase-deficient Hsc70-expressing (B) cells were analyzed, and amiloride-sensitive Isc data are expressed relative to samples incubated without Dox. Values are means ± SE for number of replicates shown above data points. *P < 0.05 vs. 0 Dox (by ANOVA).
Interestingly, ATPase-deficient Hsc70 expression induced by 1 μg/ml Dox actually caused a small, but statistically significant, increase in amiloride-sensitive Isc in Ussing chambers (Fig. 1B). As Hsc70 binding to cellular clients depends on its ATPase activity, we hypothesized that removal of this activity would inactivate the chaperone. Instead, it seems that the ATPase-deficient mutant may oppose the activity of Hsc70. Similar effects have been shown for ATPase-deficient constructs of the Hsp70-homologous molecular chaperone BiP (22, 23), as well as for Hsp70 and Hsc70 (11, 45). To investigate the mechanistic underpinnings of these effects, we chose to interrogate the changes in ENaC expression and activity using the concentrations of Dox where we saw the greatest differences in activity, namely, 5 μg/ml for Hsc70 and 1 μg/ml for ATPase-deficient Hsc70.
Increased Hsc70 expression in MDCK epithelial cells.
We next characterized the increased expression of Hsc70 in these cell lines. The epitope tag on the induced Hsc70 or ATPase-deficient Hsc70 protein reduces its electrophoretic mobility, which allows quantification of the endogenous (lower band) and overexpressed (upper band) proteins independently by immunoblot analysis. In Fig. 2A, cells were treated with Dox for increasing amounts of time, up to 24 h (the time of Dox incubation used in Fig. 1) to induce increased Hsc70 expression. As expected, Hsc70 expression increased over this time period to its highest point at 24 h, while the expression of endogenous Hsc70 (lower band) did not significantly change. All subsequent experiments were therefore performed after 24 h of incubation with Dox.
Fig. 2.

Increased expression of Hsc70 in MDCK cell lysates. A: MDCK cells were treated with 5 μg/ml Dox for 0–24 h, and Hsc70 was detected in cell lysates by immunoblot (IB). Lower and upper bands represent endogenous Hsc70 protein and Myc/His-tagged construct of Hsc70 protein, respectively. B and C: immunoblot determination of Hsc70 and ATPase-deficient Hsc70 expression after 24 h of incubation with the indicated concentration of Dox. This immunoreactivity was compared with that of purified Hsc70 protein to quantify Hsc70 expression. The 0 μg/ml and 5 μg/ml and protein standard lanes in B are from noncontiguous portions of the same immunoblot. Densitometric analysis of 4 independent experiments quantifying endogenous (light bars) and overexpressed (dark bars) chaperone are shown. Error bars, SE.
To quantify the increase in Hsc70 expression, as well as total cellular Hsc70 expression, we treated cells with or without Dox and compared the Hsc70 immunoreactivity with that of commercially available, purified Hsc70 protein (Fig. 2B, representative immunoblot). This purified Hsc70 protein was also His-tagged on its COOH terminus, so its electrophoretic mobility is also slightly less than that of the native Hsc70 and is more similar to the Dox-induced, Myc/His-tagged Hsc70 in our cell lysates. Densitometric quantification of these experiments is summarized in Fig. 2B and suggests that induced Hsc70-Myc/His expression in 25 μg of total cell lysate is ∼0.05% of total lysate protein. This is lower than the endogenously expressed Hsc70, which is in the range of 0.15–0.20% of total cellular protein. While this represents only 20% increased expression of Hsc70 when cells are treated with 5 μg/ml Dox, our data above (in Fig. 1) and below (in Figs. 4–10) demonstrate that this small increase in Hsc70 expression had significant effects on ENaC trafficking and activity.
Fig. 4.

Hsc70 decreases surface expression of ENaC in MDCK cells. MDCK cells overexpressing Hsc70 were treated with 0 or 5 μg/ml Dox for 24 h. β-ENaC (A) or γ-ENaC (B) at the apical surface was detected by surface biotinylation. Representative immunoblots for β-ENaC (using an antibody to the V5 epitope tag, A) of whole cell lysates and NeutrAvidin-precipitated proteins are shown. Immunoblots for GAPDH were used as a control for protein loading and to ensure that the membrane-impermeant biotin did not label intracellular proteins. These panels are from noncontiguous lanes of the same immunoblot. Densitometric quantification of immunoblots from 3–4 independent biotinylation experiments is also shown. Error bars, SE. *P = 0.029 (n = 4 independent experiments; A) and 0.005 (n = 3 independent experiments; B) vs. 0 μg/ml Dox (by t-test).
Fig. 10.

Hsc70 diminishes interaction between newly synthesized β- and α-subunits of ENaC. A: schematic diagram of the method used in B. MDCK cells with inducible expression of Hsc70-Myc/His were incubated without or with 5 μg/ml Dox for 24 h. Newly synthesized proteins underwent pulse radiolabeling and were then chased for 0–60 min; 250 μg of cell lysates were subjected to 2 consecutive immunoprecipitation steps; 10% of the proteins precipitated with anti-V5 (for β-ENaC) were removed for resolution by SDS-PAGE and PhosphorImager analysis, and the remainder were used for the 2nd immunoprecipitation step with anti-HA (for α-ENaC) and subsequent SDS-PAGE and PhosphorImager analysis. Top: fluorograms from noncontiguous lanes of the same experimental gels. Bottom: densitometric quantification of precipitated β- and α-ENaC. Values are means ± SE of 3 independent experiments.
In Fig. 2C, we examined the induced expression of ATPase-deficient Hsc70. Similar quantification of these control cells indicates that the expression of ATPase-deficient Hsc70 with 1 μg/ml Dox was ∼0.02% of total cellular protein (Fig. 2C).
Increased Hsc70 expression does not alter whole cell expression of ENaC or Hsp70.
To test whether Hsc70 overexpression alters the whole cell expression of the three ENaC subunits, cells were treated with increasing concentrations of Dox, and whole cell lysates were subjected to immunoblot analysis for the α-, β-, and γ-ENaC subunits using antibodies directed to the epitope tags α-HA, β-V5, and γ-Myc (Fig. 3). There was no significant change in the whole cell expression of any of the ENaC subunits with any dose of Dox. There was also no significant change in the whole cell expression of Hsp70 as a function of increasing Dox (Fig. 3).
Fig. 3.

Effect of increased Hsc70 expression on whole cell ENaC and Hsp70 expression in MDCK cells. MDCK cells were treated with Dox (0–5 μg/ml) for 24 h, and expression of the ENaC subunits was determined by immunoblot analysis using antibodies directed to the epitope tags on each of the subunits. Hsp70 immunoreactivity was similarly determined, and GAPDH immunoreactivity was used as a loading control. Immunoblots are representative of 3 independent experiments.
Increased Hsc70 expression decreases ENaC surface expression.
Using a surface biotinylation assay, we examined the effect of Hsc70 overexpression on ENaC expression at the apical surface of MDCK cells grown as polarized monolayers. Expression of β-ENaC (Fig. 4A) and the lower-molecular-weight, cleaved form of γ-ENaC (Fig. 4B) at the apical surface decreased ∼50% after treatment with 5 μg/ml Dox. As a control, biotinylated GAPDH was not detected in the NeutrAvidin-precipitated proteins, suggesting that the membrane-impermeant biotin did not enter the cell and react with internal proteins. This control indicates that we only detected surface β- and γ-ENaC in these experiments. These data indicate that increased expression of Hsc70 decreases the expression of cleaved, and presumably active, ENaC at the apical cell surface.
Increased Hsc70 expression promotes retrieval of active ENaC from the apical surface.
To determine the mechanism by which Hsc70 affects ENaC activity and surface expression, we performed additional Ussing chamber assays. First, we addressed whether increased Hsc70 expression influenced the rate at which functional ENaC was retrieved from the apical surface. Increased Hsc70 expression was induced with Dox, and a baseline Isc was determined. Cells then were treated with cycloheximide to inhibit new protein synthesis, and the apparent first-order rate constant of decay of amiloride-sensitive Isc was determined (Fig. 5A). These data indicate that increased Hsc70 expression induced by 5 μg/ml Dox increases the rate at which functional ENaC is removed from the apical cell surface by ∼30% compared with cells not treated with Dox. In contrast, there was no significant change in the rate at which functional ENaC was removed from the cell surface when ATPase-deficient Hsc70 expression was induced by 1 μg/ml Dox in similar experiments (Fig. 5B).
ENaC Po is regulated by proteolysis of the extracellular loops of its α- and γ-subunits, with uncleaved and cleaved channel being found at the apical surface (25). Because uncleaved channel that has a very low Po at the surface can be activated (to Po ∼1) by cleavage with exogenous proteases such as trypsin (1, 4, 6, 7, 16, 24, 31), we tested the hypothesis that Hsc70 overexpression may decrease the fraction or proportion of cleaved ENaC at the surface as a mechanism by which ENaC activity was decreased. To determine if this was the case, after a baseline Isc was determined in Ussing chambers, the apical surface of the cells was treated with trypsin (10 μg/ml) to cleave and fully activate all ENaC channels at the apical surface and then with amiloride to determine the amiloride-sensitive Isc. Figure 6 shows that increased Hsc70 expression (with increasing Dox) decreased the amiloride-sensitive Isc that was present prior to application of trypsin (which is similar to Fig. 1). Interestingly, increased Hsc70 expression did not appear to alter the magnitude of stimulation of amiloride-sensitive Isc by trypsin, which is indicated by the difference in Isc before vs. after trypsin (Fig. 6, left). Arithmetically, this results in the fraction of total amiloride-sensitive Isc induced by trypsin increasing as a function of increased Dox (Fig. 6, right). These data thus suggest that increased Hsc70 expression does not alter the amount of uncleaved ENaC that can be activated by trypsin at the apical surface. These data also suggest that Hsc70 inhibits ENaC activity by selectively decreasing expression of the functional, cleaved, higher-Po channel at the apical surface. Taken together with the data of Fig. 5 demonstrating that increased Hsc70 increases the net retrieval of functional ENaC from the plasma membrane and the data of Fig. 4B that Hsc70 decreases the expression of cleaved γ-ENaC at the apical surface, these data suggest that Hsc70 promotes the retrieval of cleaved, higher-Po ENaC from the apical surface.
Fig. 6.

Increased Hsc70 expression increases relative expression of uncleaved vs. cleaved ENaC at the apical surface of MDCK cells. MDCK cells expressing Hsc70 were treated without or with Dox for 24 h, and a baseline Isc was established in Ussing chambers. Trypsin (10 μg/ml) was applied to the apical surface to cleave and activate uncleaved/inactive ENaC channels, and amiloride-sensitive Isc was determined. Left: amiloride-sensitive Isc before (○) and after (▽) treatment with trypsin. Right: fraction of total amiloride-sensitive Isc stimulated by trypsin, where data are presented as change in Isc with trypsin relative to total amiloride-sensitive Isc. Values are means ± SE for number of repeats indicated above each data point. *P < 0.05 vs. 0 μg/ml Dox control by 1-way ANOVA.
Increased Hsc70 expression decreases ENaC association with a recycling endosome component and increases its association with a late endosome component.
We hypothesized that Hsc70 may alter the amount of ENaC at the apical cell surface by regulating the recycling of ENaC after internalization. ENaC that is internalized by a ubiquitin-mediated mechanism (17, 32, 38, 56, 65) can be recycled back to the apical cell surface if the ubiquitin moiety is removed (5), or it can be targeted for degradation (65). Hsc70 is known to play an important role in clathrin-mediated endocytosis and lysosomal degradation of proteins (15, 42), as well as in ubiquitin-mediated degradation (3). Thus we hypothesized that Hsc70 would alter the amount of ENaC in recycling vs. lysosomal vesicles. To test this, we examined the associations between ENaC and the Rab GTPases characteristic of specific endosomal compartments (61).
We examined the interaction of ENaC with Rab7 and Rab11, which are components of late and recycling endosomes, respectively (61); late endosomal contents are typically targeted for degradation in the lysosome. Figure 7 qualitatively demonstrates the influence of Hsc70 overexpression on the colocalization of α-ENaC with Rab7 and Rab11 by immunofluorescence microscopy. We observed that Hsc70 overexpression increased the colocalization of α-ENaC with Rab7 (Fig. 7A) while decreasing its colocalization with Rab11 (Fig. 7B), suggesting that Hsc70 directs ENaC toward late endosomes and away from recycling endosome.
Fig. 7.
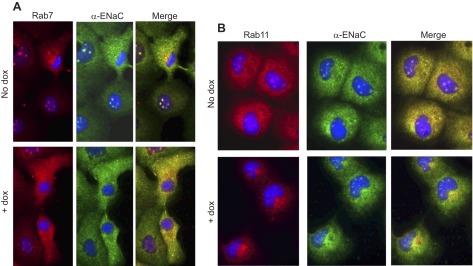
Overexpression of Hsc70 modulates colocalization between α-ENaC and Rab proteins. A: immunofluorescence detection of Rab7 (red) and α-ENaC (via HA tag, green) and their colocalization (yellow in merge image). Representative images of 4 independent experiments are shown. B: immunofluorescence detection of Rab11 (red) and α-ENaC (via HA tag, green) and their colocalization (yellow in merge image). Representative images of 4 independent experiments are shown.
We also used a coimmunoprecipitation approach (Fig. 8). Consistent with our immunofluorescence data, increased Hsc70 expression increased β-ENaC's association with Rab7 in these coimmunoprecipitation experiments (Fig. 8A). In contrast, increased Hsc70 expression decreased the interaction between β-ENaC and Rab11 (Fig. 8B). We previously published relevant control coimmunoprecipitations demonstrating that β-ENaC was not detected in the precipitated proteins when primary antibody was omitted or when an irrelevant primary antibody (α-GAPDH) was used under these experimental conditions (11). These data further suggest that Hsc70 directs ENaC toward association with Rab7 and Rab7-containing late endosomes and away from association with Rab11 and Rab11-containing recycling endosomes.
Fig. 8.

Hsc70 influences association of ENaC with endocytic vesicle components. Cells were treated with 0 or 5 μg/ml Dox for 24 h to induce increased Hsc70 expression. Samples underwent immunoprecipitation with anti-Rab7 (A) or anti-Rab11 (B), and β-ENaC was detected in precipitated proteins by immunoblot using anti-V5. Input samples represent 10% of total protein subject to immunoprecipitation. Fold change in V5-β-ENaC coprecipitated with Rab7 or Rab11 was determined by densitometric analysis of precipitated V5 from 4 independent experiments. Values (means ± SE) are presented relative to control (0 μg/ml Dox). *P = 0.029 (A) and 0.018 (B) vs. 0 μg/ml Dox (by t-test). Immunoblots of total Rab7 (A) or Rab11 (B) present in whole cell lysates are included for samples treated with or without Dox and demonstrate that Dox did not alter expression of these species.
When expression of ATPase-deficient Hsc70 was induced, the results of the coimmunoprecipitation experiments were reversed. There was a small increase in the β-ENaC-Rab11 interaction and a significant decrease in the β-ENaC-Rab7 interaction in cells expressing ATPase-deficient Hsc70 (Fig. 9).
Fig. 9.

ATPase activity of Hsc70 is required for the effect of Hsc70 on ENaC association with endocytic vesicle components. MDCK cells were treated with 0 or 1 μg/ml Dox for 24 h to induce ATPase-deficient Hsc70 expression. Samples underwent immunoprecipitation with anti-Rab7 (A) or anti-Rab11 (B), and β-ENaC was detected in precipitated proteins by immunoblot using anti-V5. Input samples represent 10% of total protein subject to immunoprecipitation. Fold change in V5-β-ENaC coprecipitated with Rab7 or Rab11 was determined by densitometric analysis of precipitated V5 from 3 independent experiments. Values (means ± SE) are presented relative to control (0 μg/ml Dox). *P = 0.029 (A); in B, P = 0.27 (by t-test).
Together, these data are consistent with the hypothesis that increased Hsc70 expression disfavors ENaC's association with a component of the recycling endosome and, therefore, likely disfavors its recycling back to the apical membrane after retrieval. Instead, increased Hsc70 expression increases ENaC's association with a component of late endosomes that may direct ENaC toward a degradative compartment. These data further suggest that Hsc70's ATPase function is required for these effects.
Hsc70 decreases the association of newly synthesized ENaC subunits.
In addition to examining how Hsc70 regulates ENaC at the apical surface, we also interrogated the role of Hsc70 in ENaC biogenesis. Hsc70 is a critical element of the ERAD of newly synthesized proteins (2, 41), leading us to examine the influence of Hsc70 overexpression on newly synthesized ENaC subunits in pulse-chase, sequential immunoprecipitation assays used in our previous publication (11). After pulse labeling, cell lysates were collected at time points up to 60 min of chase. The associations of pulse-labeled ENaC subunits were determined by immunoprecipitation first with anti-V5 (targeting β-ENaC) and then with an anti-α-ENaC. Figure 10 demonstrates that while the amount and stability of newly synthesized β-ENaC were not significantly altered by increased expression of Hsc70, the amount of ∼65-kDa (presumably cleaved) α-ENaC associated with β-ENaC at the 30-min time point after sequential immunoprecipitation decreased. In contrast, the associations of these subunits were diminished at 60 min of chase, with increased expression of Hsc70 not having a significant effect at this later time point. These data are thus consistent with increased Hsc70 expression also inhibiting the association of newly synthesized ENaC subunits early in channel biogenesis.
DISCUSSION
ENaC has critical roles in blood pressure regulation in the kidney and in the decreased mucociliary clearance and increased bacterial colonization in the CF airway (39, 51). It is clear that a more comprehensive understanding of ENaC regulation is necessary, as this may lead to therapeutics better able to combat the effects of dysregulated ENaC. Because Hsc70 is involved in clathrin-mediated endocytosis (15, 42), targeting of proteins for degradation in lysosomes (3, 12), and in the ubiquitin-dependent degradation of proteins and ERAD (40, 68), we surmised that Hsc70 might influence ENaC at multiple sites in the cell.
We became interested in the 70-kDa heat shock proteins Hsc70 and Hsp70 because of their altered expression in response to 4PBA, which improves intracellular trafficking of the most common mutation of the CFTR F508del (53, 54). 4PBA causes a 30–40% decrease in whole cell Hsc70 expression (53, 54) and a transient ∼50% increase in whole cell Hsp70 expression. We previously demonstrated that Hsp70 and Hsc70, which are ∼85% identical and typically considered to be functionally equivalent, in fact have opposite and antagonistic effects on ENaC in Xenopus oocytes, where Hsp70 promoted and Hsc70 inhibited ENaC activity and surface expression (19).
On the basis of our recent publication indicating that nearly twofold increased Hsp70 expression promotes ENaC activity in epithelial cells (11) and to further understand the mechanistic differences in Hsp70 and Hsc70's effects on ENaC, we tested the hypothesis that Hsc70 would have the opposite effect of Hsp70 in epithelial cells and inhibit ENaC activity. We again employed MDCK cells that had inducible chaperone overexpression for our studies. We demonstrated that 20% increased expression of Hsc70 (Fig. 2) decreased ENaC activity, while inducing ATPase-deficient Hsc70 expression did not have this effect (Fig. 1). While this level of increased Hsc70 expression may seem small, the endogenous Hsc70 protein represents ∼0.2% of the total cellular protein. This means that the overexpressed Hsc70, although only a small fraction of the total Hsc70 in the cell, still represents ∼0.05% of total cellular protein, and this increased expression nevertheless impacted ENaC expression and function compared with cells without increased Hsc70 expression and cells where an inactive Hsc70 was expressed at slightly lower levels.
Our heterologous expression constructs allow us to readily detect and study the ENaC subunits and the Hsc70 proteins in our cell system. In addition, these constructions allowed us to control the level and timing of increasing Hsc70 expression in our assays. An alternate method to study the effect of Hsc70 on ENaC would be to induce Hsc70 depletion by small interfering RNA (siRNA). While caveats exist in experiments employing protein overexpression, we have attempted to control for these caveats by demonstrating that expression of ATPase-deficient Hsc70 does not have the same effect as increasing Hsc70 expression. These data suggest that our observed effects are specific for increased expression of functional Hsc70. Similar and potentially more efficient methods, such as lentiviral vectors, can also be used to introduce overexpression plasmids or to induce siRNA gene depletion and might be useful in independently validating these findings. However, siRNA experiments also have difficulties, including variable transfection efficiency, as well as potential secondary effects of the gene or transfection procedure, especially when the siRNA target is a cellular stress response gene, such as Hsc70. In addition, preliminary experiments in which Hsc70 was depleted by siRNA show a compensatory increase in Hsp70 protein expression (data not shown). As we previously showed that Hsp70 and Hsc70 have opposing roles in ENaC trafficking (19), increased Hsp70 expression would confound our results. On the basis of these methodological difficulties, we believe that our overexpression system is a good method by which to study ENaC modulation by Hsc70.
Another potential caveat in these experiments, as well as in our recently published work regarding the influence of Hsp70 on ENaC (11), is our use of Dex pretreatment of our cells for 48 h. As is well described (14, 57, 58), Dex alters ENaC trafficking by increasing the function of serum and glucocorticoid-induced kinase 1, which in turn phosphorylates and decreases the activity of Nedd4-2, an E3 ubiquitin ligase that typically promotes ENaC ubiquitination and retrieval from the cell surface. However, we do not believe that this introduces significant experimental uncertainty, as all cells were identically treated with Dex for 24 h prior to induction of increased Hsc70 expression, and then over the ensuing 24 h of Hsc70 induction with Dox in the continued presence of Dex. The subsequent assay data were analyzed to compare only the influence of increased Hsc70 (or ATPase-deficient Hsc70) expression on the Dex-treated baseline.
We also believe that this 20% increase is an appropriate level of Hsc70 overexpression to examine for modulation of ENaC in these studies. While, at first glance, this might seem an insufficient change, chaperone protein expression is very tightly regulated, and, as a result, small changes in protein expression can have large effects. In fact, Hsc70 participates in many “high-volume” pathways in the cell, including ERAD and the uncoating of clathrin pits during endocytosis. Hsc70 also has a widely diverse clientele of potential binding partners within the cell. These considerations suggest that there is significant demand for Hsc70 as well, making its functional abundance more limited, especially in certain subcellular locales. In fact, published data suggest that increased Hsc70 expression and, specifically, the expression of mutant Hsc70 constructs do not affect all aspects of Hsc70 function equally and may preferentially affect clathrin-mediated endocytosis (45). Similarly, our recent work demonstrated that nearly twofold increased expression of Hsp70, without a concomitant change in Hsc70 expression, positively influenced ENaC trafficking (11). With these considerations, it becomes reasonable that small changes in Hsc70 expression, such as those studied here, may result in demonstrable effects on ENaC trafficking.
While increased Hsc70 expression did not significantly alter the whole cell expression of the ENaC subunits or Hsp70 (Fig. 3), it did result in decreased ENaC activity (Fig. 1) and a corresponding decrease in the expression of β-ENaC at the apical surface (Fig. 4). We also saw a decrease in active (cleaved) γ-ENaC at the surface of the cell when Hsc70 expression is increased (Fig. 4), suggesting that Hsc70 decreases active ENaC at the apical cell surface in MDCK cells. Because most ENaC that is present in the cell is not at the cell surface (11, 21), selective reduction of cell surface ENaC might not be expected to significantly alter ENaC's whole cell abundance. Taken together, these data and considerations suggest that Hsc70 decreases ENaC function by altering its surface expression.
ENaC activity can be regulated by modification of the total number of channels at the cell surface (change in N as a result of altered trafficking) or by cleavage of the inactive channels that were delivered to the cell surface, thus modifying Po. Because we observed a decrease in the activity and surface expression of ENaC with overexpressed Hsc70, we sought to identify which of these mechanisms were impacted by Hsc70. Our data suggest that Hsc70 overexpression selectively decreases the abundance of cleaved, higher-Po channels at the apical surface, while not altering the amount of uncleaved, low-Po ENaC at the cell surface (Fig. 6). Hsc70 overexpression does this by increasing the net retrieval of functional (presumably cleaved) ENaC from the cell surface (Figs. 4B and 5A). Together, these data therefore suggest that Hsc70 promotes the preferential removal of active, cleaved ENaC from the apical cell surface, leaving a higher proportion of uncleaved, inactive ENaC on the surface. This bias toward removal of active ENaC would lead to the increase in the fraction of channels that can be activated by trypsin observed in Fig. 6. These data are also consistent with the findings of others that cleaved, higher-Po ENaC is preferentially retrieved from the apical epithelial surface (32).
We also examined the association of β-ENaC with Rab GTPases that are characteristic components of the various compartments of the endocytic pathway. We found that increased Hsc70 expression increased the amount of α-ENaC (Fig. 7A) and β-ENaC (Fig. 8A) that interacted with Rab7, suggesting that Hsc70 directs ENaC to Rab7-containing late endosomal and lysosomal compartments of the cell (46). In contrast, increased Hsc70 expression decreased the amount of α-ENaC (Fig. 7B) and β-ENaC (Fig. 8B) associated with Rab11, a characteristic component of recycling vesicles, suggesting that ENaC recycling was impaired. This decrease in ENaC recycling may, at least in part, explain the increased net retrieval of ENaC from the apical cell surface (Fig. 5A). In contrast, ATPase-deficient Hsc70 expression had the opposite effect on β-ENaC association with Rab7 and Rab11 (Fig. 9) and promoted the association of β-ENaC with Rab11 while decreasing β-ENaC association with Rab7. This shift should promote ENaC recycling and may account for the small, but statistically significant, increase in ENaC activity with overexpression of ATPase-deficient Hsc70 (Fig. 1).
From these data, it is not clear whether the interaction between ENaC and the Rab proteins is a direct interaction or an indirect interaction as part of a larger complex. The mobility shift observed when β-ENaC was coprecipitated with the Rab GTPases in Fig. 7 was not a consistent finding in these experiments, nor was it observed in the experiments of Fig. 9. While it would be attractive to hypothesize that this mobility shift might be due to increased monoubiquitination of β-ENaC, we were not able to directly demonstrate this.
In addition to a role in endocytosis, Hsc70 also acts in the ERAD process (3, 12). To examine the possibility that Hsc70 affects ERAD of ENaC, we examined the forward trafficking of ENaC when Hsc70 expression was increased. As newly synthesized ENaC subunits undergo ERAD unless productively assembled into channels that exit the ER, we examined the rate of association of newly synthesized ENaC subunits. Our pulse-chase data (Fig. 10) show that, in the absence of increased Hsc70 expression, there was robust association of α-ENaC with β-ENaC after 30 min of chase. This association was diminished greatly when Hsc70 expression was increased, suggesting that Hsc70 antagonizes ENaC biogenesis and has the opposite effect of Hsp70, which promoted ENaC biogenesis in our previous work (11). In untreated and Dox-treated cells, there is an apparent decrease in α-ENaC association with β-ENaC after 60 min of chase; as this occurs in treated and untreated cells, this is likely not related to increased Hsc70 expression.
In summary, our data are consistent with the idea that Hsc70 decreases ENaC activity and surface expression by increasing the net retrieval of functional ENaC from the apical cell surface and, thereby, decreases the proportion of active ENaC at the apical epithelial surface. This is in agreement with our previous results in Xenopus oocytes that suggested that Hsc70 decreased ENaC activity and surface expression (19) and further distinguishes the functions and mechanisms of action of Hsc70 and its highly homologous family member Hsp70 (11). In addition, Hsc70 appears to promote ENaC association with a component of the lysosomal compartment and impairs its association with a component of the recycling compartment. Hsc70 also impairs the interaction of newly synthesized ENaC subunits early in biogenesis. Together, these data support the conclusion that small (20%) increases in Hsc70 abundance can have myriad effects on ENaC trafficking at multiple points along its cellular itinerary and that these effects alter ENaC surface expression and function. These data thus also support the hypothesis that apparently small changes in chaperone abundance can result in significant shifts in the trafficking of epithelial ion channels.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants T32 DK-07748 (R. A. Chanoux), R01 DK-58046, (R. C. Rubenstein), and R01 DK-73185 (R. C. Rubenstein).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.A.C., A.R., L.S., and R.C.R. are responsible for conception and design of the research; R.A.C., C.B.S., A.R., L.S., and R.C.R. performed the experiments; R.A.C., C.B.S., A.R., L.S., and R.C.R. analyzed the data; R.A.C., C.B.S., A.R., L.S., and R.C.R. interpreted the results of the experiments; R.A.C., A.R., L.S., and R.C.R. prepared the figures; R.A.C., L.S., and R.C.R. drafted the manuscript; R.A.C., L.S., and R.C.R. edited and revised the manuscript; R.A.C., C.B.S., A.R., L.S., and R.C.R. approved the final version of the manuscript.
REFERENCES
- 1.Adebamiro A, Cheng Y, Johnson JP, Bridges RJ. Endogenous protease activation of ENaC: effect of serine protease inhibition on ENaC single channel properties. J Gen Physiol 126: 339–352, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arndt V, Daniel C, Nastainczyk W, Alberti S, Hohfeld J. BAG-2 acts as an inhibitor of the chaperone-associated ubiquitin ligase CHIP. Mol Biol Cell 16: 5891–5900, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bercovich B, Stancovski I, Mayer A, Blumenfeld N, Laszlo A, Schwartz AL, Ciechanover A. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J Biol Chem 272: 9002–9010, 1997 [DOI] [PubMed] [Google Scholar]
- 4.Bruns JB, Carattino MD, Sheng S, Maarouf AB, Weisz OA, Pilewski JM, Hughey RP, Kleyman TR. Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J Biol Chem 282: 6153–6160, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Butterworth MB, Edinger RS, Ovaa H, Burg D, Johnson JP, Frizzell RA. The deubiquitinating enzyme UCH-L3 regulates the apical membrane recycling of the epithelial sodium channel. J Biol Chem 282: 37885–37893, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Caldwell RA, Boucher RC, Stutts MJ. Neutrophil elastase activates near-silent epithelial Na+ channels and increases airway epithelial Na+ transport. Am J Physiol Lung Cell Mol Physiol 288: L813–L819, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Caldwell RA, Boucher RC, Stutts MJ. Serine protease activation of near-silent epithelial Na+ channels. Am J Physiol Cell Physiol 286: C190–C194, 2004 [DOI] [PubMed] [Google Scholar]
- 8.Canessa CM, Merillat AM, Rossier BC. Membrane topology of the epithelial sodium channel in intact cells. Am J Physiol Cell Physiol 267: C1682–C1690, 1994 [DOI] [PubMed] [Google Scholar]
- 9.Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367: 463–467, 1994 [DOI] [PubMed] [Google Scholar]
- 10.Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet 12: 248–253, 1996 [DOI] [PubMed] [Google Scholar]
- 11.Chanoux RA, Robay A, Shubin CB, Kebler C, Suaud L, Rubenstein RC. Hsp70 promotes epithelial sodium channel functional expression by increasing its association with coat complex II and its exit from endoplasmic reticulum. J Biol Chem 287: 19255–19265, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246: 382–385, 1989 [DOI] [PubMed] [Google Scholar]
- 13.Choo-Kang LR, Zeitlin PL. Induction of HSP70 promotes ΔF508 CFTR trafficking. Am J Physiol Lung Cell Mol Physiol 281: L58–L68, 2001 [DOI] [PubMed] [Google Scholar]
- 14.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J 20: 7052–7059, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeLuca-Flaherty C, McKay DB, Parham P, Hill BL. Uncoating protein (hsc70) binds a conformationally labile domain of clathrin light chain LCa to stimulate ATP hydrolysis. Cell 62: 875–887, 1990 [DOI] [PubMed] [Google Scholar]
- 16.Donaldson SH, Hirsh A, Li DC, Holloway G, Chao J, Boucher RC, Gabriel SE. Regulation of the epithelial sodium channel by serine proteases in human airways. J Biol Chem 277: 8338–8345, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Flores SY, Debonneville C, Staub O. The role of Nedd4/Nedd4-like dependent ubiquitylation in epithelial transport processes. Pflügers Arch 446: 334–338, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Garty H. Molecular properties of epithelial, amiloride-blockable Na+ channels. FASEB J 8: 522–528, 1994 [DOI] [PubMed] [Google Scholar]
- 19.Goldfarb SB, Kashlan OB, Watkins JN, Suaud L, Yan W, Kleyman TR, Rubenstein RC. Differential effects of Hsc70 and Hsp70 on the intracellular trafficking and functional expression of epithelial sodium channels. Proc Natl Acad Sci USA 103: 5817–5822, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grunder S, Firsov D, Chang SS, Jaeger NF, Gautschi I, Schild L, Lifton RP, Rossier BC. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO J 16: 899–907, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanwell D, Ishikawa T, Saleki R, Rotin D. Trafficking and cell surface stability of the epithelial Na+ channel expressed in epithelial Madin-Darby canine kidney cells. J Biol Chem 277: 9772–9779, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Hendershot L, Wei J, Gaut J, Melnick J, Aviel S, Argon Y. Inhibition of immunoglobulin folding and secretion by dominant negative BiP ATPase mutants. Proc Natl Acad Sci USA 93: 5269–5274, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hendershot LM, Wei JY, Gaut JR, Lawson B, Freiden PJ, Murti KG. In vivo expression of mammalian BiP ATPase mutants causes disruption of the endoplasmic reticulum. Mol Biol Cell 6: 283–296, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hughey RP, Bruns JB, Kinlough CL, Harkleroad KL, Tong Q, Carattino MD, Johnson JP, Stockand JD, Kleyman TR. Epithelial sodium channels are activated by furin-dependent proteolysis. J Biol Chem 279: 18111–18114, 2004 [DOI] [PubMed] [Google Scholar]
- 25.Hughey RP, Bruns JB, Kinlough CL, Kleyman TR. Distinct pools of epithelial sodium channels are expressed at the plasma membrane. J Biol Chem 279: 48491–48494, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Hughey RP, Mueller GM, Bruns JB, Kinlough CL, Poland PA, Harkleroad KL, Carattino MD, Kleyman TR. Maturation of the epithelial Na+ channel involves proteolytic processing of the α- and γ-subunits. J Biol Chem 278: 37073–37082, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Hummler E. Epithelial sodium channel, salt intake, and hypertension. Curr Hypertens Rep 5: 11–18, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Iordache C, Duszyk M. Sodium 4-phenylbutyrate upregulates ENaC and sodium absorption in T84 cells. Exp Cell Res 313: 305–311, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Ishikawa T, Marunaka Y, Rotin D. Electrophysiological characterization of the rat epithelial Na+ channel (rENaC) expressed in MDCK cells. Effects of Na+ and Ca2+. J Gen Physiol 111: 825–846, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jasti J, Furukawa H, Gonzales EB, Gouaux E. Structure of acid-sensing ion channel 1 at 1.9 A resolution and low pH. Nature 449: 316–323, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Jovov B, Berdiev BK, Fuller CM, Ji HL, Benos DJ. The serine protease trypsin cleaves C termini of β- and γ-subunits of epithelial Na+ channels. J Biol Chem 277: 4134–4140, 2002 [DOI] [PubMed] [Google Scholar]
- 32.Kabra R, Knight KK, Zhou R, Snyder PM. Nedd4-2 induces endocytosis and degradation of proteolytically cleaved epithelial Na+ channels. J Biol Chem 283: 6033–6039, 2008 [DOI] [PubMed] [Google Scholar]
- 33.Kamynina E, Debonneville C, Bens M, Vandewalle A, Staub O. A novel mouse Nedd4 protein suppresses the activity of the epithelial Na+ channel. FASEB J 15: 204–214, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Kamynina E, Debonneville C, Hirt RP, Staub O. Liddle's syndrome: a novel mouse Nedd4 isoform regulates the activity of the epithelial Na+ channel. Kidney Int 60: 466–471, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Kashlan OB, Mueller GM, Qamar MZ, Poland PA, Ahner A, Rubenstein RC, Hughey RP, Brodsky JL, Kleyman TR. Small heat shock protein αA-crystallin regulates epithelial sodium channel expression. J Biol Chem 282: 28149–28156, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knight KK, Olson DR, Zhou R, Snyder PM. Liddle's syndrome mutations increase Na+ transport through dual effects on epithelial Na+ channel surface expression and proteolytic cleavage. Proc Natl Acad Sci USA 103: 2805–2808, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu C, Pribanic S, Debonneville A, Jiang C, Rotin D. The PY motif of ENaC, mutated in Liddle syndrome, regulates channel internalization, sorting and mobilization from subapical pool. Traffic 8: 1246–1264, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Malik B, Price SR, Mitch WE, Yue Q, Eaton DC. Regulation of epithelial sodium channels by the ubiquitin-proteasome proteolytic pathway. Am J Physiol Renal Physiol 290: F1285–F1294, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell 95: 1005–1015, 1998 [DOI] [PubMed] [Google Scholar]
- 40.Matsumura Y, David LL, Skach WR. Role of Hsc70 binding cycle in CFTR folding and endoplasmic reticulum-associated degradation. Mol Biol Cell 22: 2797–2809, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meacham GC, Patterson C, Zhang W, Younger JM, Cyr DM. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol 3: 100–105, 2001 [DOI] [PubMed] [Google Scholar]
- 42.Morgan JR, Prasad K, Jin S, Augustine GJ, Lafer EM. Uncoating of clathrin-coated vesicles in presynaptic terminals: roles for Hsc70 and auxilin. Neuron 32: 289–300, 2001 [DOI] [PubMed] [Google Scholar]
- 43.Morris RG, Schafer JA. cAMP increases density of ENaC subunits in the apical membrane of MDCK cells in direct proportion to amiloride-sensitive Na+ transport. J Gen Physiol 120: 71–85, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mueller GM, Kashlan OB, Bruns JB, Maarouf AB, Aridor M, Kleyman TR, Hughey RP. Epithelial sodium channel exit from the endoplasmic reticulum is regulated by a signal within the carboxyl cytoplasmic domain of the α-subunit. J Biol Chem 282: 33475–33483, 2007 [DOI] [PubMed] [Google Scholar]
- 45.Newmyer SL, Schmid SL. Dominant-interfering Hsc70 mutants disrupt multiple stages of the clathrin-coated vesicle cycle in vivo. J Cell Biol 152: 607–620, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ng EL, Gan BQ, Ng F, Tang BL. Rab GTPases regulating receptor trafficking at the late endosome-lysosome membranes. Cell Biochem Funct 30: 515–523, 2012 [DOI] [PubMed] [Google Scholar]
- 47.Pradervand S, Vandewalle A, Bens M, Gautschi I, Loffing J, Hummler E, Schild L, Rossier BC. Dysfunction of the epithelial sodium channel expressed in the kidney of a mouse model for Liddle syndrome. J Am Soc Nephrol 14: 2219–2228, 2003 [DOI] [PubMed] [Google Scholar]
- 48.Prince LS, Launspach JL, Geller DS, Lifton RP, Pratt JH, Zabner J, Welsh MJ. Absence of amiloride-sensitive sodium absorption in the airway of an infant with pseudohypoaldosteronism. J Pediatr 135: 786–789, 1999 [DOI] [PubMed] [Google Scholar]
- 49.Pruliere-Escabasse V, Planes C, Escudier E, Fanen P, Coste A, Clerici C. Modulation of epithelial sodium channel trafficking and function by sodium 4-phenylbutyrate in human nasal epithelial cells. J Biol Chem 282: 34048–34057, 2007 [DOI] [PubMed] [Google Scholar]
- 50.Rossi E, Farnetti E, Debonneville A, Nicoli D, Grasselli C, Regolisti G, Negro A, Perazzoli F, Casali B, Mantero F, Staub O. Liddle's syndrome caused by a novel missense mutation (P617L) of the epithelial sodium channel β-subunit. J Hypertens 26: 921–927, 2008 [DOI] [PubMed] [Google Scholar]
- 51.Rossier BC. The epithelial sodium channel: activation by membrane-bound serine proteases. Proc Am Thorac Soc 1: 4–9, 2004 [DOI] [PubMed] [Google Scholar]
- 52.Rossier BC, Pradervand S, Schild L, Hummler E. Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu Rev Physiol 64: 877–897, 2002 [DOI] [PubMed] [Google Scholar]
- 53.Rubenstein RC, Lyons BM. Sodium 4-phenylbutyrate downregulates HSC70 expression by facilitating mRNA degradation. Am J Physiol Lung Cell Mol Physiol 281: L43–L51, 2001 [DOI] [PubMed] [Google Scholar]
- 54.Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of ΔF508-CFTR. Am J Physiol Cell Physiol 278: C259–C267, 2000 [DOI] [PubMed] [Google Scholar]
- 55.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Jr, Ulick S, Milora RV, Findling JW, et al Liddle's syndrome: heritable human hypertension caused by mutations in the β-subunit of the epithelial sodium channel. Cell 79: 407–414, 1994 [DOI] [PubMed] [Google Scholar]
- 56.Snyder PM. The epithelial Na+ channel: cell surface insertion and retrieval in Na+ homeostasis and hypertension. Endocr Rev 23: 258–275, 2002 [DOI] [PubMed] [Google Scholar]
- 57.Snyder PM, Olson DR, Thomas BC. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem 277: 5–8, 2002 [DOI] [PubMed] [Google Scholar]
- 58.Snyder PM, Steines JC, Olson DR. Relative contribution of Nedd4 and Nedd4-2 to ENaC regulation in epithelia determined by RNA interference. J Biol Chem 279: 5042–5046, 2004 [DOI] [PubMed] [Google Scholar]
- 59.Staub O, Dho S, Henry P, Correa J, Ishikawa T, McGlade J, Rotin D. WW domains of Nedd4 bind to the proline-rich PY motifs in the epithelial Na+ channel deleted in Liddle's syndrome. EMBO J 15: 2371–2380, 1996 [PMC free article] [PubMed] [Google Scholar]
- 60.Staub O, Gautschi I, Ishikawa T, Breitschopf K, Ciechanover A, Schild L, Rotin D. Regulation of stability and function of the epithelial Na+ channel (ENaC) by ubiquitination. EMBO J 16: 6325–6336, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10: 513–525, 2009 [DOI] [PubMed] [Google Scholar]
- 62.Stewart AP, Haerteis S, Diakov A, Korbmacher C, Edwardson JM. Atomic force microscopy reveals the architecture of the epithelial sodium channel (ENaC). J Biol Chem 286: 31944–31952, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suaud L, Miller K, Panichelli AE, Randell RL, Marando CM, Rubenstein RC. 4-Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT-3 in cystic fibrosis epithelial cells. J Biol Chem 286: 45083–45092, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Valentijn JA, Fyfe GK, Canessa CM. Biosynthesis and processing of epithelial sodium channels in Xenopus oocytes. J Biol Chem 273: 30344–30351, 1998 [DOI] [PubMed] [Google Scholar]
- 65.Wiemuth D, Ke Y, Rohlfs M, McDonald FJ. Epithelial sodium channel (ENaC) is multi-ubiquitinated at the cell surface. Biochem J 405: 147–155, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wright JM, Zeitlin PL, Cebotaru L, Guggino SE, Guggino WB. Gene expression profile analysis of 4-phenylbutyrate treatment of IB3-1 bronchial epithelial cell line demonstrates a major influence on heat-shock proteins. Physiol Genomics 16: 204–211, 2004 [DOI] [PubMed] [Google Scholar]
- 67.Zeissig S, Bergann T, Fromm A, Bojarski C, Heller F, Guenther U, Zeitz M, Fromm M, Schulzke JD. Altered ENaC expression leads to impaired sodium absorption in the noninflamed intestine in Crohn's disease. Gastroenterology 134: 1436–1447, 2008 [DOI] [PubMed] [Google Scholar]
- 68.Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, Brodsky JL. Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol Biol Cell 12: 1303–1314, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]