

Abstract
LKB1 and its downstream targets of the AMP-activated protein kinase family are important regulators of many aspects of skeletal muscle cell function, including control of mitochondrial content and capillarity. LKB1 deficiency in skeletal and cardiac muscle (mLKB1-KO) greatly impairs exercise capacity. However, cardiac dysfunction in that genetic model prevents a clear assessment of the role of skeletal muscle LKB1 in the observed effects. Our purposes here were to determine whether skeletal muscle-specific knockout of LKB1 (skmLKB1-KO) decreases exercise capacity and mitochondrial protein content, impairs accretion of mitochondrial proteins after exercise training, and attenuates improvement in running performance after exercise training. We found that treadmill and voluntary wheel running capacity was reduced in skmLKB1-KO vs. control (CON) mice. Citrate synthase activity, succinate dehydrogenase activity, and pyruvate dehydrogenase kinase content were lower in KO vs. CON muscles. Three weeks of treadmill training resulted in significantly increased treadmill running performance in both CON and skmLKB1-KO mice. Citrate synthase activity increased significantly with training in both genotypes, but protein content and activity for components of the mitochondrial electron transport chain increased only in CON mice. Capillarity and VEGF protein was lower in skmLKB1-KO vs. CON muscles, but VEGF increased with training only in skmLKB1-KO. Three hours after an acute bout of muscle contractions, PGC-1α, cytochrome c, and VEGF gene expression all increased in CON but not skmLKB1-KO muscles. Our findings indicate that skeletal muscle LKB1 is required for accretion of some mitochondrial proteins but not for early exercise capacity improvements with exercise training.
Keywords: liver kinase B1, adenosine 5′-monophosphate-activated prokine kinase, mitochondria, exercise training, skeletal muscle
exercise training results in a host of adaptations in skeletal muscle that contribute to an enhanced capacity for subsequent exercise performance. Among the most significant of these is an increase in mitochondrial content and enzyme activity (14). Although the mechanisms involved in mitochondrial biogenesis are not fully defined, existing evidence suggests that AMP-activated protein kinase (AMPK) plays an important role in promoting this process (1, 51). AMPK is potently activated under conditions of low cellular energy, when ATP levels decline and AMP/ADP levels rise (4, 52). In skeletal muscle, this occurs during exercise or muscle work (50). Upon activation, AMPK generally signals the allocation of cellular resources to catabolism and energy production while reducing growth and proliferation signals. In line with these broad functions, chronic activation of AMPK, using the AMP mimetic AICAR, increases mitochondrial enzyme protein content and activity (18, 51). This is likely due, at least in part, to increased peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) content (18, 27) and phosphorylation-mediated PGC-1α-dependent transcriptional activation (19).
Activation of AMPK is mediated by both allosteric mechanisms and phosphorylation at Thr172 on the catalytic α-subunit by an upstream AMPK kinase (13, 15). Liver kinase B1 (LKB1) is the predominant upstream kinase mediating AMPK activation in skeletal muscle, as evidenced by a dramatically reduced AMPK activation in muscle-specific LKB1-knockout (mLKB1-KO) mice during exercise and/or contraction (25, 41, 47). In addition to the two catalytic AMPK isoforms (AMPKα1 and AMPKα2), 12 other AMPK family members are phosphorylated and activated by LKB1 (31). The role that these other AMPK family members play in skeletal muscle is very poorly defined, but recent data suggest that there may be some overlap in activation and/or functionality between them and AMPK itself (8, 26).
Efforts to characterize the role of LKB1 in skeletal muscle have been carried out by us and by other laboratories using mLKB1-KO mice in which LKB1 is lacking in both skeletal and cardiac muscle (3, 20, 25, 32, 41, 45–47). These mice exhibit decreased ability to run voluntarily, increased skeletal muscle fatigue (3, 47), and skeletal muscle wasting and dysfunction (46). Additionally, it was found that PGC-1α levels and mitochondrial content were decreased in skeletal muscle from mLKB1-KO mice (3, 25, 47), consistent with the idea that LKB1 regulates mitochondrial content via AMPK regulation of PGC-1α. Although these findings are valuable, the role of LKB1 specifically in skeletal muscle remains unclear because mLKB1-KO hearts fail at a young age (17, 21, 46). Heart failure may independently lead to many of the defects observed in mLKB1-KO mice, including mitochondrial defects in skeletal muscle (43). Thus, using those mice, it is impossible to distinguish between the effects of LKB1 ablation in skeletal muscle and those potentially brought on by heart failure.
Thus, although LKB1/AMPK signaling very likely plays an important role in the regulation of mitochondrial content, the specific role and necessity of skeletal muscle LKB1 for basal and exercise training-induced mitochondrial protein expression are still unclear. It is also not known whether LKB1 is required for improved exercise capacity after chronic training. Therefore, the purpose of this study was to determine whether the skeletal muscle-specific knockout of LKB1 (skmLKB1-KO) results in 1) decreased exercise capacity and mitochondrial protein content in sedentary muscle, 2) impaired accretion of mitochondrial proteins after chronic exercise training, and 3) attenuated improvement in exercise capacity after chronic exercise training.
MATERIALS AND METHODS
Animal care and generation of knockout mice.
All experimental procedures involving animals were approved prior to experimentation by the Institutional Animal Care and Use Committee of Brigham Young University. Mice were bred and housed under a 12:12-h light-dark cycle and temperature of 21–22°C. Skeletal muscle-specific LKB1 knockout mice (skmLKB1-KO) were generated by crossing LKB1 conditional control (CON) mice that express an LKB1 gene flanked by LoxP sites (provided by R. DePinho and N. Bardeesy, Dana-Farber Cancer Institute, Boston, MA) with myf6-Cre transgenic mice (12) heterozygously expressing Cre recombinase specifically in skeletal muscle under the Myf6 (MRF4) promoter (kindly provided by M. R. Capecchi, University of Utah, Salt Lake City, UT). The expression of Cre then drives the recombination of the LoxP sites and deletion of the LKB1 gene specifically from skeletal muscle. Genotyping was performed via polymerase chain reaction using primers for Cre and floxed LKB1, as described previously (47). Genotypes and the specificity of the skeletal muscle knockout were verified by Western blotting for LKB1 protein, as described below. CON and littermate skmLKB1-KO mice were 3–5 mo old at the time of experimentation.
AICAR treatment.
To confirm the efficacy of the knockout and that LKB1 is required for AMPK activation by AICAR treatment in skeletal muscle, CON and skmLKB1-KO mice (n = 6/group) were injected subcutaneously with AICAR dissolved in warm (37°C) sterile saline (1 mg/g body wt, 50 mg AICAR/ml) or an equivalent volume of plain saline. Thirty minutes after injection, mice were anesthetized with 2–3% isoflurane in 100% oxygen. After 30 min of anesthesia, gastrocnemius, soleus, red quadriceps, white quadriceps, heart, liver, and kidney were removed from the mice, quickly frozen between liquid nitrogen-chilled clamps, and stored at −90°C.
Sciatic nerve stimulation.
For initial experiments to determine whether exercise-induced AMPK activation is ablated in muscles from skmLKB1-KO mice, CON and skmLKB1-KO mice (n = 6/group) were anesthetized to a surgical plane with an injection of pentobarbital sodium (0.08 mg/g body wt). At least 20 min after injection, the gastrocnemius was removed from the right hindlimb. The left sciatic nerve was then isolated and electrically stimulated for 5 min to elicit contractions of the left gastrocnemius muscle (stimulation rate: 1 pulse/s; pulse duration: 10 ms). During the contraction bout, the foot was held at ∼90° to the tibia. Immediately after the contraction bout, the gastrocnemius was clamp-frozen and stored at −90°C until further analysis for AMPK activation.
Another cohort of mice was anesthetized with isoflurane, as described above. Hindlimb muscles were unilaterally stimulated via the sciatic nerve for 15 min at 0.5 pulses/s and 5-ms pulse duration. Muscles from the unstimulated hindlimb served as resting controls (CON REST). After stimulation, the resting and stimulated gastrocnemius-plantaris-soleus and tibialis anterior-extensor digitorum longus complexes were removed immediately (for analysis of signaling protein phosphorylation; n = 8/genotype) or 2 or 3 h (for mRNA expression analysis; n = 8/genotype) after the cessation of stimulation and clamp-frozen at the temperature of liquid nitrogen. Mice in the 2- and 3-h groups were maintained under isoflurane anesthesia, with the incision above the sciatic nerve closed by surgical staples the entire time, until the muscles were harvested.
Ambulatory activity monitoring.
Mice were individually housed in cages within an infrared beam-based activity monitoring system (Columbus Instruments, Columbus, OH). The number of beam breaks per hour was tracked by computer and averaged over 2 days.
Voluntary running.
CON (n = 9 males and 8 females) and skmLKB1-KO mice (n = 9 males and 7 females) were individually housed in cages equipped with in-cage activity wheels (Lafayette Instruments, Lafayette, IN) for 21 days. The distance run on the voluntary wheels was monitored by computer for the duration of the experiment.
Treadmill exercise testing and training.
Female mice were acclimatized to treadmill running for 3 days (day 1: nonmoving treadmill for 5 min + 5 min at 10 m/min; day 2: 10 m/min for 10 min; day 3: 10 m/min for 5 min + 15 m/min for 5 min). On the day after the final acclimatization bout, the mice were subjected to a maximal exercise test that consisted of 2 min at 12 m/min and 3 min at 15 m/min, followed by an increase of 1 m/min every minute until exhaustion. Exhaustion was defined as unresponsiveness to motivational stimuli (brushing the tail with a soft-bristled brush and puffs of air) for 10 s. Total running distance to fatigue was recorded as a measurement of running capacity. Beginning 3 days after pretesting, the mice were trained twice/day, 4 days/wk, for 3 wk. Duration and intensity of the training bouts for both genotypes were determined by the exercise capacity of the skmLKB1-KO mice so that the training stimulus was equal. Two cohorts of mice were trained separately and independently. The initial training bout was at 16 m/min for 16–20 min, depending on the cohort, at which time the first knockout mouse became fatigued. Speed and duration were increased for subsequent bouts, as tolerated by the skmLKB1-KO mice to a maximum 20–22 m/min for 50–60 min, depending on the cohort. Tissues were harvested from the mice under isoflurane anesthesia 48–56 h after the final training bout to allow acute effects of exercise to dissipate. At least 20 min after anesthetization, muscle tissue was removed from the mice, clamp-frozen, and stored at −90°C until analysis.
Tissue homogenization.
Muscles were glass-on-glass homogenized in 19 volumes of homogenization buffer (50 mM Tris·HCl, pH 7.4, 250 mM mannitol, 50 mM NaF, 5 mM sodium pyrophosphate; 1 mM EDTA; 1 mM EGTA, 1% Triton X-100, 50 mM B-glycerophosphate, 1 mM sodium orthovanadate, 1 mM DTT, 1 mM benzamidine, 0.1 mM phenylmethanesulfonyl fluoride, and 5 μg/ml soybean trypsin inhibitor), except for the soleus muscle, which was homogenized in 29 volumes of homogenization buffer. Homogenates were frozen at −90°C and thawed three times to ensure disruption of intracellular membranes, vortexed vigorously, and then centrifuged at 10,000 g for 20 min. The supernatants were analyzed for protein content (DC Protein Assay; Bio-Rad Laboratories, Hercules, CA) and then stored at −90°C for later analysis.
Western blotting.
Homogenates were diluted in 2× sample buffer (125 mm Tris·HCl, pH 6.8, 20% glycerol, 4% SDS, 5% β-mercaptoethanol, and 0.01% bromphenol blue) and then loaded on Tris-glycine gels (Bio-Rad Criterion System,; Bio-Rad Laboratories), and proteins were separated at 200 V for 55 min. Proteins were transferred to polyvinylidene difluoride membranes that were then probed for specific proteins via immunodetection. The antibodies used were as follows: phospho-AMPKα (no. 2535), AMPKα (no. 2532), phospho-Erk (no. 4370), Erk (no. 4695), phospho-p38 (no. 4511), p38 (no. 9212), phospho-CaMKII (no. 3361), and CaMKII (no. 3362) from Cell Signaling Technology (Danvers, MA); cytochrome c (no. sc-13156), pyruvate dehydrogenase kinase 4 (PDK4; no. sc-130841), and VEGF (no. sc-152) from Santa Cruz Biotechnology (Dallas, TX); LKB1 (no. 07-694) and PGC-1α (no. AB3242) from EMD Millipore (Billerica, MA); and OXPHOS antibody cocktail (no. 457999) from Life Technologies (Carlsbad, CA).
LKB1 activity assay.
LKB1 was immunoprecipitated overnight from 50 μl of CON and skmLKB1-KO gastrocnemius muscle homogenates (n = 6/genotype) with LKB1 antibody (sc-5640; Santa Cruz Biotechnology) bound to protein G Sepharose. Immune complexes were washed twice with wash buffer A (homogenization buffer described above + 0.5 M NaCl) and then twice with wash buffer B (40 mM HEPES, 80 mM NaCl, 8% glycerol, 0.8 mM EDTA, 5 mM MgCl2, 0.8 mM DTT). Thirty-five microliters of reaction cocktail {40 mM HEPES, 80 mM NaCl, 8% glycerol, 0.8 mM EDTA, 0.8 mM DTT, 5 mM MgCl2, 0.2 mM ATP, 10 μCi/μl [γ-32P]ATP, and 0.2 mM LKBtide (LSNLYHQGKFLQTFCGSPLYRRR)} was then added to the immune complex and incubated for 15 min at 37°C with vigorous shaking. Forty microliters of the reaction was then transferred to a half-piece of Whatman P81 filter paper (2.5 cm) and allowed to absorb for 20 s before the reaction was stopped in phosphoric acid. Incorporation of the labeled phosphate into LKB tide was then measured by scintillation counting and expressed as picomoles of phosphate incorporated per milligram of tissue per minute.
AMPK activity assay.
The activity of immunoprecipitated AMPKα1 and -α2 subunits was determined in gastrocnemius homogenates by measuring the incorporation of radiolabeled phosphate into SAMS peptide (HHMRSAMSGLHLVKRR-OH) expressed as picomoles phosphate of incorporated per gram of tissue per minute, as described previously (37a).
Citrase synthase activity.
Citrate synthase activity was measured on homogenates from gastrocnemius muscles from sedentary and treadmill-trained CON and skmLKB1-KO mice, as described by Srere (42).
Cytochrome c oxidase activity.
Activity of cytochrome c oxidase (COX) was determined for gastrocnemius muscle homogenates from treadmill-trained and sedentary mice using a cytochrome c oxidase assay kit (CYTOCOX1; Sigma) and following the manufacturer's instructions.
Mitochondrial DNA content.
Genomic DNA was isolated from red quadriceps muscles from treadmill-trained and sedentary mice using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI), following the manufacturer's instructions. DNA was quantified by spectrophotometry and diluted to 6 ng/μl; 28.5 ng of total DNA was used as the template for real-time PCR determination of cytochrome b (mitochondria encoded; forward primer 5′-TTCGCAGTCATAGCCACAG-3′, reverse primer 5′-TGCCATCCACAACAAG-3′) and β-actin (nuclear encoded; forward primer 5′-TCACCCACAATGTGCCCATCTACGA-3′, reverse primer 5′CAGCGGAACCGCTCATTGCCAATGG-3′). Cytochrome b DNA was normalized to β-actin DNA as an index of mitochondrial DNA content in the muscle using the 2−ΔΔCT calculation.
Tissue metabolite concentration.
Assays for the spectrophotometric determination of glycogen, ATP, and phosphocreatine (PCr) concentrations in tibialis anterior muscles after sciatic nerve stimulation were performed as described previously (35).
RNA isolation and real-time PCR.
Gastrocnemius-soleus-plantaris muscles were ground to powder under liquid nitrogen. RNA was isolated from the powdered muscle using Trizol (Life Technologies, Carlsbad, CA) and then cleaned up with Qiagen RNeasy columns, following the manufacturer's directions. RNA concentration and purity (260:280 ratio >1.9) were assessed by spectrophotometry. RNA integrity was verified by agarose electrophoresis. Synthesis of cDNA was performed from 500 ng of RNA using iScript Reverse Transciption Supermix (Bio-Rad, Hercules, CA). Real-time PCR was performed using KiCqStart SYBR Green quantitative PCR ReadyMix (Sigma) or SsoFast EvaGreen Supermix (Bio-Rad) according to the manufacturer's instructions, using a CFX96 real-time detection system (Bio-Rad). Primer sequences were obtained from published literature and are shown in Table 1. Amplification efficiency was verified prior to experimentation and was between 90 and 105% for all primer sets. Melt curve analysis was also performed to verify the generation of a single transcript. Gene expression relative to CON REST was performed using the 2−ΔΔCT method using muscle creatine kinase (MCK) for normalization.
Table 1.
Primer sequences used for real-time PCR
| Target (Ref. No) | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| MCK (34) | CGAGACTGGCCCGATGC | TCACCCACACAAGGAAGCTTT |
| PGC-1α (48) | GAGTCTGAAAGGGCCAAGC | GTAAATCACACGGCGCTCTT |
| TFAM (40) | GCTAAACACCCAGATGCAAAA | CGAGGTCTTTTTGGTTTTCC |
| Cytochrome c (16) | GCTTATCGGCCACCCAAGTG | GGGCTGCTGAGAGCTTGTTC |
| Cytochrome b (30) | CCCTAGCAATCGTTCACCTC | TCTGGGTCTCCTAGTATGTCTGG |
| VEGF (17) | CAGGCTGCTGTAACGATGAA | GCATTCACATCTGCTGTGCT |
| HIF-1 (22) | ATAGCTTCGCAGAATGCTCAGA | CAGTCACCTGGTTGCTGCAA |
MCK, muscle creatine kinase; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; TFAM, mitochondrial transcription factor A; HIF-1, hypoxia-inducible factor-1.
Succinate dehydrogenase staining.
Eight-micrometer sections from the tibialis anterior muscle were cut onto glass slides and air-dried. Slides were immersed in substrate solution [20 mM sodium succinate (no. 2378; Sigma), 10% nitrotetrazolium blue (no. 6639; Sigma) in 0.2 M phosphate buffer, pH 7.6] at 37°C for 40 min and then washed for 3 × 1 min in distilled water, air-dried, mounted with a coverslip, and imaged. Each condition was represented side by side on each slide to ensure equal treatment. Succinate dehydrogenase (SDH) staining intensity of the muscle fibers for the entire tibialis anterior muscle was determined using Image J software (National Institutes of Health).
Capillary immunofluorescence.
Eight-micrometer sections from the tibialis anterior muscle were cut onto glass slides, air-dried, and then fixed in acetone at 4°C for 10 min. Sections were permeabilized with 0.3% Triton X-100 in PBS for 10 min at 4°C, blocked in blocking buffer (5% normal goat serum in PBS) for 30 min at room temperature, and then incubated in a 1:50 dilution of CD31 primary antibody (no. MCA2388GA; ABD Serotech) in blocking buffer overnight at 4°C. Sections were washed for 3 × 5 min with PBS and then incubated for 30 min at room temperature in a 1:100 dilution of secondary antibody (Cy3-conjugated goat anti-rat IgG; Jackson Immunoresearch) in the dark at room temperature. Sections were washed again for 3 × 5 min in the dark at room temperature. Excess moisture was allowed to dry, after which coverslips were mounted with Fluoromount G (Southern Biotech, Birmingham, AL) and visualized with fluorescence microscopy. Exposure was adjusted such that brightly stained capillaries were visible along with light background fluorescence of muscle fibers. Capillaries and fibers were counted in blinded fashion. Data were presented as capillaries per muscle fiber.
Statistics.
Statistical comparisons for the AICAR and treadmill training experiments were made using ANOVA. ANOVA with repeated measurements was used for the electrical stimulation experiments. Fisher's least significant differene post hoc analysis was used when significant differences were detected by ANOVA. Analyses were performed using NCSS statistical software (Kaysville, UT), with significance set at P < 0.05. The data are reported as means ± SE.
RESULTS
skmLKB1-KO.
skmLKB1-KO mice were generated by crossing LKB1 conditional mice expressing floxed LKB1 (CON), with mice heterozygously expressing Cre recombinase under the Myf6 promoter. Genotyping of the resultant skmLKB1-KO mice was positive for Cre and floxed LKB1 genes but not for wild-type LKB1. Genotyping of CON mice was positive for floxed LKB1 but not for Cre or wild-type LKB1 (data not shown). LKB1 protein expression was measured by Western blotting to verify the specificity of the LKB1 knockout. Accordingly, LKB1 protein content was ≤10% in skmLKB1-KO vs. CON soleus, gastrocnemius, red quadriceps, and white quadriceps muscles (Fig. 1, A and C). LKB1 protein expression in heart, liver, and kidney was not significantly altered in skmLKB1-KO mice (Fig. 1, B and C). LKB1 activity was 89% lower in skmLKB1-KO vs. CON gastrocnemius muscles (Fig. 1D).
Fig. 1.
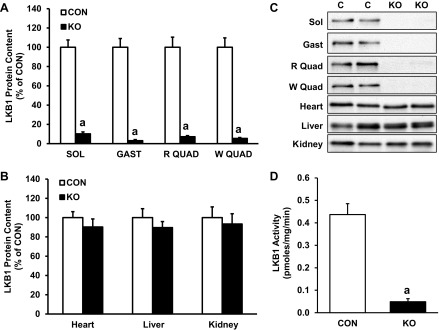
Skeletal muscle-specific knockout of liver kinase B1 (LKB1). A: LKB1 protein content in resting gastrocnemius muscles from control (CON) and skeletal muscle-specific LKB1-knockout (KO) mice. B: LKB1 protein content in heart, liver, and kidney from CON and KO mice. C: representative immunoblots for LKB1 corresponding to data in A and C. LKB1 band is at ∼57 kDa. D: LKB1 activity in resting gastrocnemius samples from CON and KO mice, as determined by the rate of incorporation of phosphate from radiolabeled ATP into LKBtide. Data are presented as means ± SE; n = 6–12. aP < 0.05 vs. corresponding CON muscle. SOL, soleus; GAST, gastrocnemius, R QUAD, red quadriceps; W QUAD, white quadriceps.
AMPK activation in LKB1-KO muscle.
Next, we verified that activation of AMPK by AICAR is dependent on skeletal muscle LKB1. AICAR is taken up into the cell and phosphorylated to form ZMP (5-aminoimidazole-4-carboxamide-1-β-d-ribosyl-5-monophosphate), an AMP mimetic that binds to AMPK and leads to its phosphorylation and activation. AMPKα2 activity in saline-treated skmLKB1-KO gastrocnemius muscles was 73% lower than that in corresponding CON muscles (Fig. 2B). Furthermore, 1 h after AICAR treatment, AMPKα2 activity increased by 200% in CON muscles but was not significantly altered in skmLKB1-KO muscles (Fig. 2B). AMPKα1 activity, on the other hand, increased modestly in both CON and skmLKB1-KO muscles with AICAR treatment (by 72 and 39%, respectively), but no significant effect of genotype was observed, although there was a distinct trend (P = 0.06) for lower AMPKα1 activity in the skmLKB1-KO muscles (Fig. 2A). Consistent with the AMPKα2 activity data, content of AMPK phosphorylated at Thr172 was 72% lower in saline-treated skmLKB1-KO vs. CON gastrocnemius muscles. Phospho-AMPK levels increased 203% with AICAR treatment in CON but not in AICAR-treated skmLKB1-KO muscles (Fig. 2, C and D).
Fig. 2.
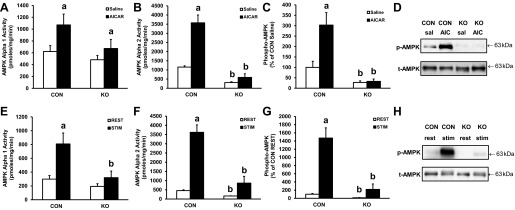
AMP-activated protein kinase (AMPK) activation in CON and skeletal muscle-specific LKB1-KO (skmLKB1-KO) gastrocnemius muscles after AICAR treatment and muscle contraction. A: AMPKα1 activity after saline or AICAR injection. B: AMPKα2 activity after saline or AICAR injection. C and D: AMPKα phosphorylation after saline or AICAR injection. E: AMPKα1 activity in resting (REST) and electrically stimulated (STIM) muscle. F: AMPKα2 activity in REST and STIM muscles. G and H: AMPKα phosphorylation in REST and STIM muscles. Data are presented as means ± SE; n = 5–6. aP < 0.05 vs. corresponding saline-treated (A–C) or resting muscle (E–G). bP < 0.05 vs. corresponding CON muscle.
We likewise verified that AMPK activation by vigorous muscle contraction is dependent on skeletal muscle LKB1 in our mice. Contractions of the hindlimb musculature were elicited by electrical stimulation of the sciatic nerve for 5 min. Immediately after the contraction bout, AMPKα1 activity increased by 170% in CON muscles but was not significantly higher in skmLKB1-KO muscles (Fig. 2E). Likewise, AMPKα2 activity increased (by 690%) with contraction in CON, but not significantly in skmLKB1-KO muscles despite a trend for such (Fig. 2F). AMPK phosphorylation increased robustly with contraction in CON muscles, but not significantly in skmLKB1-KO muscles (Fig. 2, G and H).
Ambulatory activity and voluntary running performance.
To determine whether skeletal muscle LKB1 is a contributing factor to normal physical activity in mice, we monitored their in-cage ambulatory activity using an infrared beam-based activity monitoring system for 2 days. Average activity of the CON and skmLKB1-KO mice was not different (Fig. 3A). However, when placed in cages with voluntary running wheels for 21 days, skmLKB1-KO mice ran 40% less per day on average over the course of the training period than CON littermates (Fig. 3B).
Fig. 3.

Ambulatory activity and voluntary running in CON and skmLKB1-KO mice. A: daily ambulatory activity in CON and skmLKB1-KO mice (n = 10–14). B: daily voluntary wheel running activity over 3 wk in CON and KO mice (n = 8). Data are presented as means ± SE.
Treadmill training and improvements in exercise capacity.
Next, we determined whether LKB1 is required for training-induced alterations in exercise capacity. Treadmill running performance was determined for untrained mice (pretest) using a progressive treadmill-running protocol, as described in materials and methods. Running distance to fatigue was 62% lower in untrained skmLKB1-KO vs. CON mice (266 ± 58 vs. 709 ± 67 m, respectively; Fig. 4A). The mice were then treadmill trained for 40–60 min/bout, twice/day, 4 days/wk, for 3 wk. CON and skmLKB1-KO mice were trained at equal speed and duration based on the capacity of the skmLKB1-KO mice, as described in materials and methods. After the training period, the mice were tested again for treadmill-running performance (posttest). Absolute distance to fatigue increased significantly after training to 875 ± 90 m for CON and 391 ± 54 m for skmLKB1-KO mice (Fig. 4A). Although the distance to fatigue was still substantially and significantly lower in skmLKB1-KO vs. CON mice posttraining, the average relative increase in running distance within each mouse from pre- to posttesting was significantly greater for the skmLKB1-KO mice vs. CON (61 ± 16 vs. 25 ± 11% increase, respectively; Fig. 4B). A separate cohort of skmLKB1-KO and CON mice was pre- and posttested at the same time as the trained mice but was not subjected to exercise training between tests and served as sedentary controls. The percent change in running distance from pre- to posttesting did not improve in these untrained mice of either genotype and actually tended to decline in the untrained skmLKB1-KO mice (2.4 ± 6.8 and 19.9 ± 5.6% decline in distance to fatigue from pre- to posttesting for CON and skmLKB1-KO mice, respectively; Fig. 4B), indicating that the modest increase in running performance in the trained cohort was due to the training and not to a learning effect of testing.
Fig. 4.

Maximal treadmill running capacity of CON and skmLKB1-KO mice before and after 3 wk of treadmill run training. A: total running distance to fatigue during a progressive treadmill testing protocol before and after treadmill training. B: %change in running distance to fatigue from pretesting to posttesting in sedentary and trained mice. Data are presented as means ± SE (n = 8–9). aP < 0.05 vs. corresponding pretraining (A) or sedentary mice (B); bP < 0.05 vs. corresponding CON mice.
Treadmill training and mitochondrial protein content.
PCG-1α protein content tended to be lower in skmLKB1-KO vs. CON muscles, but not significantly so (Fig. 5A). Exercise training failed to significantly increase PGC-1α content in either genotype. SDH and citrate synthase activities and protein content for PDK4 were all significantly lower in skmLKB1-KO vs. CON muscles (Fig. 5, B–E). SDH activity and PDK4 protein content did not increase with training in either genotype, although there was a trend for such in PDK4 content (Fig. 5, B and E). However, citrate synthase activity did increase with training in both CON and skmLKB1-KO muscles without any difference between genotypes (Fig. 5D). Interestingly, content of cytochrome c and components of complexes I, II, IV, and V of the electron transport chain (Fig. 5F), as well as activity of cytochrome c oxidase (Fig. 5G), were all increased with training in CON but not skmLKB1-KO muscles (Fig. 5, H and I). Mitochondrial DNA content was not significantly different between genotypes or treatment conditions (Fig. 5J).
Fig. 5.
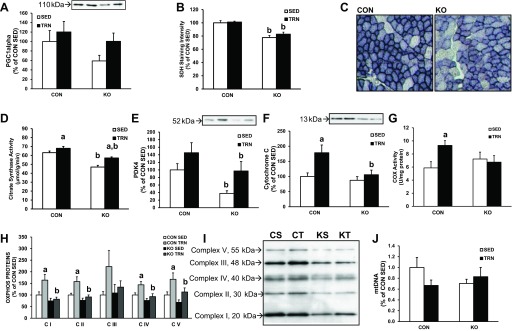
Mitochondrial protein expression in sedentary (SED) and 3-wk trained (TRN) skeletal muscle from CON and skmLKB1-KO mice. A: peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) protein content. B: succinate dehydrogenase (SDH) activity in tibialis anterior muscle. C: representative SDH-stained cross-sections for CON and KO tibialis anterior muscles. D: citrate synthase activity in gastrocnemius muscle. E: pyruvate dehydrogenase kinase 4 (PDK4) protein content in gastrocnemius muscle. F: cytochrome c protein content in gastrocnemius muscle. G: cytochrome c oxidase (COX) activity in gastrocnemius muscles. H: content of representative protein constituents of complexes I, II, III, IV, and V of the mitochondrial electron transport chain (OXPHOS proteins) in gastrocnemius muscle. I: representative blot for OXPHOS proteins corresponding to means in G. J: mitochondrial DNA content in red quadriceps muscles. Data are presented as means ± SE (n = 8–9, except for B, where n = 6–8). aP < 0.05 vs. corresponding SED muscle; bP < 0.05 vs. corresponding CON muscle. CS, control sedentary; CT, control trained; KS, skmLKB1-KO sedentary; KT, skmLKB1-KO trained.
Capillarity of the skmLKB1-KO muscle was significantly lower compared with CON muscles, but the training stimulus was not sufficient to increase this measurement (Fig. 6A). Likewise, vascular endothelial growth factor-A (VEGF) protein content was 46% lower in skmLKB1-KO vs. CON sedentary muscles (Fig. 6B). Interestingly, however, VEGF content increased by 53% in skmLKB1-KO but not significantly in CON muscles with training.
Fig. 6.

Capillarity and VEGF protein content in SED and TRN skeletal muscle from CON and skmLKB1-KO mice. A: capillaries per muscle fiber as determined by immunofluorescent detection of CD31 in tibialis anterior muscles. B: VEGF content of gastrocnemius muscles. Data are presented as means ± SE (n = 8–9). aP < 0.05 vs. corresponding SED muscle; bP < 0.05 vs. corresponding CON muscle.
Signaling pathway activation and gene expression after muscle contraction.
We next desired to determine the acute effects of a bout of muscle contractions on cell signaling and gene expression in muscle from skmLKB1-KO mice. Intermittent contraction of the mouse hindlimb was elicited unilaterally by sciatic nerve stimulation for 15 min. The gastrocnemius-plantaris-soleus complex was removed from the stimulated and contralateral resting limb either immediately or 2 or 3 h after stimulation. Consistent with the results of the 5-min contraction bout, AMPK phosphorylation increased substantially immediately after the contraction bout in CON but not in skmLKB1-KO muscles (Fig. 7A). P38 (Fig. 7B), Erk (Fig. 7C), and CaMKII (Fig. 7D) phosphorylation, on the other hand, increased similarly in both CON and skmLKB1-KO muscles immediately after stimulation. By 2 h postcontraction, AMPK (Fig. 7E), p38 (Fig. 7F), and CaMKII (Fig. 7H) phosphorylation were at resting levels in muscles from both genotypes, although phosphorylation of p38 tended, nonsignificantly, to be elevated in stimulated skmLKB1-KO muscles (P = 0.21; Fig. 7E). Erk phosphorylation, on the other hand, remained significantly elevated 2 h after stimulation in skmLKB1-KO muscles but not in CON muscles (Fig. 7G).
Fig. 7.
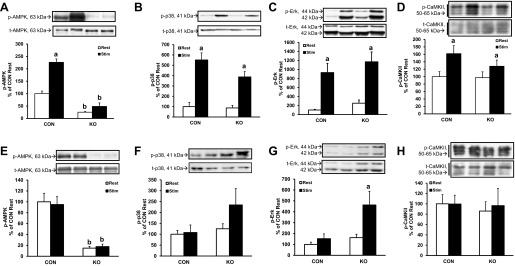
Phosphorylation of AMPK, p38 MAPK, and Erk 0 and 2 h postcontraction in CON and skmLKB1-KO mice. A: phosphorylation of AMPK at Thr172 immediately after STIM contractions. B: phosphorylation of p38 MAPK at Thr180/Tyr182 immediately after STIM contractions. C: phosphorylation of Erk (p42/p44 MAPK) at Thr202/Tyr204 immediately after STIM contractions. D: phosphorylation of CaMKIII at Thr286 immediately after STIM contractions. E: phosphorylation of AMPK at Thr172 2 h after STIM contractions. F: phosphorylation of p38 MAPK at Thr180/Tyr182 2 h after STIM contractions. G: phosphorylation of Erk (p42/p44 MAPK) at Thr202/Tyr204 2 h after STIM contractions. H: phosphorylation of CaMKIII at Thr286 2 h after STIM contractions; n = 8. Data are presented as means ± SE. aP < 0.05 vs. corresponding REST muscle; bP < 0.05 vs. corresponding CON muscle.
Expression of mitochondrial transcription factor A (TFAM) and cytochrome b, but not PGC-1α, cytochrome c, VEGF, and hypoxia-inducible factor-1 (HIF-1) mRNA was lower basally in skmLKB1-KO vs. CON muscles. Three hours after the contraction bout, mRNA expression of PGC-1α, cytochrome c, and VEGF was increased by 181, 95, and 142% above rest, respectively, in CON muscles (Fig. 8, A, C, and E) but was unaffected by contraction in skmLKB1-KO muscles. Expression of TFAM (Fig. 8B), cytochrome b (Fig. 8D), and HIF-1 (Fig. 8F) was not altered by contraction at this time point in CON or skmLKB1-KO muscles.
Fig. 8.
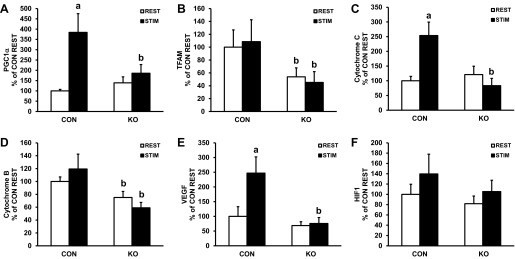
Gene expression in gastrocnemius muscle from CON and skmLKB1-KO mice 3 h after contractions. Expression of mRNA for PGC-1α (A), mitochondrial transcription factor A (TFAM; B), cytochrome c (C), cytochrome b (D), VEGF (E), and hypoxia inducible factor-1α (HIF-1α; F) 3 h after a 15-min STIM contraction bout (n = 6). Data are presented as means ± SE. aP < 0.05 vs. corresponding REST muscle; bP < 0.05 vs. corresponding CON muscle.
To determine whether substrate use during the stimulation bout was altered by LKB1-KO, we measured glycogen, ATP, and PCr levels in muscles from CON and skmLKB1-KO mice. We were unable to use gastrocnemius muscles for these assays since that tissue was already consumed for other measurements. Therefore, we used the tibialis anterior muscle, which is also activated during sciatic nerve stimulation. The stimulation bout decreased glycogen (Fig. 9A), ATP (Fig. 9B), and phosphocreatine (Fig. 9C) levels in both CON and skmLKB1-KO tibialis anterior muscles without any differences between genotypes, indicating that the energetic strain from stimulation was similar for CON and skmLKB1 muscles.
Fig. 9.

Concentration of glycogen (A), ATP (B), and phosphocreatine (PCr; C) immediately after STIM contractions or REST in tibialis anterior muscles from CON and skmLKB1-KO mice; n = 6–8s/group. Data are presented as means ± SE. aP < 0.05 vs. corresponding resting muscle.
DISCUSSION
The major purpose of this study was to determine whether skeletal muscle LKB1 is required for exercise-induced adaptations in exercise capacity and mitochondrial content. We found that upregulation of several key genes involved in mitochondrial biogenesis and angiogenesis is impaired after a bout of contractions in skmLKB1-KO muscle. Likewise, upregulation of proteins involved in mitochondrial oxidative phosphorylation with training is impaired in skmLKB1-KO muscle. However, these effects of LKB1 knockout do not prevent or impair improvements in exercise capacity after chronic exercise training.
Previously, we (20, 47) observed that running capacity was impaired in mice in which LKB1 was selectively knocked out in skeletal and cardiac muscle. Since the lack of LKB1 in heart resulted in cardiac dysfunction (17, 21, 46), it cannot be determined using that model, whether the impairment in exercise capacity is due to skeletal or cardiac muscle defects. Therefore, we generated skeletal (and not cardiac) muscle-specific LKB1-knockout mice in which Cre recombinase expression and subsequent excision of floxed LKB1 are driven by the Myf6 promoter. LKB1 expression in these mice was unaltered in nonskeletal muscle, tissue including the heart, but reduced by ∼90% or more in skeletal muscle, and this correlated with a substantial reduction in AMPK phosphorylation and AMPKα2 subunit activity and confirms previous findings showing that LKB1 is the major AMPKα2 kinase in skeletal muscle (25, 41, 47). AMPKα1 activity, on the other hand, was not significantly reduced vs. CON muscle in untreated or AICAR-treated skmLKB1-KO muscles. Our findings are consistent with previous findings from mLKB1-KO mice (25) and with very recent data from skeletal muscle-specific LKB1-dominant negative (skmLKB1-DN) mice (33). The persistent AMPKα1 activity in skmLKB1-KO muscle is likely due to 1) the presence of nonskeletal muscle cells (endothelial cells, neurons, adipocytes) in the tissue sample that highly express AMPKα1 and do not lack LKB1 and 2) the potential presence of an alternate AMPK kinase, such as calcium/calmodulin-dependent protein kinase kinase (CaMKK), that is able to phosphorylate AMPK in the absence of LKB1. Although not significant, phosphorylation and activity of AMPKα2 tended to increase slightly with electrical stimulation in the skmLKB1-KO muscle. This also supports the idea of an LKB1-independent AMPK kinase in skeletal muscle.
Consistent with previous findings in mLKB1-KO mice (20, 47), both voluntary exercise capacity and maximal treadmill exercise capacity were decreased in skmLKB1-KO mice, demonstrating that skeletal muscle LKB1 is required for optimal exercise capacity. Our findings agree with those of Miura, et al. (33), who very recently showed a similar impairment in exercise tolerance in a skmLKB1-dominant negative mice. Ambulatory cage activity, however, was not altered in skmLKB1-KO mice, indicating that differences between genotypes in untrained mice are not due to a chronic decrease in physical activity. The impairment in exercise capacity is almost certainly due in part to the lack of AMPK activity that we observed, since the double-knockout of AMPK β-subunits causes a similar decrease in performance (36). The magnitude of impairment in voluntary running after 21 days of training was less in the skmLKB1-KO mouse (∼45% less distance/day) than in previous reports for the mLKB1-KO mouse (∼75% less distance/day) (47). Likewise, maximal treadmill running speed was previously shown to be more than 50% lower in mLKB1-KO mice than in controls (20). We show here that the total distance run by the skmLKB1-KO mice was 75% lower than CON, but when expressed as the maximal treadmill speed, as reported previously for the mLKB1-KO mice, the skmLKB1-KO reached a speed that was 35% lower than CON (40 vs. 26 m/min, respectively). Therefore, it appears that the loss of LKB1 in both heart and skeletal muscle results in a greater impairment of exercise capacity than the loss of LKB1 in skeletal muscle only.
We next desired to determine whether skeletal muscle LKB1 is required for improvements in mitochondrial content, muscle vascularity, and exercise capacity with chronic training. To test this, we trained CON and skmLKB1-KO mice for 3 wk at equal speed and distance for both genotypes based on the running capacity of the skmLKB1-KO mice. Training speeds were quite low initially since the skmLKB1-KO mice ran so poorly. In fact, our first experiments (data not shown) showed that running once/day at the low speeds sustained by the skmLKB1-KO mice was not sufficient to induce adaptations in the CON mice. Therefore, we ran the mice twice daily, 4 days/wk, starting at just 16 m/min for 20 min/exercise bout, which was sufficient to completely fatigue the skmLKB1-KO mice. However, the skmLKB1-KO mice improved substantially with time such that they could endure 50–60 min at 20–22 m·min−1·bout−1 by the end of the 3-wk training period. We found that this training protocol improved running performance in both the CON and skmLKB1-KO mice, with the relative increase in running distance with training being greater in the skmLKB1-KO vs. CON mice. This clearly shows that LKB1 is not required for early improvements in exercise capacity with training. It is not known whether performance improvements would persist in skmLKB1-KO mice with continued training such that they would reach the levels seen in CON mice. Some caution should be exercised in interpreting the observed differences in the magnitude of the training response between genotypes. In our study, the genotypes were run at the same absolute intensity and volume. However, this represented a higher relative workload for the skmLKB1-KO mice, since their maximal running capacity was lower. Thus, training responses in the skmLKB1-KO mice may have been lower had they been trained at a relative workload similar to the CON mice.
The activity of citrate synthase and protein content of cytochrome c was previously shown to be lower in skeletal muscle of mLKB1-KO mice, as was the content of PGC-1α, the transcriptional cofactor thought to play a major role in the regulation of mitochondrial volume (20, 47). In agreement with those findings, we observed decreased citrate synthase activity, succinate dehydrogenase activity, and PDK4 content in skmLKB1-KO vs. CON mice, suggestive of lower mitochondrial volume, whereas PGC-1α, mtDNA, cytochrome c, and the other components of the oxidative phosphorylation system that we measured were unaffected basally by genotype. It should be noted, however, that for PGC-1α in particular, the statistical power was quite low (0.44), due to variability in the data set, such that no difference was observed between genotypes despite the mean value being 41% lower in sedentary skmLKB1-KO vs. CON muscles. Thus, it seems likely that for this measurement, statistical significance would be achieved with a larger sample size.
PGC-1α content, mtDNA content, PDK4 content, and SDH activity were not altered with training in CON or skmLKB1-KO muscles. This lack of responsiveness of some markers of mitochondrial biogenesis is not terribly surprising given the relatively low intensity of training necessitated by the poor running ability of the skmLKB1-KO mice. Regardless, citrate synthase activity as well as the protein content for cytochrome c and other mitochondrial proteins involved in oxidative phosphorylation increased with training in CON mice. The lack of correlation between mtDNA increase and mitochondrial enzyme increases is consistent with previous findings in humans where moderate exercise and weight loss resulted in increased oxidative capacity, expansion of the inner mitochondrial membrane, and increased activity of electron transport proteins despite a lack of increased mtDNA content. We were also somewhat surprised by the lack of significant response in PGC-1α content with training since it is widely considered to be a “master regulator” of mitochondrial biogenesis. However, it should be kept in mind that PGC-1α expression may not be required for exercise-induced adaptations in mitochondrial gene expression, as increased expression of cytochrome c and ALAS1 and increased content of mitochondrial proteins cytochrome c, COX-1, and ALAS1 occur with treadmill training in PGC-1α-knockout mice (28). Therefore, the lack of PGC-1α accretion in the present study would not necessarily prevent increases in mitochondrial gene or protein expression. Citrate synthase activity also increased with training in skmLKB1-KO mice, but the proteins involved in oxidative phosphorylation, including cytochrome c, did not increase with training in the skmLKB1-KO muscles, indicating that LKB1 plays an important role in the upregulation of some mitochondrial proteins with chronic increases in activity. LKB1 likely works in this regard at least partly through targets other than AMPKα2, since previous work has shown that AMPKα2 knockout does not prevent changes in mitochondrial markers with exercise training (23, 39). This is also consistent with recent findings that LKB1 regulates fat metabolism in skeletal muscle through non-AMPK-dependent pathways (20).
Exercise (2) and activation of AMPK (37, 53) promote angiogenesis and VEGF expression in skeletal muscle. Thus, it might be expected that training-induced angiogenesis and VEGF expression would be impaired in skmLKB1-KO muscles due to a lack of AMPK activation. This is consistent with our finding that VEGF gene expression after acute contractions was impaired in skmLKB1-KO muscles. Although we did observe lower capillarity and VEGF protein expression in sedentary skmLKB1-KO vs. CON muscles, our training bout was not sufficient to increase capillarity or VEGF protein expression in the CON mice. Paradoxically, however, VEGF protein content increased significantly with treadmill training in skmLKB1-KO muscles, indicating that not only is skeletal muscle LKB1 not required for VEGF accretion with training, it may even impair exercise-induced VEGF synthesis and may eventually impair increases in capillarity. This finding fits with earlier indications that LKB1-null fibroblasts and LKB1-null embryos have increased VEGF expression and that knockdown of LKB1 leads to increased VEGF expression and vascularity in cancer cells (6). Similarly, Zwetsloot et al. (53) found that VEGF expression after exercise was actually enhanced in AMPK-dominant negative mice. Thus, it would appear that LKB1's role in the regulation of VEGF is different in basal conditions vs. stressed conditions and that it may play both proangiogenic and antiangiogenic roles, perhaps through different downstream targets, depending on the intensity of stimulation.
A bout of muscle contractions results in altered expression of many genes. When repeated chronically, these changes lead to measurable changes in protein expression and muscle function that contribute to the performance adaptations that characterize the exercise training response. To determine whether acute exercise-induced changes in gene expression are dependent on LKB1, we induced a 15-min bout of electrically stimulated hindlimb muscle contractions in CON and skmLKB1-KO mice and assessed the expression of several genes 3 h after the contraction bout. Of the genes that we measured, PGC-1α, cytochrome c, and VEGF were upregulated in CON muscles after contraction, consistent with similar findings postexercise in previous reports (7, 9, 11, 24, 28). These genes were unaffected by exercise in skmLKB1-KO muscles, indicating that LKB1 is required for their expression after muscle contraction at this 3-h postexercise time point. Although protein and/or mRNA content of TFAM (10) and cytochrome b (49) are known to increase with chronic contractile activity, we observed no difference in their expression at 3 h poststimulation. Indeed, their expression may not increase until later time points after contraction (10, 38). The lack of responsiveness of HIF-1α expression to contraction is consistent with previous research (44). Interestingly, Jørgensen et al. (24) observed no difference in postexercise expression of PGC-1α between wild-type and AMPKα2-KO mice. These findings, together with ours, suggest that LKB1 can regulate exercise-induced gene expression through pathways separate from that of its best-characterized target, AMPKα2. In resting muscles, expressions of cytochrome b and TFAM were significantly lower in skmLKB1-KO mice, but no other differences between genotypes were observed for the genes that we measured, although there was a nonsignificant trend for decreased expression of VEGF. These findings in resting muscles are in general agreement with those of Miura et al. (33), who observed significantly lower gene expressions of COX-2, COX-4, and VEGF and a trend for decreased TFAM but no difference in PGC-1α expression in skmLKB1-DN mice.
The contraction bout resulted in a similar decrease in ATP, glycogen, and PCr between genotypes. Miura et al. (33) also reported a similar decrease in glycogen after treadmill running between skmLKB1-DN and WT mice, although in contrast to our findings they observed lower glycogen in resting muscles of skmLKB1-DN vs. WT mice (33). To determine how the impaired gene expression at 3 h postexercise in skmLKB1-KO muscles may have been affected by altered intracellular signaling, we measured phosphorylation of AMPK, p38 MAPK, Erk, and CaMKII. Each of these are well-characterized mediators of contraction-induced muscle adaptation. As expected, AMPK phosphorylation was elevated immediately after contraction only in CON but not skmLKB1-KO muscles. Although activation of AMPK has been reported to induce p38 and Erk phosphorylation and activation in skeletal muscle (5, 29), we observed no significant attenuation of either p38 or Erk phosphorylation immediately after contraction in skmLKB1-KO muscles, indicating that initial activation of these pathways is unaffected by LKB1 knockout. Interestingly, at 2 h postcontraction, Erk phosphorylation was significantly elevated in skmLKB1-KO but not CON muscles. Phosphorylation of p38 displayed a similar but nonsignificant (P = 0.17) trend. The potential hyperactivation of these signaling pathways after contraction may be a compensatory mechanism that could minimize the effects of impaired AMPK phosphorylation. Although this did not appear to play a major role in the expression of PGC-1α, cytochrome c, and VEGF at 3 h postexercise, expression of these and other exercise-induced genes would likely be affected by the enhanced or prolonged MAPK signaling at later time points. Together, these findings suggest that impaired upregulation of gene expression by muscle contraction at 3 h was likely due to AMPK and/or other LKB1-dependent pathways and not due to downregulation of Erk, p38, or CaMKII in skmLKB1-KO muscles.
In conclusion, skeletal muscle-specific knockout of LKB1 results in decreased exercise capacity and impaired ability to upregulate specific genes after acute exercise training. It likewise impairs accretion of mitochondrial genes involved in oxidative phosphorylation with longer-term training. However, accretion of VEGF with exercise training may normally be inhibited by LKB1. Despite the impairments in mitochondrial protein expression, the lack of LKB1 in skeletal muscle does not prevent or impair improvements in exercise capacity with training.
GRANTS
This work was supported by National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant R01-AR-051928.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.B.T. and D.M.T. contributed to the conception and design of the research; C.B.T., S.R.M., D.M.H., D.M.G., T.M.M., S.E.H., M.R.H., D.R.A., and D.M.T. performed the experiments; C.B.T., S.R.M., D.M.H., D.M.G., T.M.M., S.E.H., M.R.H., D.R.A., and D.M.T. analyzed the data; C.B.T., S.R.M., D.M.G., and D.M.T. interpreted the results of the experiments; C.B.T., S.R.M., D.M.H., D.M.G., D.R.A., and D.M.T. prepared the figures; C.B.T. and D.M.T. drafted the manuscript; C.B.T., S.R.M., D.M.H., D.M.G., T.M.M., S.E.H., M.R.H., D.R.A., and D.M.T. approved the final version of the manuscript; T.M.M., S.E.H., and D.M.T. edited and revised the manuscript.
ACKNOWLEDGMENTS
We are grateful to Dr. Mario Capecci and his laboratory for generously providing the Myf6-Cre mice used in this study. We are also grateful to Steven Anderson and Austin Finch for their assistance with animal husbandry.
REFERENCES
- 1.Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab 281: E1340–E1346, 2001 [DOI] [PubMed] [Google Scholar]
- 2.Breen EC, Johnson EC, Wagner H, Tseng HM, Sung LA, Wagner PD. Angiogenic growth factor mRNA responses in muscle to a single bout of exercise. J Appl Physiol 81: 355–361, 1996 [DOI] [PubMed] [Google Scholar]
- 3.Brown JD, Hancock CR, Mongillo AD, Benjamin Barton J, DiGiovanni RA, Parcell AC, Winder WW, Thomson DM. Effect of LKB1 deficiency on mitochondrial content, fibre type and muscle performance in the mouse diaphragm. Acta Physiol (Oxf) 201: 457–466, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carling D, Zammit VA, Hardie DG. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett 223: 217–222, 1987 [DOI] [PubMed] [Google Scholar]
- 5.Chen HC, Bandyopadhyay G, Sajan MP, Kanoh Y, Standaert M, Farese RV, Jr, Farese RV. Activation of the ERK pathway and atypical protein kinase C isoforms in exercise- and aminoimidazole-4-carboxamide-1-beta-d-riboside (AICAR)-stimulated glucose transport. J Biol Chem 277: 23554–23562, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Duivenvoorden WC, Beatty LK, Lhotak S, Hill B, Mak I, Paulin G, Gallino D, Popovic S, Austin RC, Pinthus JH. Underexpression of tumour suppressor LKB1 in clear cell renal cell carcinoma is common and confers growth advantage in vitro and in vivo. Br J Cancer 108: 327–333, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Egan B, Carson BP, Garcia-Roves PM, Chibalin AV, Sarsfield FM, Barron N, McCaffrey N, Moyna NM, Zierath JR, O'Gorman DJ. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J Physiol 588: 1779–1790, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fisher JS, Ju JS, Oppelt PJ, Smith JL, Suzuki A, Esumi H. Muscle contractions, AICAR, and insulin cause phosphorylation of an AMPK-related kinase. Am J Physiol Endocrinol Metab 289: E986–E992, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gavin TP, Westerkamp LM, Zwetsloot KA. Soleus, plantaris and gastrocnemius VEGF mRNA responses to hypoxia and exercise are preserved in aged compared with young female C57BL/6 mice. Acta Physiol (Oxf) 188: 113–121, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Gordon JW, Rungi AA, Inagaki H, Hood DA. Effects of contractile activity on mitochondrial transcription factor A expression in skeletal muscle. J Appl Physiol 90: 389–396, 2001 [DOI] [PubMed] [Google Scholar]
- 11.Gustafsson T, Puntschart A, Kaijser L, Jansson E, Sundberg CJ. Exercise-induced expression of angiogenesis-related transcription and growth factors in human skeletal muscle. Am J Physiol Heart Circ Physiol 276: H679–H685, 1999 [DOI] [PubMed] [Google Scholar]
- 12.Haldar M, Hancock JD, Coffin CM, Lessnick SL, Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell 11: 375–388, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271: 27879–27887, 1996 [DOI] [PubMed] [Google Scholar]
- 14.Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 242: 2278–2282, 1967 [PubMed] [Google Scholar]
- 15.Hong SP, Leiper FC, Woods A, Carling D, Carlson M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA 100: 8839–8843, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda S, Kizaki T, Haga S, Ohno H, Takemasa T. Acute exercise induces biphasic increase in respiratory mRNA in skeletal muscle. Biochem Biophys Res Commun 368: 323–328, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Ikeda Y, Sato K, Pimentel DR, Sam F, Shaw RJ, Dyck JR, Walsh K. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J Biol Chem 284: 35839–35849, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Irrcher I, Adhihetty PJ, Sheehan T, Joseph AM, Hood DA. PPARγ coactivator-1α expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am J Physiol Cell Physiol 284: C1669–C1677, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA 104: 12017–12022, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeppesen J, Maarbjerg SJ, Jordy AB, Fritzen AM, Pehmøller C, Sylow L, Serup AK, Jessen N, Thorsen K, Prats C, Qvortrup K, Dyck JR, Hunter RW, Sakamoto K, Thomson DM, Schjerling P, Wojtaszewski JF, Richter EA, Kiens B. LKB1 regulates lipid oxidation during exercise independently of AMPK. Diabetes 62: 1490–1499, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jessen N, Koh HJ, Folmes CD, Wagg C, Fujii N, Lofgren B, Wolf CM, Berul CI, Hirshman MF, Lopaschuk GD, Goodyear LJ. Ablation of LKB1 in the heart leads to energy deprivation and impaired cardiac function. Biochim Biophys Acta 1802: 593–600, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, Shah YM, Gonzalez FJ. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60: 2484–2495, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jørgensen SB, Treebak JT, Viollet B, Schjerling P, Vaulont S, Wojtaszewski JF, Richter EA. Role of AMPKα2 in basal, training-, and AICAR-induced GLUT4, hexokinase II, and mitochondrial protein expression in mouse muscle. Am J Physiol Endocrinol Metab 292: E331–E339, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Jørgensen SB, Wojtaszewski JF, Viollet B, Andreelli F, Birk JB, Hellsten Y, Schjerling P, Vaulont S, Neufer PD, Richter EA, Pilegaard H. Effects of alpha-AMPK knockout on exercise-induced gene activation in mouse skeletal muscle. FASEB J 19: 1146–1148, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li Y, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR, Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol 26: 8217–8227, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koh HJ, Toyoda T, Fujii N, Jung MM, Rathod A, Middelbeek RJ, Lessard SJ, Treebak JT, Tsuchihara K, Esumi H, Richter EA, Wojtaszewski JF, Hirshman MF, Goodyear LJ. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proc Natl Acad Sci USA 107: 15541–15546, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leick L, Fentz J, Bienso RS, Knudsen JG, Jeppesen J, Kiens B, Wojtaszewski JF, Pilegaard H. PGC-1α is required for AICAR-induced expression of GLUT4 and mitochondrial proteins in mouse skeletal muscle. Am J Physiol Endocrinol Metab 299: E456–E465, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Leick L, Wojtaszewski JF, Johansen ST, Kiilerich K, Comes G, Hellsten Y, Hidalgo J, Pilegaard H. PGC-1α is not mandatory for exercise- and training-induced adaptive gene responses in mouse skeletal muscle. Am J Physiol Endocrinol Metab 294: E463–E474, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Lemieux K, Konrad D, Klip A, Marette A. The AMP-activated protein kinase activator AICAR does not induce GLUT4 translocation to transverse tubules but stimulates glucose uptake and p38 mitogen-activated protein kinases alpha and beta in skeletal muscle. FASEB J 17: 1658–1665, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Li L, Pan R, Li R, Niemann B, Aurich AC, Chen Y, Rohrbach S. Mitochondrial biogenesis and peroxisome proliferator-activated receptor-gamma coactivator-1alpha (PGC-1alpha) deacetylation by physical activity: intact adipocytokine signaling is required. Diabetes 60: 157–167, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 23: 833–843, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGee SL, Mustard KJ, Hardie DG, Baar K. Normal hypertrophy accompanied by phosphoryation and activation of AMP-activated protein kinase alpha1 following overload in LKB1 knockout mice. J Physiol 586: 1731–1741, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miura S, Kai Y, Tadaishi M, Tokutake Y, Sakamoto K, Bruce CR, Febbraio MA, Kita K, Chohnan S, Ezaki O. Marked phenotypic differences of endurance performance and exercise-induced oxygen consumption between AMPK and LKB1 deficiency in mouse skeletal muscle: changes occurring in the diaphragm. Am J Physiol Endocrinol Metab 305: E213–E229, 2013 [DOI] [PubMed] [Google Scholar]
- 34.Moore C, Leu M, Muller U, Brenner HR. Induction of multiple signaling loops by MuSK during neuromuscular synapse formation. Proc Natl Acad Sci USA 98: 14655–14660, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakken GN, Jacobs DL, Thomson DM, Fillmore N, Winder WW. Effects of excess corticosterone on LKB1 and AMPK signaling in rat skeletal muscle. J Appl Physiol 108: 298–305, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Neill HM, Maarbjerg SJ, Crane JD, Jeppesen J, Jørgensen SB, Schertzer JD, Shyroka O, Kiens B, van Denderen BJ, Tarnopolsky MA, Kemp BE, Richter EA, Steinberg GR. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci USA 108: 16092–16097, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ouchi N, Shibata R, Walsh K. AMP-activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle. Circ Res 96: 838–846, 2005 [DOI] [PubMed] [Google Scholar]
- 37a.Park SH, Gammon SR, Knippers JD, Paulsen SR, Rubink DS, Winder WW. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J Appl Physiol 92: 2475–2482, 2002 [DOI] [PubMed] [Google Scholar]
- 38.Park JY, Wang PY, Matsumoto T, Sung HJ, Ma W, Choi JW, Anderson SA, Leary SC, Balaban RS, Kang JG, Hwang PM. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ Res 105: 705–712, 11 p following 712, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rockl KS, Hirshman MF, Brandauer J, Fujii N, Witters LA, Goodyear LJ. Skeletal muscle adaptation to exercise training: AMP-activated protein kinase mediates muscle fiber type shift. Diabetes 56: 2062–2069, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Rodriguez-Cuenca S, Monjo M, Gianotti M, Proenza AM, Roca P. Expression of mitochondrial biogenesis-signaling factors in brown adipocytes is influenced specifically by 17β-estradiol, testosterone, and progesterone. Am J Physiol Endocrinol Metab 292: E340–E346, 2007 [DOI] [PubMed] [Google Scholar]
- 41.Sakamoto K, McCarthy A, Smith D, Green KA, Grahame Hardie D, Ashworth A, Alessi DR. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO J 24: 1810–1820, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Srere PA. Citrate synthase. Methods Enzymol 13: 3–11, 1969 [Google Scholar]
- 43.Sullivan MJ, Green HJ, Cobb FR. Skeletal muscle biochemistry and histology in ambulatory patients with long-term heart failure. Circulation 81: 518–527, 1990 [DOI] [PubMed] [Google Scholar]
- 44.Tang K, Breen EC, Wagner H, Brutsaert TD, Gassmann M, Wagner PD. HIF and VEGF relationships in response to hypoxia and sciatic nerve stimulation in rat gastrocnemius. Respir Physiol Neurobiol 144: 71–80, 2004 [DOI] [PubMed] [Google Scholar]
- 45.Thomson DM, Brown JD, Fillmore N, Condon BM, Kim HJ, Barrow JR, Winder WW. LKB1 and the regulation of malonyl-CoA and fatty acid oxidation in muscle. Am J Physiol Endocrinol Metab 293: E1572–E1579, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Thomson DM, Hancock CR, Evanson BG, Kenney SG, Malan BB, Mongillo AD, Brown JD, Hepworth S, Fillmore N, Parcell AC, Kooyman DL, Winder WW. Skeletal muscle dysfunction in muscle-specific LKB1 knockout mice. J Appl Physiol 108: 1775–1785, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomson DM, Porter BB, Tall JH, Kim HJ, Barrow JR, Winder WW. Skeletal muscle and heart LKB1 deficiency causes decreased voluntary running and reduced muscle mitochondrial marker enzyme expression in mice. Am J Physiol Endocrinol Metab 292: E196–E202, 2007 [DOI] [PubMed] [Google Scholar]
- 48.Wadley GD, Choate J, McConell GK. NOS isoform-specific regulation of basal but not exercise-induced mitochondrial biogenesis in mouse skeletal muscle. J Physiol 585: 253–262, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams RS, Salmons S, Newsholme EA, Kaufman RE, Mellor J. Regulation of nuclear and mitochondrial gene expression by contractile activity in skeletal muscle. J Biol Chem 261: 376–380, 1986 [PubMed] [Google Scholar]
- 50.Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol Endocrinol Metab 270: E299–E304, 1996 [DOI] [PubMed] [Google Scholar]
- 51.Winder WW, Holmes BF, Rubink DS, Jensen EB, Chen M, Holloszy JO. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J Appl Physiol 88: 2219–2226, 2000 [DOI] [PubMed] [Google Scholar]
- 52.Xiao B, Sanders MJ, Underwood E, Heath R, Mayer FV, Carmena D, Jing C, Walker PA, Eccleston JF, Haire LF, Saiu P, Howell SA, Aasland R, Martin SR, Carling D, Gamblin SJ. Structure of mammalian AMPK and its regulation by ADP. Nature 472: 230–233, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zwetsloot KA, Westerkamp LM, Holmes BF, Gavin TP. AMPK regulates basal skeletal muscle capillarization and VEGF expression, but is not necessary for the angiogenic response to exercise. J Physiol 586: 6021–6035, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]