

Abstract
In the present study, we examine the hypothesis that the nitric oxide (NO) produced by endothelial NO synthase (eNOS) plays a protective role in the development of ANG II-induced hypertension and renal injury by minimizing oxidative stress and the inflammation induced by TNF-α. Systolic blood pressure (SBP) and renal injury responses to chronic infusions of ANG II (via implanted minipumps) were evaluated for 2 wk in wild-type (WT) and in eNOS knockout mice (KO) cotreated with or without a superoxide (O2−) scavenger, tempol (400 mg/l in the drinking water), or a TNF-α receptor blocker, etanercept (5 mg/kg/day ip). In study 1, when ANG II was given at a dose of 25 ng/min, it increased mean SBP in WT mice (Δ36 ± 3 mmHg; n = 7), and this effect was attenuated in mice pretreated with tempol (Δ24 ± 3 mmHg; n = 6). In KO mice (n = 9), this dose of ANG II resulted in severe renal injury associated with high mortality. To avoid this high mortality in KO, study 2 was conducted with a lower dose of ANG II (10 ng/min) that increased SBP slightly in WT (Δ17 ± 7 mmHg; n = 6) but exaggeratedly in KO (Δ48 ± 12 mmHg, n = 6) associated with severe renal injury. Cotreatment with either tempol (n = 6) or etanercept (n = 6) ameliorated the hypertensive, as well as the renal injury responses in KO compared with WT. These data demonstrate a protective role for eNOS activity in preventing renal inflammatory injury and hypertension induced by chronic increases in ANG II.
Keywords: endothelial nitric oxide synthase activity, tumor necrosis factor-α, superoxide, angiotensin II, hypertension, renal injury
hypertension is considered to be a low-grade inflammatory condition induced by various proinflammatory cytokines including TNF-α (35, 43, 44). Recent studies have implicated the involvement of TNF-α in the development of renal injury in hypertension induced by ANG II (12, 43, 44). A relationship between the renin-angiotensin system and the production of TNF-α, and its potential role in regulating cardiovascular function are increasingly evident from the findings of many recent studies (10, 18, 35, 49). TNF-α has been implicated in the development of glomerulonephritis (42) as well as salt-sensitive hypertension (12) induced by ANG II. It has been observed that chronic ANG II infusion fails to cause hypertensive responses in knockout mice lacking the gene for TNF-α (48) or its source, T-lymphocytes (10, 18). ANG II induced up-regulation of the protein expressions of NAD(P)H oxidase enzyme subunit, gp91phox, and endothelial nitric oxide synthase (eNOS) enzyme in the myocardium was absent in TNF-α gene knockout mice (49). These findings suggest an important modulatory role for this cytokine in the nitrosative and oxidative stress mechanisms induced by ANG II.
Redox equilibrium is critical for many aspects of cellular metabolism, signaling, and survival. Recent experimental evidence indicates that endogenous nitric oxide (NO) plays a critical role in the maintenance of this equilibrium state in cellular and organ function, particularly in the kidney (32). Most of this evidence suggests that there is a balance between the production of NO and superoxide (O2−) that regulates normal kidney function by opposing each other's action in the kidney. Any imbalance in the NO-O2− interaction leads to derangements in kidney function. Chronic administration of a subpressor dose of ANG II leads to the development of hypertension, and increases in oxidative stress in rats (26, 38, 41, 42). It is known that ANG II increases the production of O2− by activation of the NAD(P)H oxidase enzyme, which is an important source of O2− in the body (41). Although it has been postulated that oxidative stress plays a role in the development of ANG II-dependent hypertension, the exact mechanism involved in this pathophysiology is not yet clearly defined. ANG II administration also results in the production of NO primarily by activating AT2 receptors (6). Previous studies reported upregulation of eNOS activity in the renal tissue of ANG II-induced hypertensive rats (8). These findings strongly emphasize that the elucidation of complete interactions between ANG II, O2−, and NO is essential for understanding the renal mechanisms involved in the pathophysiology of ANG II-dependent hypertension.
We hypothesize that an increase in NO production by enhanced eNOS activity provides a protective role in the development of ANG II-induced hypertension and renal tissue injury by minimizing oxidative stress, and the proinflammatory activity of TNF-α. To examine this hypothesis, hypertensive and renal tissue injury responses to chronic administration of ANG II were evaluated in eNOS knockout mice (KO) with or without the coadministration of a O2− scavenger, tempol, or a TNF-α receptor blocker, etanercept. ANG II was administered at a dose of 25 ng/min in mice examined in the first series of experiments (study 1). However, this high dose of ANG II caused a high mortality rate in eNOS KO mice. Thus, the dose of ANG II was reduced to 10 ng/min in a later series of experiments (study 2).
METHODS
All of the experimental procedures described in this study were approved by and performed in accordance with the guidelines and practices established by the Tulane University Animal Care and Use Committee. This study was performed on male mice (8–10 wk of age) lacking the gene for eNOS, B6.129P2-NOSIII (KO), and their genetic background wild-type strain, C57BL/6J (WT), which were purchased from Jackson Laboratories (Bar Harbor, ME). These animals were housed in a temperature- and light-controlled room and allowed free access to a standard diet (Ralston-Purina, St. Louis, MO). The mice were kept in the facility for ∼7 days to become acclimatized prior to the start of the experimental protocol.
As mentioned earlier, two sets of experiments were conducted in these mice: In study 1, ANG II was administered at a dose of 25 ng/min in both KO and WT mice. However, this dose of ANG II caused marked deterioration and high mortality in KO mice (six out of nine mice died within 10 days of ANG II administration). Therefore, in the experiments for study 2, which were conducted later, the dose of ANG II was reduced to 10 ng/min to avoid the high mortality rate in the KO mice. This low dose of ANG II did not cause mortality in KO mice and, thus, allowed us to complete the protocol. This low dose of ANG II was administered in both WT and KO mice with or without the coadministration of a O2− scavenger, tempol (400 mg/l in the drinking water) or a TNF-α receptor blocker, etanercept (5 mg·kg−1·day−1 ip). The experimental mice are grouped as follows: In study 1 (n = 6 in each group), ANG II was given at a high (25 ng/min) dose (25 ng/min) in these mice: 1) WT+ANG II (25 ng/min), 2) WT+ANG II (25 ng/min) + tempol, 3) KO +ANG II (25 ng/min), and 4) KO+ANG II (25 ng/min)+ tempol. Most of the KO mice treated with ANG II alone had died during the treatment period, so three more mice were added to that group (n = 9). In study 2 (n = 6 in each group), ANG II was given at a low (10 ng/min) dose (10 ng/min) in these mice: 1) WT+ANG II (10 ng/min), 2) WT+ANG II (10 ng/min) + tempol, 3) WT+ANG II (10 ng/min) + etanercept, 4) WT+etanercept, 5) KO+ANG II (10 ng/min), 6) KO+ANG II (10 ng/min) + tempol, 7) KO+ANG II (10 ng/min) + etanercept, and 8) KO+etanercept.
Administration of ANG II doses.
ANG II was administered for 14 days to both WT and KO mice via placement of osmotic minipumps, (Durect, Cupertino, CA). Mice were anesthetized with isoflurane administered via inhalation. A 1-cm dorsal skin incision was performed, and the fascia was separated to facilitate subdural placement of the minipumps. The skin was sutured, and topical antiseptic was applied. Mice were placed on 12:12-h light-dark cycles and received food and water ad libitum throughout the study.
Blood pressure measurements in conscious mice.
Blood pressure measurements in these experimental mice in both study 1 and study 2 were performed using mainly the tail-cuff plethysmography technique. However, to compare the values obtained by this noninvasive technique with the values obtained by the surgically invasive technique of radiotelemetry, we implanted radiotelemeter probes in a separate series of experimental mice used in study 2. Figure 3 illustrates the comparison of these MAP measurements using these two devices in the different groups of mice. The results indicate that the values of the blood pressures recorded by these two techniques are comparable. For the measurements with the tail-cuff plethysmography technique, the mice were acclimatized for 3–5 days to the tail-cuff device (Visitech Systems, Apex, NC) prior to the implantation of the minipumps for ANG II infusion. Average mean arterial pressure (MAP) measured two consecutive days prior to minipump implantation was used as the control MAP value (considered as 0 day value). MAP was then measured on days 3, 7, 10, and 13 of the ANG II infusion period. For continuous radiotelemetry recordings of MAP, mice were implanted with radiotransmitters (TA11PA-C10; Data Sciences International, New Brighton, MN). For this implantation, mice were anesthetized with isoflurane administered by inhalation. A midline skin incision 2 cm long from chin to manubrium was performed to isolate the common carotid artery (29). A blunt trocar was passed from the neck incision to the abdominal region through the lateral aspect under the skin. The transmitter's catheter was placed into the common carotid artery. The transmitter body was placed under the skin in the abdominal region. The skin was sutured, and topical antiseptic was applied. Mice were placed on a 12:12-h light-dark cycle and received food and water ad libitum throughout the study (27). After 8–10 days of recovery, systolic blood pressure (SBP) and heart rate were monitored continuously using the telemetry data acquisition system (Data Sciences International).
Fig. 3.
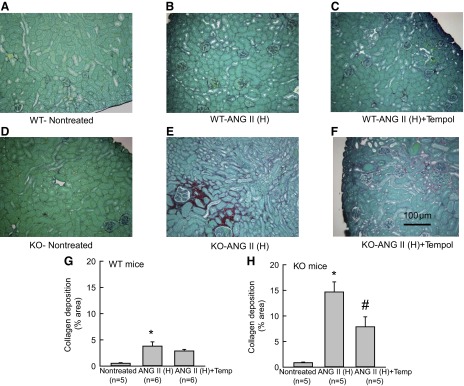
A–F: representative samples of Sirius-red staining of the renal sections collected from WT and KO mice treated with ANG II (H) (ANG II high dose; 25 ng/min). Microphotograph was taken using a 40× objective (Nikon Eclipse 50i), and images were captured with a digital camera (Nikon DS-Fil). G and H: mean optical density values of Sirius-red staining (extent of collagen deposition) of the renal sections collected from WT (G) and KO (25 ng/min) (H) mice. *P < 0.05 vs. corresponding nontreated group. #P < 0.05 vs. ANG II-treated group.
Urine collection in conscious mice.
24-h urine samples were collected in conscious mice using metabolic cages on the day before the start of the treatment (0 day) to measure baseline excretory parameters and then on the 7th and 13th days of the experimental period. Animals were housed individually in metabolic cages, and urine was collected for 24 h into sterile tubes. The drinking water mixed with tempol was changed every day, and covered bottles were used to minimize degradation by light. Urine volumes were determined from each urine collection, and samples were centrifuged (3,000 rpm/5 min; 4°C) and preserved for analysis. Urine samples were collected into tubes containing butylated hydroxytoluene (10 μl of 5 mg/ml solution in ethanol per 1-ml sample) to prevent further oxidative production of 8-isoprostane (20). Urine samples were preserved with 2-propanol (6.5%) to prevent the growth of bacteria that may degrade the nitrate/nitrite in the stored samples (20).
Renal tissue collection at the end of treatment period.
At the end of the 2-wk treatment period, mice were killed by an excess anesthetic dose of inactin (300 mg/kg), and the kidneys were isolated to collect tissue sections for histological examination. Upon sacrificing the mice, the kidneys were removed and washed two times in PBS. After the second wash, the kidneys were fixed in formalin (1:10 buffered dilution). The fixed kidneys were then embedded in paraffin blocks. These paraffin blocks were then used to cut sections that were mounted onto slides for histological analysis. The kidneys from the KO mice treated with a high dose of ANG II (study 1), which died prior to 2 wk of treatment, were collected within an hour of their death and used for histological measurements.
Analytical methods and statistics.
Urinary concentrations of sodium and potassium were assessed by flame photometry. The extent of glomerulosclerosis (GS) was determined by periodic acid Schiff (PAS) staining of these tissue sections (14). Image analysis was performed using a computer software program (NIS Elements software; Nikon Instruments, Melville, NY). Renal interstitial fibrosis (collagen deposition) was determined by either Sirius-red staining (study 1) or Gömöri's trichrome staining (study 2) of renal tissues. For Sirius-red staining, tissue slides were stained in saturated picric acid with 0.1% Sirius red F3BA (Aldrich Chemical, St. Louis, MO) (16). For trichrome staining, slides were stained in Weigert's iron hematoxylin solution. Image analysis was performed using a computer software program (NIS Elements software, Nikon Instruments).
Results are expressed as means ± SE. Statistical analysis was conducted by using Sigma-Stat software (Systat, Chicago, IL). Statistical analysis within groups was conducted by the use of the repeated-measures ANOVA and Dunnett's multiple comparison test. Percentage changes in the responses within the groups were compared with paired t-test. P < 0.05 is considered as significant.
RESULTS
Study 1: responses to administration of ANG II (25 ng/min) in WT and KO mice.
Basal mean SBP was higher in KO (n = 9) than WT mice (n = 6) (135 ± 2 vs. 121 ± 3 mmHg; P < 0.05). ANG II infusion at the high dose (H; 25 ng/min) caused gradual increases in SBP (157 ± 2 mmHg at 13th day) in WT mice. Figure 1, A and C illustrates the changes in SBP and mortality rate in these WT mice due to chronic ANG II (25 ng/min) administration. Overall, this dose was well tolerated with no apparent deterioration in the condition of the WT mice and with no significant changes in body weight during the 2 wks of treatment. Co-administration of tempol with ANG II (25 ng/min) in WT mice (n = 6) slightly but significantly (P < 0.05) attenuated this increase in SBP (146 ± 3 mmHg), and all the mice survived in good condition. However, ANG II (25 ng/min) infusion caused a high mortality rate in KO mice within the 2-wk treatment period as mentioned earlier. Beginning on the 3rd day of ANG II (25 ng/min) infusion, the KO mice gradually became lethargic, their body weight decreased markedly, and they began to die from the 6th day onward. By the end of the 2-wk treatment period, seven out of nine KO mice had died. Although SBP was measured in all surviving mice, measurement by the tail-cuff plethysmography was not possible in some of the mice due to their extremely deteriorated condition. Figure 1, A–D illustrates the changes in SBP and mortality rates in these mice. It was observed that ANG II (25 ng/min) failed to cause significant increases in SBP in the KO mice that survived during the 2-wk period. Urine output was reduced as the ANG II (25 ng/min)-treated KO mice could not take food and water due to their deteriorated condition. Therefore, urine collection from the metabolic cages was hampered in some of the mice with extremely deteriorated condition. Interestingly, cotreatment with tempol markedly reduced the mortality rate in ANG II (25 ng/min)-treated KO mice; only 1 out of 6 mice died by the 12th day of treatment. Fig. 1, C and D illustrates the changes in SBP and mortality rates in these tempol-treated mice. Tempol cotreatment also improved food and water intake with increased urinary output in these mice. The average values of renal excretory parameters are depicted in Table 1. Chronic ANG II (25 ng/min) infusion in WT mice increased urine flow and sodium excretion on the 13th day, which might be related to an increase in SBP in mice. Tempol cotreatment in these ANG II (25 ng/min)-treated WT mice attenuated the rise in SBP, and thus, there are insignificant changes in urine flow or sodium excretion. However, ANG II (25 ng/min) treatment in KO mice with or without tempol cotreatment did not cause any significant changes in the urine flow or sodium excretion on the 7th or 13th days.
Fig. 1.

Responses to chronic (2 wks) administration of high-dose (25 ng/min) of ANG II with or without tempol on systolic blood pressure (SBP) and mortality rate in wild-type (WT; A and B) and endothelial NOS knockout (eNOS KO; C and D). *P < 0.05, #P < 0.1 vs. WT-ANG II group.
Table 1.
Renal excretory responses to chronic infusion of a high dose (25 ng/min) of ANG II (ANG II-H) with or without tempol (Temp) in wild-type and eNOS knockout mice
| Animal groups | WT–ANG II-H |
WT–ANG II-H +Temp |
KO–ANG II-H |
KO–ANG II-H +Temp |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day of Treatment | 0 | 7th | 13th | 0 | 7th | 13th | 0 | 7th | 13th | 0 | 7th | 13th |
| Urine flow, mL/day | 1.82 ± 0.19 | 1.67 ± 0.39 | 4.49 ± 1.42* | 1.42 ± 0.22 | 2.20 ± 0.20* | 2.12 ± 0.40 | 1.22 ± 0.16 | 1.36 ± 0.41 | No urine- All animals died | 1.56 ± 0.28 | 2.09 ± 0.22 | 1.01 ± 0.28 |
| UNaV, mmol/day | 0.48 ± 0.04 | 0.33 ± 0.07 | 0.81 ± 0.26* | 0.17 ± 0.02 | 0.19 ± 0.02 | 0.21 ± 0.04 | 0.31 ± 0.03 | 0.26 ± 0.08 | 0.15 ± 0.03 | 0.20 ± 0.03 | 0.09 ± 0.03 | |
| UKV, mmol/day | 1.11 ± 0.12 | 0.60 ± 0.13 | 0.84 ± 0.21 | 0.27 ± 0.03 | 0.56 ± 0.12 | 0.63 ± 0.14 | 0.64 ± 0.06 | 0.43 ± 0.15 | 0.21 ± 0.04 | 0.26 ± 0.04 | 0.23 ± 0.05 | |
UNaV, urinary sodium excretion; UKV, urinary potassium excretion; WT, wild type; KO, knockout.
P < 0.05 vs. baseline values at −1 day.
Histological analysis of the kidneys from these mice revealed that there was severe renal injury (glomerulosclerosis and interstitial fibrosis) in ANG II (25 ng/min)-treated KO but not in WT. This renal injury was markedly prevented in the tempol-treated group (Fig. 2 and 3). The extent of GS was assessed by PAS staining and measured as the percent area of glomerulus covered by the stain. Compared with the untreated WT group, the percent area of GS was more than 10-fold higher in the ANG II (25 ng/min)-treated WT group (1.3 ± 0.23 vs. 15.0 ± 0.76; P < 0.001). Tempol cotreatment lowered this GS index (4.36 ± 0.41; P < 0.01) in the ANG II (25 ng/min)-treated WT group. The baseline GS value was slightly higher in the untreated KO (2.45 ± 0.17; P < 0.05) group compared with the untreated WT group. However, ANG II (25 ng/min) caused massive increases in GS (Fig. 2) in the kidney though SBP was not altered significantly (Fig. 1C). In the ANG II (25 ng/min)-treated KO group, the percent GS value was markedly increased to 33.42 ± 2.32 (P < 0.001). Tempol cotreatment reduced it to 11.80 ± 0.26 (P < 0.001). The extent of collagen deposition (interstitial fibrosis) in the renal tissue was assessed by Sirius red staining and measured as the percent area covered by the color staining of the collagen present in the tissue (Fig. 3). Compared with the untreated group, the average percent area was significantly higher in the ANG II (25 ng/min)-treated WT group (0.53 ± 0.11 to 3.8 ± 0.81; P < 0.01). Tempol cotreatment with ANG II (25 ng/min) did not significantly lower this value (2.87 ± 0.30) from the values in the group treated with ANG II (25 ng/min) alone. However, the baseline value was not significantly higher in the untreated KO (0.86 ± 1.2) group compared with that in the untreated WT group. In the ANG II (25 ng/min)-treated KO group, this percent staining was much higher (14.68 ± 1.97; P < 0.001) than in the ANG II (25 ng/min)-treated WT group. This staining was greatly reduced (7.88 ± 1.94; P < 0.05) in the ANG II (25 ng/min) and tempol cotreated KO mice.
Fig. 2.
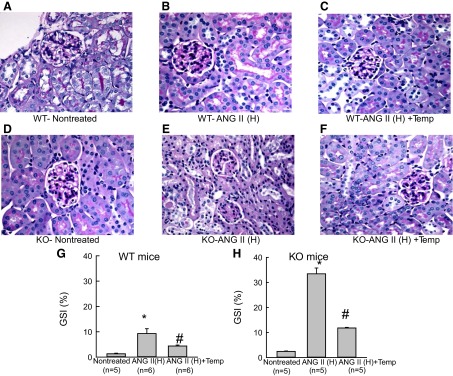
A–F: Representative samples of periodic acid Schiff (PAS) staining of the renal sections collected from wild-type (WT) and eNOS knockout (KO) mice treated with ANG II (H) (ANG II high dose, 25 ng/min). Microphotograph was taken using a 40× objective (Nikon Eclipse 50i), and images were captured with a digital camera (Nikon DS-Fil). G and H: mean value of glomerulosclerosis index (GSI) determined from PAS staining of the renal sections collected from WT (G) and KO (25 ng/min) (H) mice. *P < 0.05 vs. corresponding nontreated group; #P < 0.05 vs. ANG II-treated group.
Study 2: responses to administration of ANG II (10 ng/min) in WT and KO mice.
Figures 4, A and B illustrate the response of mean systemic arterial pressure (MAP) to a lower dose of ANG II (10 ng/min) in these mice. The MAP values depicted in Fig. 4A were taken from the experiments using tail-cuff methods, and the values depicted in Fig. 4B were taken from the experiments using radio telemetry devices in mice. The MAP values obtained from these two methods were more or less identical. Thus, in all other experiments, MAP was measured using the tail-cuff method, as this made it possible to perform collections of 24-h urine samples on the day before the start (0 day), 7th, and 13th days of the treatment period using metabolic cages. As illustrated in both Fig. 4, A and B, basal MAP was slightly higher in KO than WT (111 ± 1 vs. 100 ± 2 mmHg; P < 0.05). However, with this low dose, MAP increased modestly in WT (100 ± 2 to 117 ± 6 mmHg; P < 0.05; n = 6) but increased markedly in KO mice (111 ± 1 to 160 ± 13 mmHg, P < 0.001; n = 6). The percent changes in MAP from the baseline values at 0 day of treatment in different groups of mice were illustrated in Fig. 4A (responses in WT mice) and in Fig. 4B (responses in KO mice). Cotreatment with tempol or etanercept during ANG II (10 ng/min) administration ameliorated this hypertensive response in KO mice (tempol group: 107 ± 2 to 114 ± 5 mmHg; n = 6 and etanercept group: 110 ± 1 to 108 ± 8 mmHg; n = 6). Tempol treatment alone did not cause any appreciable changes in the WT or KO mice. Although etanercept treatment alone also did not cause any significant change in SBP in WT mice, SBP was significantly decreased in KO mice (111 ± 1 to 91 ± 2 mmHg; P < 0.01; n = 4) after 2 wk of etanercept treatment. The average values of renal excretory parameters are depicted in Tables 2 and 3. Although ANG II treatment in WT mice at the high dose (25 ng/min) increases urine flow and sodium excretion on day 13, the low dose (10 ng/min) treatment did not cause such changes, as there were smaller increases in MAP in these mice (Fig. 5). It was noted that etanercept treatment significantly attenuated V and UNaV on the 13th day in WT mice. Although the changes were not statistically significant, a similar trend in attenuation of V and UNaV was also seen with etanercept treatment in KO mice. As TNF-α induces diuresis and natriuresis in the kidney (44), such responses to etanercept treatment were not entirely unexpected. Etanercept treatment also showed significant attenuation of ANG II-induced UNoxV in WT but not in KO mice. The results also showed that etanercept treatment reduced UIsoV responses to chronic ANG II (25 ng/min) in KO but not in WT mice. It is noted that there are some variations in the basal levels of UNoxV or UIsoV in different groups of mice, as these were examined separately at different time periods. However, the responses to drug treatments in each group are considered by comparing the experimental values with the baseline values in the same group.
Fig. 4.

Comparison of the responses on mean arterial pressure (MAP) in WT and KO mice treated with ANG II low dose [10 ng/min; ANG II (L)], recorded by tail-cuff plethysmography (A) and implanted radiotelemetry (B). Note that the measurements recorded by these two methods are mostly identical. *P < 0.05 vs. corresponding baseline values prior to the start of drug treatment (at −1 day). #P < 0.05 between baseline values these two strains (WT and KO mice).
Table 2.
Renal excretory responses to chronic infusion of a low dose (10 ng/min) of ANG II-L in wild-type mice
| Animal groups | WT–ANG II-L (n = 6) |
WT–ANG II-L+Temp (n = 6) |
WT–ANG II-L+ETN (n = 6) |
WT–ETN (n = 6) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day of Treatment | 0 | 7th | 13th | 0 | 7th | 13th | 0 | 7th | 13th | 0 | 7th | 13th |
| Urine flow, ml/day | 1.69 ± 0.23 | 1.30 ± 0.29 | 1.60 ± 0.30 | 1.31 ± 0.14 | 1.30 ± 0.14 | 1.17 ± 0.14 | 1.74 ± 0.15 | 1.14 ± 0.17 | 1.12 ± 0.48 | 1.84 ± 0.17 | 1.15 ± 0.11 | 0.65 ± 0.13* |
| UNaV, mmol/day | 0.15 ± 0.015 | 0.15 ± 0.03 | 0.12 ± 0.02 | 0.16 ± 0.01 | 0.18 ± 0.01 | 0.15 ± 0.02 | 0.20 ± 0.02 | 0.17 ± 0.03 | 0.08 ± 0.01* | 0.20 ± 0.02 | 0.17 ± 0.03 | 0.08 ± 0.01* |
| UKV, mmol/day | 0.43 ± 0.13 | 0.37 ± 0.07 | 0.37 ± 0.08 | 0.31 ± 0.05 | 0.41 ± 0.04 | 0.40 ± 0.05 | 0.51 ± 0.10 | 0.29 ± 0.02 | 0.12 ± 0.03 | 0.53 ± 0.04 | 0.26 ± 0.02 | 0.70 ± 0.01 |
| UNOxV, μmol/day | 0.27 ± 0.10 | 0.39 ± 0.15 | 0.74 ± 0.37* | 0.41 ± 0.09 | 0.56 ± 0.06 | 0.93 ± 0.30* | 1.25 ± 0.21 | 0.89 ± 0.35 | 0.75 ± 0.15 | 1.55 ± 0.40 | 1.81 ± 0.68 | 2.53 ± 0.57 |
| UISOV, ng/day | 1.0 ± 0.2 | 1.8 ± 0.4* | 1.5 ± 0.4* | 1.9 ± 0.1 | 2.5 ± 0.6 | 2.1 ± 0.3 | 1.7 ± 0.5 | 2.1 ± 0.7 | 3.3 ± 0.9* | 1.3 ± 0.3 | 1.5 ± 0.1 | 2.3* ± 0.3 |
Temp, tempol; ETN, etanercept; UNOxV, urinary nitrate/nitrite excretion; UISOV, urinary isoprostane excretion; Temp, tempol; ETN, etanercept.
P < 0.05 vs. baseline values at −1 day.
Table 3.
Renal excretory responses to chronic infusion of a low dose (10 ng/min) of ANG II (ANG II-L) in KO (eNOS KO mice)
| Animal groups | KO–ANG II-L (n = 6) |
KO–ANG II-L +Temp (n = 6) |
KO–ANG II-L+ETN (n = 6) |
KO–ETN (n = 4) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Day of Treatment | 0 | 7th | 13th | 0 | 7th | 13th | 0 | 7th | 13th | 0 | 7th | 13th |
| Urine flow, ml/day | 1.16 ± 0.17 | 1.12 ± 0.21 | 1.47 ± 0.19 | 1.37 ± 0.11 | 2.06 ± 0.12* | 1.76 ± 0.14 | 1.64 ± 0.31 | 1.9 ± 0.3 | 1.14 ± 0.12 | 1.08 ± 0.13 | 1.01 ± 0.17 | 0.77 ± 0.08 |
| UNaV, mmol/day | 0.13 ± 0.01 | 0.13 ± 0.02 | 0.17 ± 0.02 | 0.16 ± 0.01 | 0.22 ± 0.02 | 0.18 ± 0.01 | 0.16 ± 0.01 | 0.2 ± 0.03 | 0.15 ± 0.03 | 0.14 ± 0.003 | 0.14 ± 0.02 | 0.16 ± 0.02 |
| UKV, mmol/day | 0.33 ± 0.05 | 0.32 ± 0.06 | 0.36 ± 0.03 | 0.32 ± 0.02 | 0.41 ± 0.04 | 0.40 ± 0.05 | 0.45 ± 0.04 | 0.41 ± 0.05 | 0.37 ± 0.03 | 0.36 ± 0.05 | 0.36 ± 0.09 | 0.33 ± 0.05 |
| UNOxV, μmol/day | 0.77 ± 0.26 | 0.86 ± 0.28 | 0.63 ± 0.14 | 0.40 ± 0.06 | 0.84 ± 0.09* | 0.71 ± 0.04* | 0.78 ± 0.18 | 0.45 ± 0.10 | 0.41 ± 0.05 | 0.64 ± 0.06 | 0.57 ± 0.19 | 0.49 ± 0.08 |
| UISOV, ng/day | 1.1 ± 0.1 | 1.4 ± 0.2 | 2.0 ± 0.2* | 1.7 ± 0.2 | 2.7 ± 0.4* | 1.8 ± 0.1 | 2.0 ± 0.2 | 2.6 ± 0.1 | 2.7 ± 0.4 | 1.5 ± 0.2 | 2.3 ± 0.5 | 1.6 ± 0.3 |
P < 0.05 vs. baseline values at −1 day.
Fig. 5.

Percent changes in mean arterial pressure (MAP) from corresponding baseline values in WT (wild type; A) and KO (eNOS knockout; B) mice treated with ANG II (L) (10 ng/min), tempol and etanercept (ETN) recorded by tail-cuff plethysmography. *P < 0.05 vs. corresponding baseline values prior to the start of drug treatment (at −1 day).
In study 2, histological analysis of the kidneys from only the ANG II (10 ng/min) and etanercept-treated groups was performed. It was not possible to analyze the tissue samples collected from the tempol-treated groups due to technical faults during collection. However, as the tissue injury responses to tempol cotreatment are assessed in study 1, we did not repeat these experiments in study 2, as these would be expected to provide qualitatively similar results as in study 1. Overall, the extent of renal injury (glomerulosclerosis and interstitial fibrosis) during low-dose administration of ANG II (10 ng/min) in study 2 was somewhat lower than (though qualitatively similar to) that observed in study 1. However, it was observed that these injuries were markedly reduced in the etanercept cotreated group (Figs. 6 and 7). As illustrated in Fig. 6, the values of GS percent area were not statistically different between sham control groups of WT (1.61 ± 0.22) and KO (1.88 ± 0.33). ANG II (10 ng/min) treatment increased percent GS value in WT (12.84 ± 2.95; P < 0.001) mice but more so in KO mice (16.1 ± 1.8; P < 0.02). Cotreatment with etanercept in the ANG II (10 ng/min)-treated group markedly reduced the GS value in the WT (4.21 ± 0.96; P < 0.001) group but less so in the KO group (10.24 ± 1.09; P < 0.05). The extent of collagen deposition (interstitial fibrosis) in renal tissue was assessed by trichrome staining and measured as the percent area covered by the color staining of the collagen present in the tissue. As illustrated in Fig. 7, the average percent area was higher in the ANG II (10 ng/min)-treated WT group (0.90 ± 0.38 vs. 3.98 ± 0.42; P < 0.05) compared with the untreated group. Etanercept cotreatment with ANG II (10 ng/min) did not alter this value (3.29 ± 0.81) in WT mice. However, compared with untreated WT mice, the baseline value was slightly higher in the untreated KO mice (1.70 ± 0.53; P < 0.05). In the ANG II (10 ng/min)-treated KO mice, this percent collagen deposition was much higher (13.97 ± 1.79; P < 0.001). Etanercept cotreatment reduced this value to 6.93 ± 0.95 (P < 0.05) in the ANG II (10 ng/min) treated KO group.
Fig. 6.
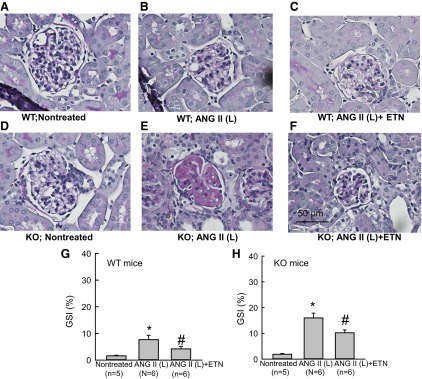
A–F: representative samples of PAS staining of the renal sections collected from WT and KO mice treated with ANG II (L) (10 ng/min). Microphotograph was taken using a 40× objective (Nikon Eclipse 50i), and images were captured with a digital camera (Nikon DS-Fil). G and H: mean value of glomerulosclerosis index (GSI) determined from PAS staining of the renal sections collected from WT (G) and KO (25 ng/min) mice. *P < 0.05 vs. corresponding nontreated group. #P < 0.05 vs. ANG II-treated group.
Fig. 7.
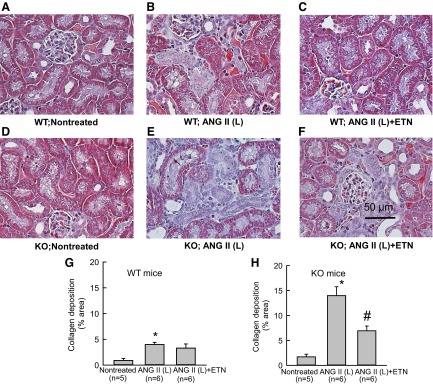
A–F: representative samples of trichrome staining of the renal sections collected from WT and KO mice treated with ANG II (L) (10 ng/min). Microphotograph was taken using a 40× objective (Nikon Eclipse 50i), and images were captured with a digital camera (Nikon DS-Fil). G and H: mean optical density values of trichrome staining (extent of collagen deposition) of the renal sections collected from WT (G) and KO (25 ng/min) (H) mice. *P < 0.05 vs. corresponding nontreated group. #P < 0.05 vs. ANG II-treated group.
DISCUSSION
The present study demonstrates that KO mice lacking the gene for eNOS have exaggerated hypertensive and renal injury (glomerulosclerosis and interstitial collagen deposition) responses to chronic administration of ANG II (both in high or low doses) compared with the corresponding dose in WT mice. ANG II at the higher dose (25 ng/min) induced a high mortality rate, but the lower dose (10 ng/min) was more tolerable in these KO mice. These exaggerated hypertensive and renal injury responses to the doses (high or low) of ANG II administration for 2 wk in KO mice were ameliorated by cotreatment with a O2− scavenger, tempol, or a TNF-α receptor blocker, etanercept. A high mortality rate (∼80%) during a higher dose of ANG II (25 ng/min) administration for 2 wk in KO mice was also prevented by coadministration of tempol. Basal blood pressure in KO mice was higher than that in WT mice, which was ameliorated by etanercept treatment alone. Thus, the results indicate that an enhancement of TNF-α activity is linked to higher MAP in eNOS KO mice. Recent findings in our laboratory (48) also show that the renal level of TNF-α is markedly higher (approximately twofold) in eNOS KO mice compared with that in WT mice. Similar to this, it was observed that the systemic NO inhibition by acute infusion of l-NAME resulted in twofold to three-fold increases in plasma as well as renal levels of proinflammatory cytokines, including TNF-α. (47). Earlier in vitro experiments also demonstrated that NOS blockade induces an increase in the production of TNF-α in the myocardium (54), as well as in cultured monocytes (55) and macrophages (22). However, it is noted that etanercept treatment caused minimal changes in blood pressure in both treated and untreated WT mice with a low dose of ANG II (10 ng/min). As endogenous TNF-α production is influenced by the status of oxidative stress (1, 29, 46, 47), which is enhanced during NO deficiency (34), it is likely that the TNF-α activity would be greater in KO than in WT mice. Thus, it is expected that the effect of etanercept treatment would be more prominent in KO mice than in WT mice, as seen in the present study. Collectively, these results demonstrate that functional eNOS activity provides a protective role in preventing oxidative stress and inflammatory responses induced by ANG II. Among the various isoforms of NOS enzymes, eNOS is considered to be the main source of ANG II-induced NO production (21). Although a renoprotective role for NO in the kidney during acute or chronic administration of ANG II was reported earlier (9, 30, 33, 39, 40), the results of the present study confirm that the eNOS isoform is the primary source of NO that exerts this protective effect.
As the hypertension induced by ANG II is attenuated by tempol or etanercept cotreatment, it could be argued that the beneficial effects on the renal injury associated with antagonism of oxidative stress or inflammation is simply due to reduction in blood pressure. Although this possibility may not be entirely ruled out, it was observed that the high doses of ANG II failed to cause significant increases in SBP in KO mice that survived (Fig. 1C), yet caused massive increases in glomerulosclerosis (Fig. 2) and collagen deposition (Fig. 3) in the kidney. Moreover, tempol cotreatment did not cause any significant alteration in SBP in the high dose ANG II (25 ng/min)-treated KO mice (Fig. 1C) but did cause marked attenuation of renal injury (Figs. 2 and 3). These findings indicate a dissociation of changes in blood pressure with changes in renal pathology in these KO mice. Such dissociation between the changes in renal perfusion pressure and in renal morphological injury was not at all unexpected as many previous studies have demonstrated that the reduction in blood pressure by hydralazine or hydrochlorothiazide failed to prevent ANG II-induced renal injury in many animal models of hypertension (1, 15, 25, 36, 37). It was also demonstrated that etanercept treatment can markedly reduce renal injury changes without affecting blood pressure in hypertensive rats induced by DOCA salt (11) or ANG II with high-salt intake (12). Thus, it is likely that the beneficial effects of tempol or etanercept in attenuating renal injury in KO mice were mostly independent of the changes in blood pressure.
In the present study, it has been observed that etanercept treatment alone deceases urine flow and sodium excretion in WT mice. This antidiuretic and antinatriuretic response to TNF-α antagonism is expected, as it has been shown earlier that TNF-α induces potent diuretic and natriuretic responses (45). However, it could be argued that eNOS-induced NO production is involved in the natriuretic response to TNF-α as etanercept has a minimal effect on sodium excretion in KO mice. This possibility is unlikely, as we have demonstrated previously that TNF-α induced natriuresis is independent of NO (45, 46) and occurs mainly via inhibiting tubular ENaC activity (31). As KO mice exhibit a higher renal level of TNF-α than WT mice (48), it is possible that a higher dose of etanercept is needed to observe the expected antinatriuretic effect in KO mice. It was also noted that the blood pressure remains mostly unaffected despite significant reductions in urine flow and sodium excretion in response to chronic etanercept treatment in WT mice. Although the reason for this lack of blood pressure effect in response to chronic salt retention is not clear, it should be emphasized that maintenance of normal blood pressure depends on the interaction between many intrarenal and extrarenal factors maintaining body homeostasis. In the present study, the possible increase in blood pressure due to enhancement of sodium retention by etanercept treatment may be counteracted by an adjustment in peripheral resistance through intact eNOS activity in WT mice.
The mechanism of how eNOS activity provides an inhibitory influence on the TNF-α activity induced by ANG II is not yet clear. However, it is known that NO has powerful anti-inflammatory properties and endogenous production of NO plays an important role in the regulation of various inflammatory cytokines, including TNF-α (7, 51). Although there is evidence to suggest that TNF-α induces eNOS activity (49), endogenous TNF-α production was also known to be influenced by the oxidative stress status in the tissue (1, 17, 43, 47). Apart from the recent findings in our laboratory demonstrating that the TNF-α level is considerably higher in mice treated with an NO inhibitor (47) and in KO mice lacking the gene for eNOS enzyme (48), reports are also available from other laboratories (2, 3, 4, 7), indicating that the production of TNF-α and other inflammatory cytokines is regulated by NOS activity. Among many cell types, monocytes and macrophages are considered to be important cellular sources for various inflammatory cytokines. Although direct evidence is not yet available, it has been reported in a recent in vitro study (54) that application of asymmetric dimethylarginine (ADMA; an endogenous NO synthase inhibitor compound) can induce TNF-α production in monocytes. In addition, inhibition of NOS by Nω-nitro-l-arginine induces TNF-α production in a dose-dependent manner in rat cardiomyocytes (54), as well as cultured macrophage cells (22). Although the underlying molecular mechanism by which NOS deficiency induces TNF-α release in response to ANG II was not evaluated in the present study, a specific role of NF-κB and p38 MAP kinase pathways has been recently implicated in this phenomenon (53, 55). ANG II has been reported to increase TNF-α production in monocytes (19), in the thick ascending limb of the Loop of Henle in the kidney (13), and in the mammalian heart (23). Increased levels of TNF-α have been reported in many forms of experimental hypertension, such as ANG II-dependent hypertension (13), Dahl salt-sensitive hypertension (14), DOCA-salt induced hypertension (11), and l-NAME-induced hypertension (5). It has also been shown that chronic ANG II administration failed to cause a hypertensive response in TNF-α knockout mice (49), as well as in T-lymphocyte-deleted mice (10, 18). Collectively, these studies suggest that TNF-α plays a key role in the pathogenesis of hypertension and cardiovascular disease. Future studies are warranted to identify the mechanistic link between eNOS activity and its inhibitory action in cells and/or organs that are prone to the production of inflammatory mediators.
An increased level of plasma TNF-α has been observed in many acute and chronic pathophysiological conditions in which endogenous NO production is generally compromised, such as chronic l-NAME-induced hypertension (5) and many other models of hypertension (12, 17, 18). A direct relationship between NOS deficiency and increases in TNF-α level has been reported in many recent in vivo (2, 4) as well as in vitro studies (54, 55). However, in experimental models representing the condition of sepsis, TNF-α has been reported to stimulate the inducible form of NOS (iNOS) or a blockade of NOS actually suppressed TNF-α production (24, 52), rather than being suppressed by endogenous NO, as observed in the present study. It should be noted here that conditions, such as septicemia (or LPS-induced sepsis), are associated with the production of NO in large amounts, which is mostly mediated by enhanced iNOS activity (24). The iNOS isoform is known to be induced only in the pathological conditions and is generally not expressed under the basal conditions. Therefore, the consequences of blockade of NOS in such conditions should be different from those in the basal condition as in the present experimental settings. Therefore, the mechanism of TNF-α production and its interactions with NOS activity may vary depending on the underlying physiological and pathophysiological conditions.
In conclusion, the data from the present experiments demonstrate that the functional eNOS activity provides a protective role in preventing oxidative stress and inflammatory cytokine-mediated renal injury and hypertension induced by increases in ANG II level.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: C.W., A.C., and M.Z.H. performed experiments; C.W. and M.Z.H. analyzed data; C.W., A.C., M.Z.H., and D.S.A.M. prepared figures; C.W. drafted manuscript; C.W. and D.S.A.M. edited and revised manuscript; C.W., A.C., M.Z.H., and D.S.A.M. approved final version of manuscript; D.S.A.M. conception and design of research; D.S.A.M. interpreted results of experiments.
ACKNOWLEDGMENTS
We acknowledge the technical help of Dr. Miguel Graciano in analyzing renal tissue PAS and Sirius-red staining employed in this study. This study was supported by National Heart Lung and Blood Institute Grant HL-66432 and Tulane Enhancement Fund.
REFERENCES
- 1.Agarwal D, Elks CM, Reed SD, Mariappan N, Majid DSA, Francis J. Chronic exercise preserves renal structures and hemodynamics in spontaneously hypertensive rats. Antioxid Redox Signal 16: 139–152, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alayan J, Ivanovski S, Gemmell E, Ford P, Hamlet S, Farah CS. Deficiency of iNOS contributes to Porphyromonas gingivalis-induced tissue damage. Oral Microbiol Immunol 21: 360–365, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Araujo M, Welch WJ. Oxidative stress and nitric oxide in kidney function. Curr Opin Nephrol Hypertens 15: 72–77, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Bougaki M, Searles RJ, Kida K, Yu J, Buys ES, Ichinose F. Nos3 protects against systemic inflammation and myocardial dysfunction in murine polymicrobial sepsis. Shock 34: 281–290, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bourraindeloup M, Adamy C, Candiani G, Cailleret M, Bourin MC, Badoual T, Su JB, Adubeiro S, Roudot-Thoraval F, Dubois-Rande JL, Hittinger L, Pecker F. N-acetylcysteine treatment normalizes serum tumor necrosis factor-α level and hinders the progression of cardiac injury in hypertensive rats. Circulation 110: 2003–2009, 2004 [DOI] [PubMed] [Google Scholar]
- 6.Carey RM. Update on the role of the AT2 receptor. Curr Opin Nephrol Hypertens 14: 67–71, 2005 [DOI] [PubMed] [Google Scholar]
- 7.Coleman JW. Nitric oxide: a regulator of mast cell activation and mast cell-mediated inflammation. Clin Exp Immunol 129: 4–10, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chin SY, Pandey KN, Shi SJ, Kobori H, Moreno C, Navar LG. Increased activity and expression of Ca2+-dependent NOS in renal cortex of ANG II-infused hypertensive rats. Am J Physiol Renal Physiol 277: F797–F804, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chin SY, Wang CT, Majid DS, Navar LG. Renoprotective effects of nitric oxide in angiotensin II-induced hypertension in the rat. Am J Physiol Renal Physiol 274: F876–F882, 1998 [DOI] [PubMed] [Google Scholar]
- 10.De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 298: R1136–R1142, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elmarakby AA, Quigley JE, Imig JD, Pollock JS, Pollock DM. TNF-α inhibition reduces renal injury in DOCA-salt hypertensive rats. Am J Physiol Regul Integr Comp Physiol 294: R76–R83, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elmarakby AA, Quigley JE, Pollock DM, Imig JD. Tumor necrosis factor alpha blockade increases renal Cyp2c23 expression and slows the progression of renal damage in salt-sensitive hypertension. Hypertension 47: 557–562, 2006 [DOI] [PubMed] [Google Scholar]
- 13.Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC. Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Physiol Renal Physiol 274: F148–F155, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Fujihara CK, Malheiros DM, Zatz R, Noronha IL. Mycophenolate mofetil attenuates renal injury in the rat remnant kidney. Kidney Int 54: 1510–1519, 1998 [DOI] [PubMed] [Google Scholar]
- 15.Fujimoto H, Satoh M, Horike H, Hatta H, Haruna Y, Kobayashi S, Namikoshi T, Arakawa S, Tomita N, Kashihara N. Olmesartan ameliorates progressive glomerular injury in subtotal nephrectomized rats through suppression of superoxide production. Hypertens Res 31: 305–313, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Grimm PC, Nickerson P, Gough J, McKenna R, Stern E, Jeffery J, Rush DN. Computerized image analysis of Sirius Red-stained renal allograft biopsies as a surrogate marker to predict long-term allograft function. J Am Soc Nephrol 14: 1662–1668, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Gu JW, Tian N, Shparago M, Tan W, Bailey AP, Manning RD., Jr Renal NF-κB activation and TNF-α upregulation correlate with salt-sensitive hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 291: R1817–R1824, 2006 [DOI] [PubMed] [Google Scholar]
- 18.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hahn AW, Jonas U, Bühler FR, Resink TJ. Activation of human peripheral monocytes by angiotensin II. FEBS Lett 347: 178–180, 1994 [DOI] [PubMed] [Google Scholar]
- 20.Haque MZ, Majid DS. High salt intake delayed angiotensin II-induced hypertension in mice with a genetic variant of NADPH oxidase. Am J Hypertens 24: 114–118, 2011 [DOI] [PubMed] [Google Scholar]
- 21.Islam MT, Agarwal D, Francis J, Matrougui K, Majid DSA. Inhibition of nitric oxide synthase enhances the production of tumor necrosis factor-α in macrophage cells. FASEB J 25: A1030.7, 2011 [Google Scholar]
- 22.Kalra D, Sivasubramanian N, Mann DL. Angiotensin II induces tumor necrosis factor biosynthesis in the adult mammalian heart through a protein kinase C-dependent pathway. Circulation 105: 2198–2205, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Kirkebøen KA, Strand OA. The role of nitric oxide in sepsis—an overview. Acta Anaesthesiol Scand 43: 275–288, 1999 [DOI] [PubMed] [Google Scholar]
- 24.Kobori H, Ozawa Y, Suzaki Y, Nishiyama A. Enhanced intrarenal angiotensinogen contributes to early renal injury in spontaneously hypertensive rats. J Am Soc Nephrol 16: 2073–2080, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kopkan L, Castillo A, Navar LG, Majid DS. Enhanced superoxide generation modulates renal function in angiotensin II-induced hypertensive rats. Am J Physiol Renal Physiol 290: F80–F86, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Kopkan L, Hess A, Husková Z, Cervenka L, Navar LG, Majid DS. High-salt intake enhances superoxide activity in eNOS knockout mice leading to the development of salt sensitivity. Am J Physiol Renal Physiol 299: F656–F663, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kopkan L, Husková Z, Vanourková Z, Thumová M, Skaroupková P, Cervenka L, Majid DS. Superoxide and its interaction with nitric oxide modulates renal function in prehypertensive Ren-2 transgenic rats. J Hypertens 25: 2257–2265, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Liu F, Wei CC, Wu SJ, Chenier I, Zhang SL, Filep JG, Ingelfinger JR, Chan JSD. Apocynin attenuates tubular apoptosis and tubulointestinal fibrosis in transgenic mice independent of hypertension. Kidney Int 75: 156–166, 2009 [DOI] [PubMed] [Google Scholar]
- 29.López B, Salom MG, Arregui B, Valero F, Fenoy FJ. Role of superoxide in modulating the renal effects of angiotensin II. Hypertension 42: 1150–1156, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Majid DS. Tumor necrosis factor-α, and kidney function: Experimental findings in mice. Adv Exp Med Biol 691: 471–480, 2011 [DOI] [PubMed] [Google Scholar]
- 31.Majid DS, Kopkan L. Nitric oxide and superoxide interactions in the kidney and their implication in the development of salt-sensitive hypertension. Clin Exp Pharmacol Physiol 34: 946–952, 2007 [DOI] [PubMed] [Google Scholar]
- 32.Majid DS, Nishiyama A, Jackson KE, Castillo A. Superoxide scavenging attenuates renal responses to ANG II during nitric oxide synthase inhibition in anesthetized dogs. Am J Physiol Renal Physiol 288: F412–F419, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Majid DS, Nishiyama A, Jackson KE, Castillo A. Inhibition of nitric oxide synthase enhances superoxide activity in canine kidney. Am J Physiol Regul Integr Comp Physiol 287: R27–R32, 2004 [DOI] [PubMed] [Google Scholar]
- 34.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension 48: 149–156, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Mervaala E, Müller DN, Schmidt F, Park JK, Gross V, Bader M, Breu V, Ganten D, Haller H, Luft FC. Blood pressure-independent effects in rats with human renin and angiotensinogen genes. Hypertension 35: 587–594, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Nakamura T, Obata JE, Kimura H, Ohno S, Yoshida Y, Kawachi H, Shimizu F. Blocking angiotensin II ameliorates proteinuria and glomerular lesion in progressive mesangioproliferative glomerulonephritis. Kidney Int 55: 877–889, 1999 [DOI] [PubMed] [Google Scholar]
- 37.Ortiz MC, Manriquez MC, Romero JC, Juncos LA. Antioxidants block angiotensin II-induced increases in blood pressure and endothelin. Hypertension 38: 655–659, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Patterson ME, Mullins JJ, Mitchell KD. Renoprotective effects of neuronal NOS-derived nitric oxide and cyclooxygenase-2 metabolites in transgenic rats with inducible malignant hypertension. Am J Physiol Renal Physiol 294: F205–F211, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Patzak A, Steege A, Lai EY, Brinkmann JO, Kupsch E, Spielmann N, Gericke A, Skalweit A, Stegbauer J, Persson PB, Seeliger E. Angiotensin II response in afferent arterioles of mice lacking either the endothelial or neuronal isoform of nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol 294: R429–R437, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Pech V, Sikka SC, Sindhu RK, Vaziri ND, Majid DS. Oxidant stress and blood pressure responses to angiotensin II administration in rats fed varying salt diets. Am J Hypertens 19: 534–540, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Reckelhoff JF, Romero JC. Role of oxidative stress in angiotensin-induced hypertension. Am J Physiol Regul Integr Comp Physiol 284: R893–R912, 2003 [DOI] [PubMed] [Google Scholar]
- 42.Rodríguez-Iturbe B, Franco M, Tapia E, Quiroz Y, Johnson RJ. Renal inflammation, autoimmunity and salt-sensitive hypertension. Clin Exp Pharmacol Physiol 10: 1440–1681, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruiz-Ortega M, Ruperez M, Lorenzo O, Esteban V, Blanco J, Mezzano S, Egido J. Angiotensin II regulates the synthesis of proinflammatory cytokines and chemokines in the kidney. Kidney Int Suppl 82: S12–S22, 2002 [DOI] [PubMed] [Google Scholar]
- 44.Shahid M, Francis J, Majid DSA. Tumor necrosis factor-alpha induces renal vasoconstriction as well as natriuresis in mice. Am J Physiol Renal Physiol 295: F1836–F1844, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shahid M, Francis J, Matrougui K, Majid DSA. Involvement of TNF-α in the natriuretic response to systemic infusion of nitric oxide synthase inhibitor in anesthetized mice. Am J Physiol Renal Physiol 299: F217–F224, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Singh P, Castillo A, Johnson RA, Majid DSA. Reduction in interleukin-10 level in plasma and in renal tissue during systemic inhibition of nitric oxide synthase in anesthetized mice. Hypertension 60: A182, 2012 [Google Scholar]
- 47.Singh P, Stephenson R, Majid DSA. Changes in plasma, and renal tissue levels of inflammatory cytokines during chronic high salt intake in wildtype and eNOS knockout mice. J Invest Med 61; 484: A392, 2013 [Google Scholar]
- 48.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-α in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51: 1345–1351, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stegbauer J, Kuczka Y, Vonend O, Quack I, Sellin L, Patzak A, Steege A, Langnaese K, Rump LC. Endothelial nitric oxide synthase is predominantly involved in angiotensin II modulation of renal vascular resistance and norepinephrine release. Am J Physiol Regul Integr Comp Physiol 294: R421–R428, 2008 [DOI] [PubMed] [Google Scholar]
- 50.Thomassen MJ, Buhrow LT, Connors MJ, Kaneko FT, Erzurum SC, Kavuru MS. Nitric oxide inhibits inflammatory cytokine production by human alveolar macrophages. Am J Respir Cell Mol Biol 17: 279–283, 1997 [DOI] [PubMed] [Google Scholar]
- 51.Yamaguchi H, Kidachi Y, Umetsu H, Ryoyama K. l-NAME inhibits tumor cell progression and pulmonary metastasis of r/m HM-SFME-1 cells by decreasing NO from tumor cells and TNF-α from macrophages. Mol Cell Biochem 312: 103–112, 2008 [DOI] [PubMed] [Google Scholar]
- 52.Wang HD, Xu S, Johns DG, Du Y, Quinn MT, Cayatte AJ, Cohen RA. Role of NADPH oxidase in the vascular hypertrophic and oxidative stress response to angiotensin II in mice. Circ Res 88: 947–953, 2001 [DOI] [PubMed] [Google Scholar]
- 53.Wenzel S, Rohde C, Wingerning S, Roth J, Kojda G, Schlüter KD. Lack of endothelial nitric oxide synthase-derived nitric oxide formation favors hypertrophy in adult ventricular cardiomyocytes. Hypertension 49: 193–200, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Zhang GG, Bai YP, Chen MF, Shi RZ, Jiang DJ, Fu QM, Tan GS, Li YJ. Asymmetric dimethylarginine induces TNF-α production via ROS/NF-κB-dependent pathway in human monocytic cells and the inhibitory effect of reinioside C. Vascul Pharmacol 48: 115–121, 2008 [DOI] [PubMed] [Google Scholar]