

Abstract
Aldosterone increases tubular Na+ absorption largely by increasing α-epithelial Na+ channel (αENaC) transcription in collecting duct principal cells. How aldosterone reprograms basal αENaC transcription to high-level activity in the collecting duct is incompletely understood. Promoter methylation, a covalent but reversible epigenetic process, has been implicated in the control of gene expression in health and disease. We investigated the role of promoter methylation/demethylation in the epigenetic control of basal and aldosterone-stimulated αENaC transcription in mIMCD3 collecting duct cells. Bisulfite treatment and sequencing analysis after treatment of the cells with the DNA methyltransferase (DNMT) inhibitor 5-aza-2′-deoxycytidine (5-Aza-CdR) identified clusters of methylated cytosines in a CpG island near the transcription start site of the αENaC promoter. 5-Aza-CdR treatment or small interfering RNA-mediated knockdown of DNMT3b or methyl-CpG-binding domain protein (MBD)-4 derepressed basal αENaC transcription, indicating that promoter methylation suppresses basal αENaC transcription. Aldosterone triggered a time-dependent decrease in 5mC and DNMT3b and a concurrent enrichment in 5-hydroxymethylcytosine (5hmC) and ten-eleven translocation (Tet)2 at the αENaC promoter, consistent with active demethylation. 5-Aza-CdR mimicked aldosterone by enhancing Sp1 binding to the αENaC promoter. We conclude that DNMT3b- and MBD4-dependent methylation of the αENaC promoter limits basal αENaC transcription, in part by limiting Sp1 binding and trans-activation. Aldosterone stimulates the dispersal of DNMT3b and recruitment of Tet2 to demethylate the αENaC promoter to induce αENaC transcription. These results disclose a novel epigenetic mechanism for the control of basal and aldosterone-induced αENaC transcription that adds to previously described epigenetic controls exerted by histone modifications.
Keywords: epigenetic, chromatin, transcription factor, methylcytosine, methyltransferase, epithelial Na+ channel
the epithelial na+ channel (ENaC) mediates Na+ entry across the apical membranes of salt-absorbing epithelia of the kidney, distal colon, and lung and functions as the rate-limiting step in active transepithelial Na+ and fluid absorption. In serving this role, ENaC plays important roles in the regulation of extracellular fluid volume and blood pressure (1, 20). Of the three ENaC subunits (α, β, and γ), αENaC appears to be critical to the overall salt balance, as evidenced by the finding that mice with targeted inactivation of αENaC in the connecting tubule (CNT)/collecting duct (CD) exhibit severe renal salt wasting characteristic of a pseudohypoaldosteronism type I phenotype (5). αENaC is also a molecular target of aldosterone, which stimulates its transcription in a manner that is rate limiting for the full induction of ENaC activity in the CD in animal models. Aldosterone treatment or secondary hyperaldosteronism induced by a low-Na+ diet increases the abundance of ENaC and does so by augmenting αENaC gene transcription without increasing β- or γ-subunit expression or reducing αENaC mRNA turnover (15).
In the decade since aldosterone was first discovered to stimulate αENaC transcription in CD cells (15), a complex model of transcriptional αENaC activation has evolved. A promoter-reporter study (11) of the αENaC gene in CD cells revealed the involvement of a glucocorticoid responsive element at −811 in the aldosterone response, leading to the assumption that aldosterone activation of αENaC gene transcription was solely due to the action of aldosterone, liganded to the mineralocorticoid receptor (MR), acting at this glucocorticoid responsive element. However, mice with CNT/CD-specific MR inactivation (19) failed to develop the severe salt-wasting phenotype observed with CNT/CD-specific ablation of αENaC in these same segments (5), indicating the large contribution of MR-independent pathways in αENaC gene regulation.
Subsequently, we discovered additional aldosterone-sensitive pathways for the genetic and epigenetic control of αENaC gene transcription that involve basal trans-activation mediated by the transcription factor Sp1 binding to a cis-element at +222/+229 of αENaC, counterbalanced by aldosterone-sensitive epigenetic transcriptional constraints. The epigenetic repression/derepression mechanisms thus far described include combinatorial interactions of histone methyltransferase disruptor of telomeric silencing (Dot)1a with either the putative DNA-binding protein ALL-1 fused gene with chromosome 9 (Af9) acting at an Af9 element at +78/+92 of αENaC (26–29) or sirtuin (Sirt)1 (25). Dot1a hypermethylates histone H3 K79 at the αENaC promoter to limit basal αENaC transcription in CD cells in vitro and in vivo, with the latter evidenced by the fact that CNT/CD-specific ablation of Dot1 in mice results in an upregulation of renal αENaC mRNA expression (29). Aldosterone, through suppression of Dot1a, Af9, and Sirt1 abundance and through serum- and glucocorticoid-induced kinase-1-mediated phosphorylation of Af9 and consequent disruption of the Dot1/Af9 complex at the αENaC promoter, results in the derepression of αENaC transcription in a MR-independent manner (28). At the same time, aldosterone enhances Sp1- (23) and MR-mediated (11, 25) trans-activation in CD cells. Interestingly, aldosterone enhances Sp1 occupation of the αENaC promoter without changing the nuclear abundance of the transcription factor (23), suggesting that other context-dependent factors influence its binding and/or activity at the αENaC promoter.
In the mammalian genome, ∼70% of CpG sites are methylated (6), and these sites are often clustered in CpG islands within promoter regions DNA. Promoter methylation, a covalent but reversible epigenetic process, plays critical roles in cell-specific gene expression during differentiation, development, and disease (13, 14). DNA methylation typically results in a chromatin configuration that represses transcription, and it involves DNA methyltransferase (DNMT)-mediated transfer of a methyl group from S-adenyl methionine to the fifth carbon of cytosine, forming 5-methylcytosine (5mC) (21). DNMT1, DNMT3a, and DNMT3b are the principal mediators of the establishment and maintenance of DNA methylation in mammals. DNMT3a and DNMT3b mediate de novo DNA methylation, whereas DNMT1 acts on newly synthesized DNA to maintain methylation marks (21). Transcription may be limited by an effect of promoter methylation itself to reduce the affinity for trans-activating factors and by methyl-CpG binding domain (MBD) proteins, which recognize 5′-methylated cytosine (5mC) residues, recruiting transcriptional repressors or chromatin modifiers to these sites (16) and stearically hindering transcription factor binding (12). Moreover, DNMT3b and MBD4 have been shown to physically and functionally interact in a complex with negative vitamin D response element-binding protein to initiate and maintain methylation of the cytochrome P-450 (CYP)27B1 promoter (10).
Demethylation, with its consequent transcriptional derepression, occurs via passive or active processes. Although the precise mechanisms for the erasure of cytosine methylation remain controversial, recent studies have demonstrated that target gene activation is often accompanied by the early conversion of 5mC to 5-hydroxymethylcytosine (5hmC) catalyzed by the ten-eleven translocation (Tet) family of dioxygenases. Since MBDs bind 5mC but not 5hmC (9), Tet-mediated conversion of 5mC → 5hmC limits the binding of MBDs, and potentially MBD-binding partners, such as DNMT3b, to facilitate passive demethylation. In addition, Tet-mediated generation of 5hmC has also been proposed to initiate a base excision repair process that results in demethylation (12).
Promoter methylation/demethylation represents a potential switch for the prompt conversion of basally constrained transcription to hormonally induced transcription. Our analysis of the murine αENaC promoter region using CpG Island Finder (http://dbcat.cgm.ntu.edu.tw) revealed a dense CpG island at −5/+359 within the R3 subregion of the αENaC promoter (Fig. 1), the principal region we have determined to control MR-independent αENaC transcription (23, 28, 29). This finding prompted us to hypothesize that regulation of the cytosine methylation status of the R3 subregion could contribute to the epigenetic control of basal and aldosterone-stimulated αENaC transcription in mIMCD3 cells and might help to explain the aldosterone-induced enhancement of Sp1 binding and trans-activation at the αENaC promoter we previously observed in these cells (23). We discovered that basal cytosine methylation is dependent on the actions of DNMT3b and MBD4, which serve to constrain basal αENaC transcription. Aldosterone treatment disperses DNMT3b from the αENaC R3 subregion and recruits Tet2 to convert 5mC → 5hmC at this subregion to contribute to the derepression of αENaC transcription. Aldosterone-dependent demethylation of the αENaC promoter also facilitates Sp1 binding to the promoter to allow further trans-activation of the αENaC gene. Taken together with our earlier work, these results provide new evidence for DNA-histone cross-talk in basal and aldosterone-stimulated αENaC transcription.
Fig. 1.

Map of the R3 subregion of the α-epithelial Na+ channel (αENaC) 5′-flanking region. The nucleotide span of the subregion, the CpG Island, and Sp1 and ALL-1 fused gene with chromosome 9 (Af9) elements are schematized. The methylated cytosines [5-methylcytosine (5mc)] identified by comparison of bisulfite-treatment and DNA sequence analysis of 5-aza-2′-deoxycytidine (5-Aza-CdR)-treated and untreated mIMCD3 cells (see results and Fig. 2) are also indicated.
MATERIALS AND METHODS
Reagents and plasmids.
Aldosterone and 5-aza-2′-deoxycytidine (5-Aza-CdR) were from Sigma (St. Louis, MO). SYBR GreenER quantitative (q)PCR SuperMix Universal and Lipofectamine 2000 reagent were purchased from Invitrogen (Carlsbad, CA). The plasmids pGL3-basic-1.3αENaC, which contains the murine αENaC promoter fused to the gene encoding firefly luciferase, and pcDNA3.1-Zeo-1.3αENaC-Luc, which contains the 1.3-kb αENaC promoter and luciferase-coding cassette from pGL3-basic-1.3αENaC subcloned into pcDNA3.1-Zeo, have been previously described (26–28). Antibodies used included those directed against Sp1 and Tet1 (Millipore, Temecula, CA), 5mC and 5hmC (Epigentek, Farmingdale, NY), and DNMT3b, Tet2, and Tet3 (Abcam, Cambridge, MA).
mIMCD3 cell culture and aldosterone treatment.
Wild-type mIMCD3 cells and mIMCD3 cells stably transfected with pcDNA3.1-Zeo-1.3αENaC-Luc (26–28) were cultured at 37°C in a 5% CO2 environment in DMEM-F-12 plus 10% FBS. The stable cell lines were maintained under Zeocin selection. For aldosterone treatment experiments, cells were cultured in medium of the same composition except containing 10% charcoal-stripped FBS for at least 24 h before the addition of 1 μm aldosterone or 0.01% ethanol as a vehicle control. For time-course experiments, the values for aldosterone-treated samples were normalized to those of vehicle-treated time control samples.
Transient transfection, RNA interference, and promoter activity assays.
In transient overexpression experiments, cells were transiently transfected with a MBD4 expression plasmid or its empty vector using Lipofectamine 2000 reagent (Life Technologies) as previously detailed (23, 24, 28, 29). The level of overexpression was monitored by qRT-PCR. Small interfering (si)RNA knockdown was performed using Lipofectamine 2000 reagent (Life Technologies) and control and gene-specific siRNAs for DNMT1, DNMT3a, DNMT3b, MBD4, Tet1, Tet2, and Tet3. Successful knockdown of the target genes was assayed by qRT-PCR and ranged from a 50% to 70% reduction compared with cells transfected with scrambled control siRNA (data not shown). The specificity control was GAPDH, and its expression was unaffected by any of the siRNAs. Luciferase activity of the stable mIMCD3 cell lines expressing pcDNA3.1-Zeo-1.3αENaC-Luc was assayed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI) and was normalized to cell protein content as previously detailed (23, 24, 28, 29). For transient transfection of promoter-firefly luciferase constructs, the Renilla luciferase reporter pRL-SV40 was cotransfected as an internal control. The firefly luciferase activity of each sample was normalized to its Renilla luciferase activity to generate the reported promoter activity.
Chromatin immunoprecipitation/qPCR and qRT-PCR.
Chromatin immunoprecipitation (ChIP)/qPCR and 5mC or 5hmC immunoprecipitation/qPCR were performed and analyzed essentially as previously described (23, 26, 28) except that antibodies directed against Sp1, 5mC, 5hmC, DNMT3b, Tet1, Tet2, and Tet3 were used and only the R3 subregion (−57 to +438) of the αENaC promoter was examined. RT-qPCR to measure αENaC and β-actin mRNA levels in miMCD3 cells used previously described methods and primers (18).
Sodium bisulfite modification and genomic sequencing.
mIMCD3 cells were treated with 0.5 μg/ml 5-Aza-CdR or vehicle for 24 h, and genomic DNA was extracted using the DNAeasy kit (QIAGEN) according to the manufacturer's protocol. Sodium bisulfite modification of the DNA and purification of the modified DNA was performed as we have previously described (22). The αENaC promoter region −1327 to +438 in the bisulfite-reacted DNA was PCR amplified and cloned into the pCR4-TOPO10 vector (Invitrogen). Clones with appropriate sized inserts were sequenced.
Data analysis.
Quantitative data are expressed as means ± SEM and were analyzed for statistical significance by one-way ANOVA or Student's t-test as appropriate. P values of <0.05 were taken as significant.
RESULTS
The DNA methylation inhibitor 5-Aza-CdR demethylates the αENaC promoter and basally augments endogenous αENaC mRNA expression and αENaC promoter activity.
To identify CpGs in the αENaC promoter that are methylated under basal conditions in mIMCD3 cells, we treated cells with vehicle or the DNA methylation inhibitor 5-Aza-CdR, extracted genomic DNA, and subjected the DNA to sodium bisulfite treatment and DNA sequence analysis. Sodium bisulfite deaminates unmethylated cytosine to uracil in single-stranded DNA under conditions in which 5mC remains nonreactive. Since 5mC is resistant to sodium bisulfite treatment, it remains unchanged, allowing it to be distinguished from unmethylated cytosines within the sequenced fragment. Thus, all cytosines remaining at the time of sequencing represent cytosines that were methylated (that is, all 5mCs) in the original DNA sequence. Sequencing analysis of −1327 to +438 of αENaC from the bisulfite-treated samples showed 5mC residues, clustered in a CpG island in the proximal promoter region at −99, −91, +13, +22, +29, +32, +38, +48, +215, +216, and +224, that were demethylated in the presence of 5-Aza-CdR (Figs. 1 and 2). A representative DNA sequencing graph of +238 to +207, which includes the +222/+229 Sp1 element, is shown in Fig. 2. We (23) have previously demonstrated that this Sp1 element mediates trans-activation of the αENaC promoter.
Fig. 2.

Sodium bisulfite modification and genomic sequencing of the αENaC promoter reveals cytosine methylation within and near the +222/+229 Sp1 site. mIMCD3 cells were treated with vehicle or 0.5 μg/ml 5-Aza-CdR for 24 h. Genomic DNA was harvested and exposed to sodium bisulfite to modify methylated cytosine to uracil. DNA sequencing was then performed. A representative (n = 3) sequencing graph of the +238/+207 region is shown. Arrows indicate modified and unmodified cytosines. The red boxes outline the +222/+229 Sp1 element.
Given the evidence for basal hypermethylation of the R3 subregion of the αENaC promoter and the effectiveness of 5-Aza-CdR in erasing cytosine methylation, we hypothesized that hypermethylation of this subregion could constrain basal αENaC gene expression and be relieved by demethylation. Accordingly, we used qRT-PCR to measure basal and aldosterone-stimulated αENaC mRNA levels in mIMCD3 cells that had been treated with vehicle or 5-Aza-CdR. As shown in Fig. 3A, 5-Aza-CdR-treated mIMCD3 cells exhibited a basal level of αENaC mRNA that was 56% greater than control cells. The trend toward higher levels of αENaC mRNA in aldosterone- and 5-Aza-CdR-treated cells compared with vehicle- and 5-Aza-CdR-treated cells did not reach statistical significance (Fig. 3A). To determine whether the stimulatory effect of 5-Aza-CdR on αENaC mRNA expression was mediated at the transcriptional level, previously characterized mIMCD3 cell lines stably expressing pcDNA3.1-Zeo-1.3αENaC-Luc were treated with vehicle or aldosterone with or without 5-Aza-CdR, and αENaC promoter activity was measured. In agreement with the mRNA results, cells treated with 5-Aza-CdR exhibited basal αENaC promoter activity that was 48% greater than control cells (Fig. 3B). The numeric differences in aldosterone-stimulated αENaC promoter activity between vehicle- and 5-Aza-CdR-treated cells did not achieve statistical significance (Fig. 3B). These data suggest that promoter hypermethylation represses and hypomethylation derepresses basal αENaC promoter activity in mIMCD3 cells.
Fig. 3.

The DNA methylation inhibitor 5-Aza-CdR augments αENaC mRNA expression and αENaC promoter activity in mIMCD3 cells. A: mIMCD3 cells were treated with vehicle or 0.5 μg/ml 5-Aza-CdR, together with vehicle (−Aldo) or 1 μM aldosterone (+Aldo) for 24 h. A: RNA was then prepared for quantitative (q)RT-PCR to measure αENaC mRNA levels. n = 6. *P < 0.05 vs. vehicle (−Aldo). B: mIMCD3 cells stably transfected with pcDNA3.1-Zeo-1.3αENaC-Luc were treated as described in A, and cell lysates were then prepared for luciferase activity measurements, which were normalized to cell protein content. n = 6. *P < 0.05 vs. vehicle (−Aldo).
Fig. 5.

Small interfering (si)RNA knockdown of DNMT3b induces basal αENaC promoter activity in mIMCD3 cells. mIMCD3 cells stably transfected with pcDNA3.1-Zeo-1.3αENaC-Luc were transiently transfected with control siRNA or with DNA methyltransferase (DNMT)1-, DNMT3a-, or DNMT3b-specific siRNAs. Cells were then treated with vehicle or Aldo for 24 h, and luciferase activity, which was normalized to cell protein content, was measured. n = 6. *P < 0.05 vs. vehicle-treated control siRNA.
Cytosine methylation limits Sp1 binding to the αENaC promoter.
Cytosine methylation in the promoter region, when present within regulatory elements, could potentially interfere with the binding of specific transcription factors that bind these motifs. We (23) have previously demonstrated that +222/+229 is the exclusive site for Sp1 binding to the R3 subregion and contributes to basal and aldosterone-stimulated trans-activation of the αENaC gene. One perplexing observation in this previous study was that aldosterone triggered additional Sp1 association with the αENaC promoter without increasing Sp1 expression levels in the nucleus. Since cytosines neighboring and within the Sp1 element were methylated under basal conditions (Figs. 1 and 2), one potential explanation for the aldosterone-induced enrichment of Sp1 at the αENaC promoter would be that basal cytosine methylation limits Sp1 binding and that promoter demethylation relieves this constraint. Therefore, we tested whether 5-Aza-CdR demethylation of the αENaC promoter enhances Sp1 binding to this region in the context of chromatin. ChIP/qPCR assays for Sp1 binding to the R3 subregion were performed in mIMCD3 cells that had been treated with vehicle or 5-Aza-CdR, as in the mRNA expression, promoter, and bisulfite treatment assays. As shown in Fig. 4, cells treated with 5-Aza-CdR exhibited ∼75% enhanced enrichment of Sp1 at the R3 subregion of the αENaC promoter compared with vehicle-treated control cells, an effect equivalent to that observed with aldosterone treatment of the cells (see Fig. 8A in Ref. 23).
Fig. 4.

Cytosine methylation inhibits Sp1 binding to the αENaC promoter. mIMCD3 cells were treated in the presence of vehicle (v..) or the DNA methylation inhibitor 5-Aza-CdR (5..) as described in Fig. 3. Chromatin immunoprecipitation (ChIP)/qPCR analysis of the R3 subregion of the αENaC gene using anti-Sp1 antibodies (or nonimmune IgG as a negative control) was performed (n = 3). Error bars indicate ±SEs. *P < 0.05 vs. vehicle.
Fig. 8.
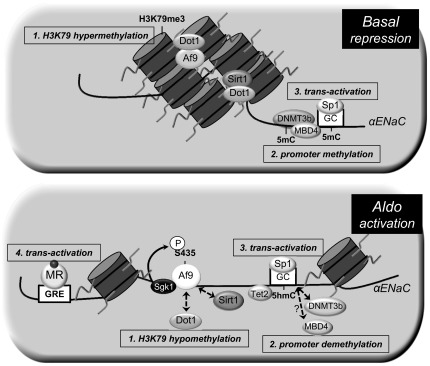
Model of the genetic and epigenetic control of basal and Aldo-stimulated transcription of the αENaC gene in mIMCD3 cells. Under basal conditions (top), the histone methyltransferase disruptor of telomeric silencing (Dot)1a is complexed with the DNA-binding protein Af9 or sirtuin (Sirt)1 in chromatin associated with specific regions of the αENaC 5′-flanking region and hypermethylates histone H3K79 (1). In addition, DNMT3b and MBD4 mediate and maintain cytosine methylation (5mC) of the αENaC promoter (2), which also constrains αENaC transcription, in part by limiting the binding and full trans-activation effect transcription factor Sp1 binding to its GC box in the αENaC promoter (3). Aldo (bottom) downregulates the expression of Dot1a, Af9, and Sirt1 (1), leading to decreased abundance of the repressor complex, and the hormone induces serum- and glucocorticoid-induced kinase 1, which phosphorylates Ser435 of Af9, causing disruption of the protein-protein interactions of Dot1a and Af9. This results in the hypomethylation of histone H3K79 and release of transcriptional repression of αENaC. In addition, DNMT3b and, presumably, MBD4 are dispersed from, and Tet2 recruited to, chromatin associated with the αENaC promoter, resulting in promoter demethylation with the conversion of 5mC to 5hmC, further de-repressing the promoter (2). The promoter demethylation maximizes Sp1 binding and trans-activation of the αENaC gene (3). Finally, the Aldo-liganded mineralocorticoid receptor (MR) binds to a glucocorticoid response element (GRE) in the αENaC promoter to further trans-activate the αENaC gene (4).
DNMT3b and MBD4 contribute to the basal repression of αENaC transcription in mIMCD3 cells.
Given the results of pharmacological inhibition of DNA methylation on αENaC promoter methylation and transcription (Figs. 2 and 3, A and B), we next tested the role of DNMT in mediating functionally relevant methylation on basal and aldosterone-stimulated αENaC promoter activity. Promoter activity in mIMCD3 cells stably expressing pcDNA3.1-Zeo-1.3αENaC-Luc was measured after transfection with control siRNA or siRNAs specific for DNMT1, DNMT3a, or DNMT3b. As shown in Fig. 5, DNMT3b knockdown resulted in an ∼70% increase in basal αENaC promoter activity, whereas knockdown of DNMT1 or DNMT3a had no measurable effect compared with similarly treated siRNA controls. None of the DNMT-specific siRNAs significantly affected aldosterone-stimulated promoter activity.
Given prior evidence in HEK-293F cells showing that DNMT3b associates with MBD4 in chromatin associated with the CYP27B1 promoter (10), we tested whether MBD4 is functionally involved in the transcriptional repression of αENaC transcription. As shown in Fig. 6A, mIMCD3 cells transfected with MBD4 siRNA exhibited doubled levels of basal αENaC mRNA compared with control cells but did not augment the levels induced by aldosterone. This result suggests that aldosterone completely negates the repressive effect of MBD4 on αENaC transcription. Indeed, overexpression of MBD4 suppressed aldosterone-stimulated αENaC promoter activity practically to the basal level (Fig. 6B). Collectively, the 5-Aza-CdR, DNMT, and MBD4 functional data indicate that hypermethylation, mediated and maintained by DNMT3b and MBD4, of the αENaC promoter inhibits its basal activity and results in suppressed basal αENaC gene transcription in mIMCD3 cells. Since aldosterone did not enhance αENaC gene transcription in cells depleted of DNMT3b and MBD4, it appears that the hormone completely overcame their repressive effects.
Fig. 6.

Methyl-CpG-binding domain protein (MBD)4 regulates αENaC gene expression in mIMCD3 cells. A: siRNA knockdown of MBD4 induces basal αENaC gene expression in mIMCD3 cells. mIMCD3 cells were transiently transfected with control siRNA or with MBD4-specific siRNA. Cells were then treated with vehicle or Aldo, and αENaC mRNA, which was normalized to β-actin mRNA, was measured. n = 6. *P < 0.05 vs. vehicle-treated control siRNA. B: mIMCD3 cells stably transfected with pcDNA3.1-Zeo-1.3αENaC-Luc were transiently transfected with control vector or a MBD4 expression plasmid. Cells were then treated with vehicle or aldosterone for 24 h, and luciferase activity, which was normalized to cell protein content, was measured. n = 6. *P < 0.05 vs. Aldo-treated control siRNA.
Aldosterone promotes the conversion of 5mC to 5hmC, loss of DNMT3b, and gain of Tet2 associated with the R3 subregion of the αENaC promoter.
Since 5-Aza-CdR mimicked the effect of aldosterone to enhance αENaC transcription, we tested whether aldosterone triggers loss of the repressive 5mC and de novo appearance of the activation mark 5hmC from the αENaC promoter R3 subregion. Anti-5mC and anti-5hmC antibodies were used for methylated and hydroxymethylated DNA immunoprecipitation followed by qPCR with primers to amplify the R3 subregion in mIMCD3 cells that had been treated with vehicle or aldosterone for 0, 1, 3, 6, or 24 h. We chose this method instead of bisulfite modification because 5hmC does not undergo a C-to-T transition after bisulfite treatment and thus cannot be distinguished from 5mC by that technique (8). As shown in Fig. 7A, 5mC was depleted and 5hmC enriched at the R3 subregion within 2 h of aldosterone treatment. These reciprocal changes, which are consistent with active demethylation, were sustained over the 24 h.
Fig. 7.

Aldo-induced reprogramming of methylation of the R3 (−57/+438) subregion of the αENaC promoter. The time course of Aldo-induced changes in methylation marks and modifiers is shown. mIMCD3 cells were cultured in DMEM-F-12 plus 10% charcoal-stripped serum for 20 h and then treated with vehicle or 1 μM Aldo for the indicated time periods. Cells were then harvested and subject to DNA immunoprecipitation/qPCR assays with antibodies specific for 5mC or 5-hydroxymethylcytosine (5hmC; A) and ChIP/qPCR for DNMT3b (B) and ten-eleven translocation (Tet)1, 2, and 3 (C). Specific primers were used to amplify the R3 subregion of αENaC. Data from Aldo-treated samples were normalized to the vehicle time-control values, which did not significantly vary over the time period. Data in A–C are presented as fold changes of the indicated time relative to their value at time 0. Error bars indicate ±SEs; n = 3. *P < 0.05 vs. the corresponding value at time 0. D: mIMCD3 cells were transiently transfected with control siRNA or with Tet2-specific siRNA. Cells were then treated with vehicle or Aldo for 24 h, and αENaC mRNA, which was normalized to β-actin mRNA, was measured. The value of Aldo-treated control siRNA-transfected cells was set at 1, and the other data were normalized to it. n = 6. *P < 0.05 vs. control siRNA.
We next analyzed the time-dependent effects of aldosterone on DNMT3b enrichment at the R3 subregion using ChIP/qPCR. Given the loss of 5mC, we hypothesized that the cytosine methyltransferase responsible for the 5mC marks, DNMT3b, would also be lost from the R3 subregion with aldosterone treatment. In agreement with our hypotheses, DNMT3b occupancy of the R3 subregion diminished beginning at 1 h (Fig. 7B). This result, coupled with the fact that DNMT3b mediated the transcriptional repression of αENaC under basal conditions (Figs. 5 and 6), suggests that aldosterone promotes derepression in part by dispersing DNMT3b from the R3 subregion.
Finally, given the reciprocal changes in 5mC and 5hmC with aldosterone treatment, we hypothesized that aldosterone prompts the enhanced enrichment of one or more Tet isoforms at the R3 subregion to oxidize 5mC to 5hmC and thereby contribute to derepression. As seen in the ChIP/qPCR data shown in Fig. 7C, aldosterone induced the recruitment of Tet2, but not Tet1 or Tet3, to the R3 subregion. To determine whether Tet2 is functionally important for the derepression of αENaC, we assayed αENaC mRNA levels in aldosterone-treated mIMCD3 cells transfected with control or Tet2-specific siRNA. As shown in Fig. 7D, aldosterone-treated mIMCD3 cells depleted of Tet2 exhibited 50% lower levels of αENaC mRNA compared with control cells.
DISCUSSION
Increasing evidence indicates that aldosterone plays a significant role in the development of hypertension and progression of cardiovascular and renal damage (1). Yet compared with other steroid hormones, relatively little is known about how it signals to chromatin to effect changes in gene transcription. With the increased clinical attention to aldosterone and MR antagonists in the pathogenesis and therapy of chronic renal and cardiovascular diseases, there is a growing gap in understanding the full range of aldosterone effects on the epigenetic and transcriptional machinery. Having already identified Dot1a as the first chromatin modifier to be regulated by aldosterone (26), the present study now identifies promoter methylation/demethylation, and specific effectors of these pathways (DNMT3b, MBD4, and Tet2), as aldosterone-responsive mediators of αENaC gene transcription in mIMCD3 cells.
The finding of a CpG island near the transcription start site of the αENaC promoter (Fig. 1) is typical of genes regulated by the control of promoter methylation status. Basal cytosine methylation in this region of the αENaC promoter was evident in the comparison of 5-Aza-CdR-treated cells versus vehicle-treated cells in bisulfite modification and DNA sequencing experiments (Fig. 2) as well as in the 5mC immunoprecipitation/qPCR assays (Fig. 7A). The fact that 5-Aza-CdR-mediated promoter demethylation enhanced αENaC mRNA expression (Fig. 3A) and αENaC promoter activity (Fig. 3B), indicates that basal αENaC transcription is constrained in part by promoter methylation. Although the stimulatory effect of 5-Aza-CdR treatment could be related to direct demethylation of the αENaC gene, or of another gene whose expression is required for αENaC transcription, the fact that knockdown of DNMT3b (but not DNMT1 or DNMT3a; Fig. 5) or MBD4 (Fig. 6A) enhanced basal αENaC gene expression indicates these proteins are largely, if not exclusively, responsible for the de novo methylation and maintenance of αENaC promoter methylation in mIMCD3 cells under basal conditions.
DNMT3b is generally considered to be active in de novo methylation. Although a recent study (3) has also demonstrated that DNMT3b can function as a redox state-dependent DNA 5hmC dehydroxymethylase (converting 5hmC to unmodified C), it does not appear to exert this activity at the αENaC promoter, since reciprocal, rather than parallel, changes in DNMT3b occupation and 5hmC at the αENaC R3 subregion were observed with aldosterone treatment (Fig. 7, A and B). DNMT3b has also been shown to interact with MBD4 via the highly conserved PWWP motif and catalytic domain of DNMT3b (4). Since the PWWP domain confers DNA-binding activity and targets DNMT3b to heterochromatin (4), this interaction may help direct MBD4 to sites of heterochromatin. The fact that overexpression of MBD4 impaired aldosterone induction of the αENaC promoter (Fig. 6B), indicates that MBD4 is not participating in active DNA demethylation initiated by the deaminase-catalyzed conversion of 5mC to thymine, as has been reported in other systems (10).
The results also demonstrate that aldosterone reprogrammed the methylation status of the R3 subregion of the αENaC promoter from a predominance of 5mC under basal conditions to a predominance of 5hmC (Fig. 7A). This change was accompanied by depletion of DNMT3b at this subregion (Fig. 7B) and enhanced enrichment of Tet2 (Fig. 7C). The fact that Tet2 knockdown limited aldosterone induction of αENaC transcription (Fig. 7D) suggests that aldosterone stimulates Tet2 action at the R3 subregion to mediate the conversion of 5mC to 5hmC for the derepression of αENaC transcription. The detailed mechanisms underlying the aldosterone-stimulated recruitment of Tet2 and dispersal of DNMT3b at the αENaC promoter in the context of chromatin remain to be discovered.
Cytosine methylation-mediated interference of Sp1 binding to its cis-elements on promoters is known to inhibit the expression of several of its target genes in various tissues (2, 7, 17). We (23) have previously demonstrated that Sp1 activates basal αENaC transcription in mIMCD3 cells and that aldosterone enhances Sp1 binding to, and trans-activation of, the αENaC promoter, without altering nuclear Sp1 protein levels. Given our new results showing that 5-Aza-CdR-mediated demethylation enhances Sp1 occupation of the αENaC promoter (Fig. 4) to a level virtually identical to that of aldosterone-stimulated cells (see Fig. 8A in Ref. 23) and that aldosterone prompts the conversion of 5mC to 5hmC in the αENaC promoter (Fig. 7A), it appears that basal αENaC promoter methylation also limits the full effect of Sp1-mediated binding and trans-activation of the αENaC promoter under basal conditions and that this constraint is relieved in the presence of aldosterone. Thus, under basal conditions, concurrent Sp1-mediated trans-activation and Dot1a-mediated histone H3K79 hypermethylation at the αENaC promoter appear to act as if accelerator and brake, respectively, were simultaneously applied, allowing the accelerator to overcome the brake sufficiently to allow αENaC transcription for normal renal salt excretion but also poised for the rapid acceleration of αENaC transcription via derepression (removal of the brake effect) in the presence of aldosterone.
Further studies with combinations of selective mutations of Af9-, Dot1a-, Sp1-, and MR-binding sites of the αENaC promoter-binding sites and knockdown/overexpression of DNMT3b, MBD4, and Tet2 will be needed to determine the integration and relative contributions of the various genetic and epigenetic controls on basal and aldosterone-stimulated αENaC transcription and to ENaC-mediated Na+ transport in the CD. Unfortunately, we and our collaborators have been unable to get consistent patch-clamp ENaC activity measurements from mIMCD3 cells after they have been subjected to transfection and charcoal-stripped serum for these types of experiments. We do know, however, that a 50% reduction in αENaC mRNA achieved by overexpression of Dot1a (see Fig. 4A in Ref. 18), which is comparable to that observed with basal DNA methylation (vehicle-treated cells; Fig. 3A) versus demethylation (5-Aza-CdR-treated cells; Fig. 3A), resulted in ∼50% decrements in benzamil-sensitive intracellular Na+ concentration, as assessed with sodium-binding benzofuran isophthalate-acetoxymethyl ester in single cell fluorescence imaging, an indirect assessment of ENaC activity (see Fig. 8E in Ref. 18). Thus, we feel it is probable that the large changes in αENaC expression we observed in this report are physiologically relevant.
Aldosterone plays a significant role in the development of hypertension and progression of cardiovascular and renal damage. Yet compared with other steroid hormones, relatively little is known about how it signals to chromatin or alters the methylation status of gene promoters to effect changes in gene transcription. The present results build on a complex model (Fig. 8) of aldosterone-induced αENaC transcription that includes trans-activation mediated by the binding of the liganded MR to a glucocorticoid responsive element (11) and recruitment of Sp1 to its cis-element (23) and at least two epigenetic mechanisms for derepression: disruption of Dot1a-mediated histone H3K79 methylation (28) and the targeted demethylation of the αENaC promoter reported here. The control of αENaC transcription through epigenetic modifications of both promoter DNA and histones, in addition to classical trans-activation mechanisms by transcription factors, lends considerable versatility to control of renal salt excretion under basal conditions and in response to aldosterone.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-075065 (to B. C. Kone).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Z.Y. and B.C.K. conception and design of research; Z.Y. and Q.K. performed experiments; Z.Y., Q.K., and B.C.K. analyzed data; Z.Y., Q.K., and B.C.K. interpreted results of experiments; Z.Y. and B.C.K. prepared figures; Z.Y., Q.K., and B.C.K. edited and revised manuscript; Z.Y., Q.K., and B.C.K. approved final version of manuscript; B.C.K. drafted manuscript.
REFERENCES
- 1.Bubien JK. Epithelial Na+ channel (ENaC), hormones, and hypertension.J Biol Chem 285: 23527–23531, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ceccarelli V, Racanicchi S, Martelli MP, Nocentini G, Fettucciari K, Riccardi C, Marconi P, Di Nardo P, Grignani F, Binaglia L, Vecchini A. Eicosapentaenoic acid demethylates a single CpG that mediates expression of tumor suppressor CCAAT/enhancer-binding protein delta in U937 leukemia cells. J Biol Chem 286: 27092–27102, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen CC, Wang KY, Shen CK. DNA 5-methylcytosine demethylation activities of the mammalian DNA methyltransferases. J Biol Chem 288: 9084–9091, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen T, Tsujimoto N, Li E. The PWWP domain of Dnmt3a and Dnmt3b is required for directing DNA methylation to the major satellite repeats at pericentric heterochromatin. Mol Cell Biol 24: 9048–9058, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christensen BM, Perrier R, Wang Q, Zuber AM, Maillard M, Mordasini D, Malsure S, Ronzaud C, Stehle JC, Rossier BC, Hummler E. Sodium and potassium balance depends on alphaENaC expression in connecting tubule. J Am Soc Nephrol 21: 1942–1951, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehrlich M, Gama-Sosa MA, Huang LH, Midgett RM, Kuo KC, McCune RA, Gehrke C. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10: 2709–2721, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo D, Wu B, Yan J, Li X, Sun H, Zhou D. A possible gene silencing mechanism: hypermethylation of the Keap1 promoter abrogates binding of the transcription factor Sp1 in lung cancer cells. Biochem Biophys Res Commun 428: 80–85, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLos One 5: e8888, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res 38: e125, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, Matsumoto T, Fujiyama S, Shirode Y, Yamaoka I, Kitagawa H, Takeyama K, Shibuya H, Ohtake F, Kato S. DNA demethylation in hormone-induced transcriptional derepression. Nature 461: 1007–1012, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Kohler S, Pradervand S, Verdumo C, Merillat AM, Bens M, Vandewalle A, Beermann F, Hummler E. Analysis of the mouse Scnn1a promoter in cortical collecting duct cells and in transgenic mice. Biochim Biophys Acta 1519: 106–110, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Kondo E, Gu Z, Horii A, Fukushige S. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol Cell Biol 25: 4388–4396, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, Gnirke A, Jaenisch R, Lander ES. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454: 766–770, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mick VE, Itani OA, Loftus RW, Husted RF, Schmidt TJ, Thomas CP. The α-subunit of the epithelial sodium channel is an aldosterone-induced transcript in mammalian collecting ducts, and this transcriptional response is mediated via distinct cis-elements in the 5′-flanking region of the gene. Mol Endocrinol 15: 575–588, 2001 [DOI] [PubMed] [Google Scholar]
- 16.Otani J, Arita K, Kato T, Kinoshita M, Kimura H, Suetake I, Tajima S, Ariyoshi M, Shirakawa M. Structural basis of the versatile DNA recognition ability of the methyl-CpG binding domain of methyl-CpG binding domain protein 4. J Biol Chem 288: 6351–6362, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paonessa F, Latifi S, Scarongella H, Cesca F, Benfenati F. Specificity protein 1 (Sp1)-dependent activation of the synapsin I gene (SYN1) is modulated by RE1-silencing transcription factor (REST) and 5′-cytosine-phosphoguanine (CpG) methylation. J Biol Chem 288: 3227–3239, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reisenauer MR, Wang SW, Xia Y, Zhang W. Dot1a contains three nuclear localization signals and regulates the epithelial Na+ channel (ENaC) at multiple levels. Am J Physiol Renal Physiol 299: F63–F76, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ronzaud C, Loffing J, Bleich M, Gretz N, Grone HJ, Schutz G, Berger S. Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol 18: 1679–1687, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Schild L. The epithelial sodium channel and the control of sodium balance. Biochim Biophys Acta 1802: 1159–1165, 2010 [DOI] [PubMed] [Google Scholar]
- 21.Turek-Plewa J, Jagodzinski PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett 10: 631–647, 2005 [PubMed] [Google Scholar]
- 22.Yu Z, Kone BC. Hypermethylation of the inducible nitric-oxide synthase gene promoter inhibits its transcription. J Biol Chem 279: 46954–46961, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Yu Z, Kong Q, Kone BC. Sp1 trans-activates and is required for maximal aldosterone induction of the αENaC gene in collecting duct cells. Am J Physiol Renal Physiol 305: F653–F662, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu Z, Li M, Zhang D, Xu W, Kone BC. Sp1 trans-activates the murine H+-K+-ATPase α2-subunit gene. Am J Physiol Renal Physiol 297: F63–F70, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang D, Li S, Cruz P, Kone BC. Sirtuin 1 functionally and physically interacts with disruptor of telomeric silencing-1 to regulate α-ENaC transcription in collecting duct. J Biol Chem 284: 20917–20926, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W, Xia X, Jalal DI, Kuncewicz T, Xu W, Lesage GD, Kone BC. Aldosterone-sensitive repression of ENaCα transcription by a histone H3 lysine-79 methyltransferase. Am J Physiol Cell Physiol 290: C936–C946, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCα in an aldosterone-sensitive manner. J Biol Chem 281: 18059–18068, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest 117: 773–783, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang W, Yu Z, Wu H, Chen L, Kong Q, Kone BC. An Af9 cis-element directly targets Dot1 to mediate transcriptional repression of the αENaC gene. Am J Physiol Renal Physiol 304: F367–F375, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]