

Abstract
The vacuolar H+-ATPase (V-ATPase) in intercalated cells contributes to luminal acidification in the kidney collecting duct and nonvolatile acid excretion. We previously showed that the A subunit in the cytoplasmic V1 sector of the V-ATPase (ATP6V1A) is phosphorylated by the metabolic sensor AMP-activated protein kinase (AMPK) in vitro and in kidney cells. Here, we demonstrate that treatment of rabbit isolated, perfused collecting ducts with the AMPK activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) inhibited V-ATPase-dependent H+ secretion from intercalated cells after an acid load. We have identified by mass spectrometry that Ser-384 is a major AMPK phosphorylation site in the V-ATPase A subunit, a result confirmed by comparing AMPK-dependent phosphate labeling of wild-type A-subunit (WT-A) with that of a Ser-384-to-Ala A subunit mutant (S384A-A) in vitro and in intact HEK-293 cells. Compared with WT-A-expressing HEK-293 cells, S384A-A-expressing cells exhibited greater steady-state acidification of HCO3−-containing media. Moreover, AICAR treatment of clone C rabbit intercalated cells expressing the WT-A subunit reduced V-ATPase-dependent extracellular acidification, an effect that was blocked in cells expressing the phosphorylation-deficient S384A-A mutant. Finally, expression of the S384A-A mutant prevented cytoplasmic redistribution of the V-ATPase by AICAR in clone C cells. In summary, direct phosphorylation of the A subunit at Ser-384 by AMPK represents a novel regulatory mechanism of the V-ATPase in kidney intercalated cells. Regulation of the V-ATPase by AMPK may couple V-ATPase activity to cellular metabolic status with potential relevance to ischemic injury in the kidney and other tissues.
Keywords: AMPK, kidney, mass spectrometry, V-ATPase, Intercalated cells
kidney type a intercalated cells participate in nonvolatile acid secretion (1). Dysfunction of these cells can lead to distal renal tubular acidosis, a condition associated with severe pathological consequences throughout the organism, especially in kidney and bone (15). Type A intercalated cells express high levels of the V-ATPase at their apical membrane (1, 20, 21, 24, 31, 47, 48). The V-ATPase subunits are divided into two sectors: the V1 peripheral domain, which includes the A subunit of the V-ATPase that catalyzes ATP hydrolysis (reviewed in Ref. 15) and the V0 integral membrane domain, which mediates the translocation of 2 H+ for each ATP consumed (15). The V-ATPase requires a constant supply of ATP to maintain acid secretion (15). Therefore, cells expressing high levels of V-ATPase such as type A intercalated cells are likely to use several regulatory mechanisms to link the activity of this transport protein complex to the metabolic status of the cell. In epithelial cells, enzymes of the glycolytic pathway such as aldolase and phosphofructokinase regulate V-ATPase activity (28–30, 49). We have demonstrated that in kidney intercalated cells and in epididymal clear cells, which share a common developmental origin in the Wolffian duct, the V-ATPase redistributes from the apical membrane to the cytosol with stimulation of the metabolic sensor AMPK (16, 18). In our previous studies we first identified the V-ATPase A subunit as an AMPK substrate using the unbiased “MudSeeK” proteomic screening method and subsequently showed that the V-ATPase A subunit is phosphorylated directly by AMPK in kidney cells (18, 52). AMPK is an important cell energy-sensing kinase that has been shown to downregulate several kidney membrane transport proteins (17, 39). We have shown that AMPK reduces apical V-ATPase accumulation acutely in kidney intercalated cells, and that AMPK activation antagonizes the ability of PKA to increase V-ATPase localization at the apical pole of type A intercalated cells (16).
Phosphatidylinositol 3-kinase and PKA are additional kinases that regulate the function of the V-ATPase in the mammalian kidney (16, 41, 43). PKA agonists also regulate the V-ATPase in other animal models (42, 54). However, it was not until recently that work by our group linked the direct phosphorylation of the V-ATPase by PKA at Ser-175 to increases in V-ATPase plasma membrane activity in mammalian cells (2). Ser-175 A subunit phosphorylation is likely to occur downstream of acid-base-sensing pathways, which require the presence of active carbonic anhydrase, the soluble adenylyl cyclase (sAC), and increases in cellular cAMP (16, 18, 38).
We have proposed a V-ATPase-regulatory model where in the acute setting PKA couples V-ATPase activity to acid-base status, and AMPK couples the activity of the pump to cellular metabolic status (39). Coregulation of the proton pump by these two kinases could be critical in scenarios where distal nephron proton secretion is needed to maintain or restore acid-base homeostasis in conjunction with a decrease in kidney perfusion. For example, renal hypoperfusion during shock should activate AMPK acutely (33), which would inhibit V-ATPase-mediated proton secretion. Conversely, increased plasma CO2 levels would act to increase collecting duct proton secretion (44) via acute activation of sAC and downstream activation of PKA (16, 18, 57). Therefore, if both kidney hypoperfusion and increased plasma CO2 occur concomitantly, the net effect on proton secretion may depend on the integrated response of V-ATPase activity to AMPK and PKA.
In the current study, we set out to elucidate the mechanisms by which AMPK induces changes in V-ATPase subcellular localization and activity. We first confirmed that V-ATPase-mediated H+ secretion in isolated, perfused rabbit kidney outer medullary collecting ducts (OMCDs) decreases in the presence of an AMPK activator. In addition, we have identified by mass spectrometry that Ser-384 is a major AMPK phosphorylation site in the V-ATPase A subunit, and we have characterized its functional relevance. This residue has been previously identified as a phosphorylation site in the mouse brain, although the particular kinase involved was not identified (34). Our findings may inform a better understanding of the regulation of acid-base status during metabolic stress, such as during acute kidney injury due to ischemia or sepsis.
MATERIALS AND METHODS
Reagents and chemicals.
All reagents and chemicals used were purchased from Sigma (St. Louis, MO) or Fisher Scientific (Pittsburgh, PA) unless otherwise stated. The cell-permeant AMPK activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) and the PKA-specific inhibitor myristoylated PKI (mPKI) were obtained from Biomol (Toronto Research Chemicals, Toronto, Canada). The anti-FLAG and anti-phospho-Thr-172-AMPK-α (pThr-172-AMPK-α) antibodies were obtained from Cell Signaling Technology (Danvers, MA). Paraformaldehyde was obtained from Electron Microscopy Sciences (Hatfield, PA). The cell-permeant compound bafilomycin A1 (LC Laboratories, Woburn, MA), a potent and specific inhibitor of the V-ATPase (6), was prepared as a 1 μM stock solution in DMSO and diluted to a final concentration of 1 nM (2). N-methyl-d-glucamine (NMDG+) was used to substitute for Na+ in some of the solutions used for pH measurements. The fluorescent membrane-permeant pH indicator BCECF-AM was obtained from Molecular Probes (Sigma). The principal cell marker rhodamine-labeled Dolichos biflorus agglutinin (DBA) was obtained from Vector Laboratories (Burlingame, CA) (14). Nigericin (2 mM stock solution) was diluted to a final 10 μM in each standard intracellular pH (pHi) calibration solution (9).
Animal studies.
Adult (>6 wk) female New Zealand White rabbits (Covance, Princeton, NJ) were housed at the Center for Comparative Medicine, Icahn School of Medicine at Mount Sinai (ISMMS). All animals were allowed free access to tap water and standard rabbit chow. Animals were euthanized in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Animal protocols were approved by the Institutional Animal Care and Use Committee at the ISMMS.
Microperfusion of isolated rabbit tubules and measurement of pHi in intercalated cells.
These ex vivo experiments were performed using previously described methods (9). Rabbit kidneys were removed via a midline incision. Single OMCDs were dissected freehand in 4°C Na+-containing Ringer solution (NaR) containing (in mM) 135 NaCl, 2.5 K2HPO4, 2.0 CaCl2, 1.2 MgSO4, 4.0 lactate, 6.0 l-alanine, 5.0 HEPES, and 5.5 d-glucose, pH 7.4, and 290 ± 2 mosmol/kgH2O, as previously described (9). A single OMCD from each animal was immediately transferred to a temperature-controlled specimen chamber assembled with a no. 1 coverslip (Corning, Tewksbury, MA) painted with a 1-μl drop of poly-d-lysine hydrobromide 0.01% (BD Biosciences, San Jose, CA), set on the stage of a Nikon inverted epifluorescence microscope (Eclipse TE300 or Diaphot; Nikon, Melville, NY) linked to a Cascade 512F camera (Photometrics, Tucson, AZ) or a cooled Pentamax CCD camera (Princeton Instruments, Trenton, NJ), interfaced with a digital imaging system (MetaFluor, Universal Imaging, Sunnyvale, CA). The OMCD was then mounted on concentric glass pipettes, cannulated, and perfused and bathed at 37°C with NaR (34) with or without 2 mM AICAR added to the luminal perfusate for 1 h during the equilibration period. Thereafter, 20 μM BCECF-AM was added to the bath for 15 min (in the continued presence/absence of AICAR), as originally described by Weiner and Hamm (56), and the preparation was then rinsed three times with NaR solution for 1 min. The luminal perfusate was then replaced with a Na+- and K+-free solution (0Na, 0K). Once a steady-state pHi was obtained, the BCECF-loaded OMCD was acid loaded by a 3-min peritubular exposure to a 30-mM NH4Cl solution. Rapid washout of the basolateral NH4Cl solution with 0Na, 0K solution led to a fall in pHi; these manual washes were accomplished by fully replacing the volume of the bath (∼1.5 ml) at least three times within 10 s, as previously described (9). The 490-nm-to-440-nm fluorescence intensity ratios (FIRs) were monitored in the absence of Na+ and K+ in the lumen and bath for at least 10 min, and then the bathing solution was replaced with NaR solution, which allowed for Na+/H+ exchange and pHi normalization. FIR measurements were obtained within 30 s of each change in solution, and then at 1- to 3-min intervals.
At the end of each experiment, the tubule was perfused with rhodamine-DBA to identify principal cells and then an intracellular pHi calibration was performed using the nigericin technique (9, 50).
The compositions of the solutions used for the NH4Cl prepulse technique for the acute exogenous acid loading of tubular cells and calibrations have been previously described (9). All studies were performed in the nominal absence of CO2 and HCO3−. Na+ in the Na+-free solutions was replaced with NMDG+, and pH was adjusted to 7.4. The bathing solution was continuously exchanged at a rate of 10 ml/h using a syringe pump (Razel, St. Albans, VT) during the equilibration and pHi recovery periods.
BCECF-loaded cells in the OMCDs were visualized under epifluorescence illumination using a ×40 water-immersion objective (Nikon S Fluor 40; numeric aperture 0.9, working distance 0.3) (9, 27). OMCDs were alternately excited at 490 and 440 nm using a wavelength switcher (DG-4 or LAMBDA 10–2; Sutter). Images of the fluorescence emission at 530 nm (BCECF) overlying individually identified cells residing in the lateral wall of each perfused tubule (to capture the fluorescence signal from a single cell) were acquired at intervals ranging from 2 to 15 s using MetaFluor image-acquisition software (Universal Imaging) and stored on a Digital Instruments computer. Autofluorescence was not detected at the camera gains utilized. The 490-nm-to-440-nm FIRs were subsequently calculated using the MetaFluor digital image-analysis system.
At least two intercalated cells residing in the lateral wall of the midregion of each perfused OMCD (to capture the fluorescence signal from a single cell) were randomly selected for final analysis; their FIRs were converted to pHi measurements using linear regression analysis from the nigericin calibration and standard equations. The steady-state pHi of a single cell was calculated based on the average of at least six FIR readings. All pHi results are reported as the mean of measurements in four tubules.
Mass spectrometry of wild-type mouse FLAG-A V-ATPase subunit.
The V-ATPase wild-type mouse FLAG-tagged A subunit construct (WT-A) was characterized in our previous studies (2, 40). This plasmid was transfected into HEK-293 cells, and the cell lysate products were immunoprecipitated using the M2 anti-FLAG monoclonal antibody coupled to protein A/G beads (Pierce, Rockford, IL), as described (4, 7, 16). This FLAG-tagged WT-A V-ATPase subunit (WT-A) was phosphorylated by a purified AMPK holoenzyme (36) at 37°C in kinase buffer (10 mM HEPES·Cl, pH 7.4, 200 μM ATP, 40 μM AMP, 5 mM MgCl2) supplemented with [γ-32P]-ATP for 2 h. The reaction was stopped by adding SDS-containing sample buffer and heating to 95°C for 5 min. The following steps were performed as described previously (11) with only one minor modification. Briefly, following SDS-PAGE and Coomassie blue staining, the gel band corresponding to the WT-A subunit was excised from the wet gel and subjected to in-gel digestion with trypsin. Concentrated tryptic peptides were applied to a microbore reversed-phase column connected to a capillary liquid chromatography system and equipped with a microcollection/spotting system, thus allowing the microfractionation onto a Prespotted AnchorChip (PAC) (51). After autoradiography, selected fractions of the PAC target indicating the presence of radiolabeled phosphopeptides were analyzed by matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS and MALDI-MSMS) (51).
To confirm the phosphorylation site identified by mass spectrometry analysis, the candidate phosphorylation site (Ser-384) was mutated using a Stratagene QuikChange (Stratagene, La Jolla, CA) kit according to the manufacturer's instructions using the pMO plasmid expressing the FLAG-tagged WT-A V-ATPase subunit as a template (2), which we have used for mammalian cell expression (18). All mutations were confirmed by DNA sequencing.
V-ATPase A subunit in vitro phosphorylation assays.
These studies were performed essentially as described previously (2, 4, 18). HEK-293 cells were transiently transfected using Lipofectamine 2000 (Invitrogen, Life Technologies, Carlsbad, CA) to express either FLAG-tagged V-ATPase A subunit wild-type (WT-A, mouse sequence) (2) or this subunit with a specific point mutant (Ser-384 to Ala; S384A-A) identified as the AMPK target phosphorylation site by mass spectrometry. Cells were lysed 2 days after transfection, and the WT-A and S384A-A subunits were immunoprecipitated from cell lysates using the M2 anti-FLAG monoclonal antibody (Sigma) coupled to protein A/G beads (Pierce). In vitro phosphorylation was performed using purified active AMPK holoenzyme (36) with [γ-32P]-ATP labeling, as described (2, 18). After SDS-PAGE and transfer to nitrocellulose membranes, immunoblotting for expression of FLAG-labeled V-ATPase A subunits (WT-A and S384A-A) was first performed using a horseradish peroxidase (HRP)-linked anti-FLAG antibody and quantified using a Versa-Doc Imager with Quantity One software (Bio-Rad, Hercules, CA). After the chemiluminescent signal decayed, phosphorylated bands on the membrane were identified by exposure of the same membrane to a phosphoscreen, and the detected bands were quantitated using a Bio-Rad phosphorimager with Quantity One software. The intensity of each phosphoscreen band was corrected by subtracting out the local background in the same lane.
V-ATPase A subunit phosphorylation assays in HEK-293 cells.
For these experiments, we used our previously generated and characterized cell lines derived from HEK-293 cells that allow us to modulate intracellular AMPK activity (18). These cell lines are stably transfected with a plasmid expressing either AMPK-α1 short hairpin (sh) RNA (AMPK-KD) or a control shRNA that does not target any known mammalian gene (CON) (18). Cells were seeded onto 60-mm dishes and transiently transfected with 3 μg of either WT-A or S384A-A mutant subunit plasmid DNA using Lipofectamine 2000. After transfection, cells were grown in the presence of doxycycline (1 μg/ml) to induce expression of the shRNAs for 2 days (18). [32P]orthophosphate labeling in HEK-293 cells was performed essentially as previously described (2, 18). Labeling was carried out in the presence of the specific PKA inhibitor mPKI (10 μM) to suppress phosphorylation of the A subunit by PKA at Ser-175, a site previously shown to be stimulated by AMPK knockdown (18). In addition, to assess potential differences in AMPK-mediated [32P]orthophosphate labeling in the cells across conditions, 10 μg of cell lysate for each condition (∼1.5 μl) was spotted onto a nitrocellulose membrane. The [32P]orthophosphate-labeled proteins in each spot were detected by exposing the membranes to a phosphoscreen and then quantified using the phosphorimager. Thereafter, the membranes were blocked in 5% BSA in TBST buffer and probed with an antibody recognizing the active phosphorylated AMPK-α subunit (anti-phospho-Thr-172-AMPK-α; 1:1,000; Cell Signaling Technologies) to account for potential differences in AMPK activity modulation across conditions. The intensities of the phosphoscreen spot signals were normalized to the intensity of their respective β-actin signals.
Measurement of chronic changes in extracellular pH.
HEK-293 cells were transiently transfected and treated using the experimental methods described above in the subsection “V-ATPase A subunit phosphorylation assays in HEK-293 cells” (2). Transfected cells were grown in high-glucose, HCO3−-containing Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% FBS at 37°C in the presence of 5% CO2-95% air. At 28–31 h after transfection, and before the initiation of the phosphorylation experiments described in the section above, the medium from each plate was collected, and the pH was measured using an Accumet AB15 pH meter (Fisher Scientific) after careful equilibration of the sample with 5% CO2-95% air.
Measurement of acute V-ATPase-dependent changes in extracellular pH.
Clone C cells were seeded onto 10-cm2 plates and grown at 32°C in medium containing Dulbecco's modified Eagle's medium/F-12 (Invitrogen), 10% heat-inactivated FBS, 27.6 μM hydrocortisone, 0.45% insulin-transferrin-sodium selenite media supplement, 15 mg/l epidermal growth factor, 1 mM glutamine, and 0.5% penicillin/streptomycin (Invitrogen) as previously described (13). When the cells reached ∼80% confluence, they were transiently transfected the next day with 12 μg/plate of plasmid DNA (pTracer, Invitrogen) bicistronically expressing enhanced GFP (eGFP) and either WT-A or S384A-A using Lipofectamine 2000. One day after transfection, clone C cells were seeded onto poly-l-lysine-coated 24-well plates at a density of 2.5 × 105 cells/well and grown at 40°C (in 5% CO2-95% air) (53) in Dulbecco's modified Eagle's medium with 1.8% FBS supplemented with hormones and growth factors (3). A total of six wells per transfection were plated for each plasmid.
Transfection efficiency for each well was assessed by obtaining micrographs of each of the transfected wells using a Nikon Coolpix ID 7000 camera tethered to a fluorescence inverted microscope (Nikon Eclipse TE2000-U) using a ×10 objective (×100 magnification) under identical acquisition parameters. Transfected cells were detected using a xenon lamp and fluorescence filter sets for visualizing eGFP. Cells were confluent when the images were obtained.
The total area of each micrograph was measured using Metamorph software (Downingtown, PA). The number of cells expressing green fluorescence were counted in that region of interest (ROI). For each ROI, we counted >100 eGFP-positive cells. The average transfected cell area was calculated for each well. This number was then used to calculate the number of cells present in the entire ROI and the ratio of positive to total cells.
After the transfection efficiency was ensured to be equivalent in all wells, at 28–31 h posttransfection the media from each well was removed and replaced with fresh media containing 2 mM AICAR (4 h) for three wells of cells transfected with WT-A and three wells of cells transfected with S384A-A. The remainder of the wells received fresh media containing vehicle (water). The cells were then placed in the incubator at 40°C for 4 h in the presence of 5% CO2-95% air. After the 4-h incubation, all wells were briefly washed in 1 ml of a Na+-free, low-buffering-capacity solution containing (in mM) 135 N-methyl-d-glucamine, 5 KCl, 2 CaCl2, 1.2 MgSO4, 5.5 d-glucose, 6 l-alanine, 4 lactic acid, and 1 HEPES, titrated to pH 7.43 using 1 M HCl (modified from Ref. 9) (2). The wells were then replaced with 1 ml of fresh solution (±AICAR, 2 mM) and then incubated for 20 min at 37°C before collection of the buffer for extracellular pH (pHo) measurements, which were also performed at 37°C. After these incubations, the same procedure was repeated with a 20-min incubation in the continued presence/absence of AICAR, with all wells receiving 1 μM bafilomycin A1, a specific V-ATPase inhibitor. As we have previously described (2), reported acidification rates (in pH units/min) were calculated from the pH drop of the solutions (final minus initial pH) over the 20-min incubation period. The V-ATPase-dependent rate of pHo acidification for each sample well was defined as the difference in the acidification rate measured in the absence vs. presence of bafilomycin A1 (i.e., bafilomycin A1-sensitive pHo acidification rate). Comparing pHo change rates from the same starting pH in the absence vs. presence of bafilomycin allowed us to quantify the V-ATPase-dependent extracellular acidification rate within the same tissue culture well [−(final buffer pH − initial buffer pH)/Δt]. At the end of each experiment (n = 5), cells were collected to determine total cell number, as counted with a hemocytometer, and cell viability, as assessed by Trypan blue exclusion (2).
Immunofluorescence labeling of clone C cells transfected with V-ATPase A subunit mutants and quantification of V-ATPase-associated fluorescence.
Cells were seeded onto 10-cm2 plates and transfected with either the WT-A or S384A-A subunit using the techniques described above (2). One day after transfection, the cells were trypsinized and then seeded onto 0.33-cm2 Transwells at a concentration of 1 × 106/cm2. The cells were then allowed to polarize for 5 days in media (3) before incubation in fresh media in the presence or absence of 2 mM AICAR for 4 h. Immunofluorescence labeling of these filters was performed as previously described (2), using the apical membrane marker concanavalin A coupled to CY3 (ConA; Vector Laboratories) and an anti-FLAG antibody raised in mice (M2 anti-FLAG) along with a secondary antibody coupled to fluorescein (Jackson Immunologicals, West Grove, PA). ProlongGold (Invitrogen) was used as the mounting media (2). Images from these filters were acquired with identical laser confocal microscope settings across all transfection conditions. Images were imported into Adobe Photoshop for presentation as described previously (2, 18). Three-dimensional reconstructions of confocal stacks were used to obtain X-Z or X-Y projections of the transfected cells in the monolayers, which were then imported into MetaMorph for quantification. V-ATPase apical accumulation was determined by measuring the mean pixel intensity (MPI) of FLAG-associated fluorescence in an apical ROI (ROI-1, colocalizing with CY3-concanavalin A) and a cytoplasmic ROI of identical size immediately below concanavalin A labeling (ROI-2), as we have previously described (2, 5, 18). The brightest area of the cell at the apical membrane was chosen for measuring the ROI-1 (∼200 pixels). After X-Z reconstruction, at least two additional investigators blinded as to the nature of the transfected subunits evaluated the distribution of anti-FLAG labeling in TIF images of these cells. The degree of apical accumulation of the V-ATPase FLAG A subunit mutants was determined by the ratio of apical-to-cytoplasmic ROI (ROI-1/ROI-2) of FLAG-associated fluorescence for each cell. At least three separate filters were evaluated for each condition, and we performed quantification of 20–45 cells/condition.
Evaluation of AMPK-phosphorylation status in clone C cells.
Clone C cells were transfected with the WT-A subunit plasmid and placed on filters, as above. After AICAR treatment for 4 h, immunoblotting of cell lysates was performed using the antibody against pThr-172-AMPK-α. Membranes were then stripped and reprobed with an antibody to β-actin. The pThr-172-AMPK-α signal was normalized to the β-actin signal.
Co-immunoprecipitation studies.
Clone C cells were transfected with plasmid encoding either the WT-A or S384A-A mutant subunit as described above. One day after transfection, cells were harvested in ice-cold lysis buffer using our established techniques (2, 4). We used 1 mg of precleared lysate for immunoprecipitations performed at 4°C using the M2 anti-FLAG antibody (0.5 mg/immunoprecipitation) coupled to protein A/G beads. Immunoprecipitation in the absence of the anti-FLAG antibody was also performed as a control. After three washes in lysis buffer, the immunoprecipitation samples were eluted in sample buffer and, along with the cell lysate samples, subjected to SDS-PAGE. Immunoblotting was performed with either 1) V0 a subunit antibody (1:2,000 dilution, raised in rabbits, Santa Cruz Biotechnology, Dallas, TX), followed by the appropriate secondary antibodies coupled to HRP as needed (Jackson Immunologicals); or 2) anti-FLAG antibody coupled to HRP (1:5,000 dilution, raised in chickens, Sigma) (2, 18).
Statistics.
Data shown represent means ± SE per group. In most experiments, significance was determined using two-tailed, unpaired Student's t-tests assuming unequal variances for the treatment groups, unless otherwise indicated. P values <0.05 were considered significant.
RESULTS
AMPK regulates Na+- and K+-independent changes in pHi in intercalated cells of isolated, perfused rabbit OMCDs.
The rate of AMPK-regulated V-ATPase-dependent H+ extrusion in individual intercalated cells was assessed in isolated, perfused rabbit OMCDs (9). OMCDs were loaded with the pH-sensitive dye BCECF, which allowed us to monitor changes in pHi in response to the imposition of an acute exogenous acid load via the NH4Cl prepulse technique. V-ATPase activity was determined as the rate of pHi recovery from the nadir, achieved after washout of peritubular NH4Cl, monitored in the absence of Na+ and K+ and in the nominal absence of HCO3− (9). Past studies by members of our group revealed that in the absence of Na+ and K+, H+ extrusion by the basolateral Na+/H+ exchanger (such as NHE1) and the H+/K+ ATPase (H-K-ATPase), respectively, are impaired. Moreover, in the same report the H+ extrusion pathway that was retained in intercalated cells in the absence of Na+ and K+ was inhibited by the V-ATPase-specific inhibitor bafilomycin A1 (9).
To evaluate whether AMPK activation inhibited V-ATPase function in intercalated cells, we preincubated OMCDs in the presence or absence of the AMPK activator AICAR for 60 min, followed by loading of the cells with the pHi indicator BCECF-AM. The intercalated cells were identified under the microscope by their strong fluorescence intensity signal resulting from selective accumulation of BCECF compared with that detected in adjacent principal cells (9, 56). This differential loading is largely due to the abundance of carbonic anhydrase activity in intercalated cells (12), which leads to rapid cleavage of the AM moiety from BCECF-AM. In addition, we identified principal cells by colabeling the tubules with a principal cell marker, the rhodamine-labeled lectin from D. biflorus (Fig. 1A, middle and right). Figure 1B depicts two experiments showing the general patterns of pHi recovery in several intercalated cells from control OMCDs preincubated in vehicle alone (left) compared with those from OMCDs that had been preincubated with the AMPK activator AICAR (right). The rate of pHi recovery after an acid load was significantly decreased in the intercalated cells of OMCDs incubated with AICAR (Fig. 1, B and D). These findings indicate that V-ATPase activity in intercalated cells, as measured by the pHi recovery in the absence of Na+ and K+ and nominal absence of HCO3−, is inhibited by AMPK. These results are consistent with our previous findings that AMPK induces a decrease in apical V-ATPase accumulation in epididymal clear cells and intercalated cells in kidney slices (16, 18).
Fig. 1.

The AMPK activator 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) inhibits the V-ATPase-mediated intracellular pH (pHi) recovery in rabbit isolated, perfused outer medullary collecting ducts after an acute acid load. A: fluorescence micrographs of a single microperfused rabbit outer medullary collecting duct (OMCD), each captured at a slightly different focal plane. The images show increased accumulation of the pH-sensitive dye BCECF loaded from the bath as the acetoxymethyl ester (left) in intercalated cells (IC; marked with asterisks). In the middle panel, the lectin Dolichos biflorus agglutinin coupled to rhodamine (rhodamine-DBA) decorates principal cells (PC; marked with carets). In the right panel (merge), the IC show higher fluorescence intensity (green pseudocolor), while the PC are identified by the rhodamine-DBA caps (red pseudocolor). B: representative patterns of pHi recovery observed in IC of rabbit OMCD in response to an acute in vitro acid load (NH4Cl prepulse) in the absence (left) or presence (right) of the AMPK activator AICAR (2 mM) included in the bath for 1 h before and continued throughout the experiment shown. C: under both conditions, cytosolic acidification from an initial pHi of ∼7.3 in Na+-Ringer (NaR) buffer to a nadir pHi of ∼6.3 was accomplished following a 3-min bath exposure to NH4Cl. D: partial pHi recovery was observed in the absence of Na+ and K+ (0Na/0K) and a nominal absence of HCO3−/CO2 at a rate of 0.15 ± 0.02 pH U/min in control untreated OMCD, while the pHi recovery was only 0.04 ± 0.03 pH U/min in the presence of the AICAR. Values are means ± SE. Reintroduction of Na+ to the bathing solution in all tubules led to full recovery of the pHi to ∼7.3 (n = 4 OMCD in each group. *P < 0.05 vs. control).
We also studied whether the AMPK activator AICAR affected pHi of intercalated cells during the different sections of the perfusion protocol (as shown in Fig. 1B). For example, after 1 h of incubation, the resting pHi of intercalated cells was 7.30 ± 0.06 in the control tubules incubated in Na+-Ringer buffer and 7.29 ± 0.04 in AICAR-treated tubules (n = 4) (Fig. 1C). Tubules were then exposed basolaterally for 3 min to 30 mM NH4Cl before a rapid washout to induce intracellular acidification in Na+-, K+-, and nominally HCO3−-free buffer, as described in materials and methods. Control tubules reached a nadir pHi of 6.25 ± 0.12 after the NH4Cl washout, which was not significantly different from the nadir pHi of 6.34 ± 0.13 reached by tubules incubated with AICAR (Fig. 1C). In the absence of Na+, K+, and in the nominal absence of HCO3−, the rate of pHi recovery of intercalated cells was 0.15 ± 0.02 in control tubules vs. 0.04 ± 0.03 in AICAR-treated tubules (P < 0.05) (Fig. 1D). These findings indicate that V-ATPase activity, as measured by the rate of pHi recovery after an acid load in the absence of Na+ and K+ and in the nominal absence of HCO3− (9), is inhibited by AMPK activator in OMCD intercalated cells ex vivo. This downregulation occurs within the time frame AICAR usually takes to induce AMPK activation in kidney slices ex vivo (16). The results of these experiments complement our prior immunofluorescence imaging findings that the AMPK activator AICAR induced V-ATPase redistribution to the cytosol in intercalated cells in the ex vivo kidney slices system (16). Together, these data strongly suggest that when AMPK becomes activated in the kidney collecting duct, such as under conditions of metabolic stress, this kinase inhibits V-ATPase activity, which would act to help preserve cellular energy stores by inhibiting V-ATPase-associated ATP consumption.
AMPK phosphorylates the V-ATPase A subunit at Ser-384 in vitro.
We have previously shown that the V-ATPase A subunit can be phosphorylated by AMPK (18, 52). However, the functionally relevant AMPK phosphorylation site(s) within the A subunit or other V-ATPase subunits have not yet been investigated. We identified and quantified phosphorylated peptides from the WT-A subunit phosphorylated in vitro by AMPK on a MALDI target plate before MS analysis after liquid chromatography and a MALDI-MS (LC-MALDI-MS) workflow (51). We previously used this approach to identify candidate PKA phosphorylation sites in the V-ATPase A subunit (2). We identified by this technique a major phospho-labeled tryptic peptide fragment that eluted in fraction C20 (Fig. 2, A and B). We detected in this elution peak a phosphorylated peptide corresponding to the expected molecular mass of a tryptic fragment with sequence LASFYER. Sequence confirmation was obtained by fragmentation of this peptide using MSMS (Fig. 2, C and D). In this peptide fragment sequence (underlined in Fig. 2E), only Ser-384 (bolded in Fig. 2E) fits within a loose consensus AMPK phosphorylation target motif (10). The amino acid sequence around this residue is highly conserved in the V-ATPase A subunit of vertebrates (Fig. 2F). This finding suggests that AMPK phosphorylation at this site could play a fundamental role in V-ATPase function during conditions that induce AMPK activation.
Fig. 2.

AMP-activated protein kinase (AMPK) phosphorylates the V-ATPase A subunit at Ser-384 deduced by matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS and MALDI-MSMS). FLAG-tagged wild-type V-ATPase A subunit (WT-A) was expressed in and then immunoprecipitated from HEK-293 cell lysates. Immunoprecipitated WT-A was then incubated with a substoichiometric amount of recombinant AMPK in the presence of [γ-32P]ATP. After this phosphorylation reaction, the proteins were subjected to SDS-PAGE followed by autoradiography. The band corresponding to the WT-A subunit was then excised and subjected to in-gel digestion with trypsin as described in materials and methods. A: autoradiography of a prespotted AnchorChip showing the fractionation profile of the WT-A V-ATPase subunit phosphorylated by AMPK after in-gel digestion and microfractionation. The spotting occurred from A1 to A24 and continued from B24 to B1 then C1 to C24 and D24 to D1, etc. The arrows denote the directionality of the spotting. B: analysis by densitometry of individual radioactive spots identified after the microfractionation (shown in A). The phosphorylated peptides were quantified using the appropriate standards, and the major radioactive peptide peak was eluted with fraction C20. C: characterization and identification of the V-ATPase A subunit phosphorylated peptide by MALDI-MS. The peptide of spot C20 with a mass of 965.4 Da showed predominant neutral mass losses of 80 Da (−HPO3) and 98 Da (−H3PO4). D: the MSMS fragmentation spectrum of the precursor identified the tryptic peptide to be LASFYER (with the unphosphorylated peptide having a mass of Mr 867.4 Da). E: the amino acid sequence for the mouse FLAG-tagged WT-A subunit is shown, spotlighting the highly phosphorylated peptide by AMPK (underlined C20 fraction), and Ser-384 (asterisk). F: Ser-384 (bold and asterisk) is part of a highly conserved region that appears to conform to the loose consensus AMPK target phosphorylation motif (underlined residues).
To further confirm that Ser-384 is a specific AMPK phosphorylation site, we generated a putative phosphorylation-deficient FLAG-tagged A subunit mutant with Ser-384 mutated to Ala (S384A-A) and then performed in vitro phosphorylation experiments on immunoprecipitated WT-A and S384A-A subunits (2, 18). We then compared in vitro phosphate labeling of these two V-ATPase A subunits exposed to a purified active AMPK holoenzyme in the presence of [γ-32P]-ATP (Fig. 3, A and B). The S384A-A V-ATPase mutant had a >90% reduction in 32P labeling relative to the WT-A subunit after normalization to the amount of immunoprecipitated protein, thus confirming AMPK-dependent phosphorylation at Ser-384 in vitro.
Fig. 3.

AMPK phosphorylation of the A subunit occurs at Ser-384 in vitro. A: representative phosphoscreen image (top) revealing the signal of the AMPK in vitro phosphorylated V-ATPase WT-A subunit compared with the Ser-384-to-Ala mutant. The immunoblot using an anti-FLAG antibody (bottom) confirms similar protein expression and loading of the gel for the different conditions. B: quantification of the V-ATPase A subunit phosphorylation signal normalized for protein loading as assessed by densitometry of the immunoblot. Values are means ± SE. Compared with the WT-A subunit, the phosphorylation-deficient (Ser-to-Ala) mutant showed a significant 90–95% decrease in phosphorylation by AMPK in vitro. *P < 0.05 relative to WT, unpaired t-test; n = 3.
AMPK-dependent phosphorylation of the V-ATPase A subunit occurs at Ser-384 in intact HEK-293 cells.
To determine whether Ser-384 is a target for AMPK-dependent phosphorylation in intact cells, we compared [32P]orthophosphate labeling of the WT-A and S384A-A mutant subunits transfected into HEK-293 cells with AMPK activity modulation. In these cells, AMPK activity is generally high at baseline, and AICAR treatment has little AMPK-activating effect (Hallows KR, unpublished observations). Therefore, to modulate AMPK activity we used a previously characterized cell line derived from HEK-293 cells that allows for inducible AMPK knockdown (AMPK-KD) (2, 18). This cell line was generated by stable transfection with a plasmid that expresses shRNA directed against AMPK-α1. We transfected WT-A or the S384A-A phosphorylation-deficient mutant into HEK-293 cells expressing either a nontargeting shRNA (Fig. 4A, CONTROL) or into AMPK-KD cells (Fig. 4A, AMPK-KD). After transfection, all cells were grown in the presence of doxycycline, which induces expression of anti-AMPKα shRNA in AMPK-KD cells (18).
Fig. 4.

Ser-384 is the target for AMPK-dependent phosphorylation of the V-ATPase A subunit in HEK-293 cells. Plasmids encoding FLAG-tagged WT-A or the S384A-A mutant were transfected into HEK-293 cells induced to express either an irrelevant mammalian short hairpin (sh) RNA (CONTROL) or an shRNA for AMPK-α1 knockdown (AMPK-KD) 2 days before experimentation. The cells were then incubated with [32P]orthophosphate for 2 h in the presence of a PKA inhibitor (10 μM mPKI) for the entire labeling period. Cell lysis, immunoprecipitation using an anti-FLAG antibody, SDS-PAGE, immunoblotting with anti-FLAG antibody, and exposure of the same membrane to a phosphoscreen were then performed as described in materials and methods. A: representative phosphoscreen image (top) revealing the signal of the phosphorylated A subunit in cells expressing the WT-A subunit or S384A-A subunit. The immunoblot (bottom) confirms similar protein expression and loading of the gel for the different conditions. B: quantification of the V-ATPase A subunit phosphorylation signal relative to WT-A control condition and normalized for protein expression. Values are means ± SE. *P < 0.05 relative to WT control. #P < 0.05 relative to WT control (one-tailed t-test); n = 3 replicate experiments. C: representative immunoblot analysis of cell lysates from each condition for the active form of AMPK using an antibody raised against pThr-172-AMPK-α. β-Actin was used as a loading control to normalize pThr-172-AMPK-α across conditions. D: summary of relative AMPK activity (pThr-172-AMPK-α intensity corrected to β-actin as a loading control). Values are means ± SE normalized to that of control cells expressing the WT A subunit. *P < 0.05 relative to control cells expressing WT-A. E: representative phosphoscreen image of total [32P]orthophosphate protein labeling in cellular lysates from the above experiments (top). Also shown is a representative immunoblot for the loading control β-actin (bottom). F: the relative [32P]orthophosphate signal to β-actin was not significantly different across conditions.
To minimize PKA-mediated phosphorylation of the A subunit in these studies, [32P]orthophosphate incubation was carried out in the presence of mPKI, a specific PKA inhibitor. After the [32P]orthophosphate-labeling period, WT-A and S384A-A subunits were immunoprecipitated from cell lysates and subjected to SDS-PAGE and immunoblotting. The A subunit 32P signal on the phosphoscreen (Fig. 4A, top) was normalized to the respective A subunit expression signal on the immunoblot (Fig. 4A, bottom) from the same membrane. Inhibition of cellular AMPK activity by RNA interference significantly decreased phosphorylation of the WT-A subunit by ∼60% relative to control cells expressing an irrelevant shRNA, thus confirming our previous findings that AMPK is a relevant kinase for A subunit phosphorylation in intact cells (Fig. 4B, wild-type CONTROL vs. AMPK-KD) (18). AMPK knockdown was confirmed by immunoblotting for pThr172-AMPK-α corrected for β-actin expression as a loading control and then normalized to that of control cells expressing the WT-A subunit (Fig. 4, C and D).
The phosphorylation level of the S384A-A mutant expressed in control cells was ∼60% lower than that of the WT-A subunit expressed in control cells, indicating that Ser-384 is an important residue for A subunit phosphorylation in intact cells in the presence of a PKA inhibitor (Fig. 4B). Moreover, the low levels of phosphorylation observed in S384A-A subunits expressed in CONTROL cells were similar to the levels of the same subunits expressed in AMPK-KD (Fig. 4B, right). Overall, these findings indicate that Ser-384 in the V-ATPase A subunit is the main target of AMPK phosphorylation in the intact cellular environment.
Finally, we also tested whether AMPK knockdown could interfere with orthophosphate uptake into the cells, which could affect labeling of phosphoproteins in these experiments. As shown in Fig. 4, E and F, there was no significant difference in the overall cellular orthophosphate uptake between conditions.
AMPK mediates inhibition of extracellular acidification in HEK-293 cells via Ser-384.
We then directly assessed the role of residue Ser-384 in the A subunit on the activity of the V-ATPase at the plasma membrane. We first chose HEK-293 cells for these experiments because these cells are of human kidney origin, they express the V-ATPase at the plasma membrane, and they can regulate their pHi via the V-ATPase (25). We performed transfections as described above by expressing WT-A or the S384A-A AMPK phosphorylation-deficient mutant into HEK-293 cells stably transfected to express either an irrelevant shRNA (CONTROL) or AMPK-KD cells. Transfected cells were then incubated in equal volumes of culture medium, and we measured the pHo of the medium 1 day after transfection (Fig. 5). The baseline pH of HEK-293 cell culture medium is generally in the range of 7.48 ± 0.01 at 37°C in a 5% CO2 incubator (2). In AMPK-KD cells expressing WT-A (Fig. 5, AMPK-KD, WT-A), the chronic pHo was slightly, but significantly more acidic than in control cells also expressing the WT-A (Fig. 5, CONTROL, WT-A). This result suggests that under steady-state conditions tonic AMPK activity inhibits either proton secretion or base reabsorption from HEK-293 cells. We also examined the role of Ser-384 in the A subunit on the ability of HEK-293 cells to acidify the culture medium. With control cells expressing the S384A-A mutant (Fig. 5, CONTROL, S384A-A), pHo became more acidic than with control cells expressing WT-A, which is consistent with the idea that the inability to phosphorylate Ser-384 in the A subunit chronically promotes V-ATPase activity at the plasma membrane and thus extracellular acidification.
Fig. 5.

Expression of WT and mutant V-ATPase A subunit in HEK-293 cells modulates extracellular pH (pHo). Shown is quantification of chronic pHo from media incubated with HEK-293 cells for 28–31 h. The cells expressed either an irrelevant mammalian shRNA (Control) or an shRNA targeting knockdown of AMPK-α1 (AMPK-KD) and had been transfected with either WT-A or S384A-A subunits. *P < 0.05 relative to WT-A Control. #P < 0.05 relative to WT-A AMPK-KD; n = 4.
In the setting of AMPK knockdown (AMPK-KD), cells transfected with the AMPK phosphorylation-deficient S384A-A mutant had a lower medium pHo than cells expressing WT-A. Thus the inability of Ser-384 in the A subunit to become phosphorylated in these cells appears to promote acid secretion, most likely via inhibition of the V-ATPase, more strongly than does AMPK knockdown. One potential explanation for this effect is that knockdown of AMPK activity is incomplete in these cells (Fig. 4D), so residual AMPK function may still be sufficient to dampen functional V-ATPase expression at the apical membrane. Moreover, it is possible that an additional kinase or kinases may also target Ser-384 in cells, such that complete ablation of phosphorylation at that site has a more powerful effect than (partial) knockdown of AMPK alone. The increased extracellular acidification conferred by the S384A-A mutation was not significantly affected by changes in AMPK activity (see Fig. 5, CONTROL, S384A-A vs. AMPK-KD, S384A-A), suggesting that the AMPK-dependent effect on extracellular acidification is mediated largely via V-ATPase A subunit Ser-384 phosphorylation. Overall, these findings point to a common mechanism for extracellular acidification from HEK-293 cells that is inhibited by AMPK activity and requires an intact Ser-384 residue in the V-ATPase A subunit.
AMPK regulates acid secretion by the V-ATPase via Ser-384 in clone C cells of intercalated cell origin.
In additional experiments, we monitored pHo changes under acute conditions (over 20-min intervals) in clone C cells, which are of intercalated cell origin (53), seeded at equal densities into 24-well plates and at equivalent transfection efficiencies with either the WT-A or S384-A mutant subunit. One day later, the cells were incubated for 4 h in the presence or absence of the AMPK activator AICAR. The V-ATPase-dependent extracellular buffer acidification rates were measured as described in materials and methods. We replaced the culture medium with a low-buffering-capacity solution in the continued presence or absence of AICAR for the duration of the protocol (2). This buffer was prepared without Na+ to minimize any contribution of Na+/H+ exchange to pHo, in the absence PKA agonists, and in the nominal absence of HCO3− to minimize sAC activity, which could independently increase intracellular cAMP concentration and PKA activity (37, 57). WT-A subunit-expressing cells exhibited a significant decrease in the V-ATPase dependent acidification rate {expressed as [−(final buffer pH − initial buffer pH)/Δt]} following AICAR treatment (Fig. 6). In contrast, the extracellular acidification rate of cells expressing the S384A-A subunit was unaffected by AICAR treatment. Taken together, these results suggest that Ser-384 in the A subunit is required for the AMPK-mediated downregulation of V-ATPase activity in intercalated cells.
Fig. 6.

The Ser-384 A subunit mutant modulates bafilomycin-sensitive, V-ATPase-dependent extracellular acidification with AMPK activation in clone C cells. Clone C IC were transiently transfected with either the WT-A or S384A-A subunit. Twenty-four hours posttransfection, the cells were treated ± AICAR (2 mM) for 4 h and then pHo measurements were performed. The rate of extracellular acidification under each indicated condition was measured in a low-buffering-capacity solution before and after the addition of bafilomycin A1, a specific V-ATPase inhibitor (see materials and methods). The bafilomycin-sensitive rate of extracellular acidification [−(final buffer pH − initial buffer pH)/Δt] was obtained for cells incubated either with or without the AMPK activator AICAR (n = 5 for each transfection and treatment condition). Values are means ± SE. The transfection efficiency in each well was calculated as described in materials and methods, and no significant differences were observed across all conditions. *P < 0.01 relative to untreated cells transfected with the WT-A subunit.
Ser-384 is required for V-ATPase cytosolic redistribution of the V-ATPase during AMPK activation in clone C cells of intercalated cell origin.
To examine whether the role of Ser-384 in V-ATPase-mediated proton secretion correlated with changes in its subcellular localization, we performed immunofluorescence labeling experiments in clone C cells transfected with either WT-A or S384A-A constructs and grown on filter supports (2). In clone C cells, we have shown that transfected WT-A subunits incorporate into endogenous V-ATPase complexes (2). In contrast to HEK-293 cells, clone C cells also form monolayers when grown on filter supports. Moreover, the apical membrane of polarized clone C cells can be labeled with the lectin concanavalin A, coupled to the fluorophore CY3 (ConA-CY3), before fixation (2). Clone C cells transfected with either WT-A or S384-A were grown on filter supports and treated with either the AMPK activator AICAR or vehicle control for 4 h. For each of the four conditions, three to five filters were processed for immunofluorescence labeling (Fig. 7, A and B) while another three filters per condition were used to prepare cell lysates to measure the effects of AICAR treatment on AMPK activation, as measured by pThr-172-AMPK-α immunoblots (Fig. 7, C and D). After normalization of the p-AMPK to the β-actin loading control (Fig. 7C, bottom), we found that AICAR treatment significantly increased the activated form of AMPK to ∼300 ± 50% of control levels in this cell line (Fig. 7D).
Fig. 7.
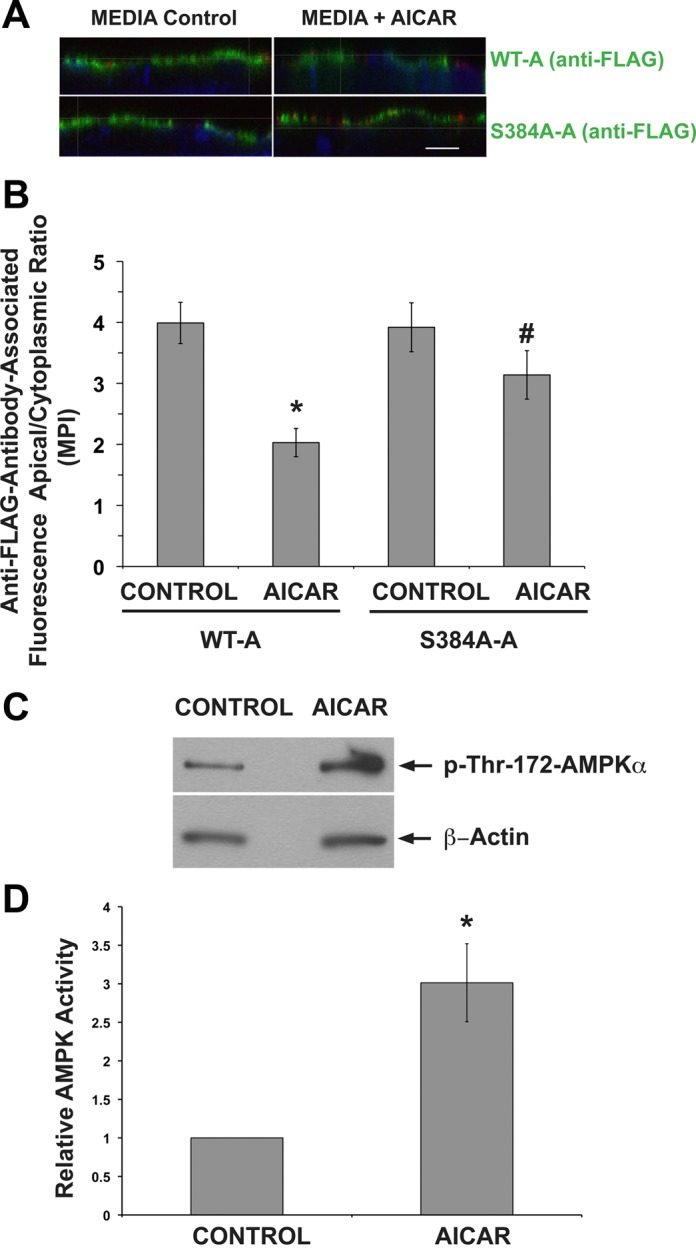
The AMPK phosphorylation-deficient S384A V-ATPase A subunit mutant remains at the apical membrane of IC in response to an AMPK activator. Clone C cells were transiently transfected with either the WT-A or S384A-A subunit. A: 1 day after transfection with either the WT-A (top) or S384A-A mutant subunit (bottom), clone C cells were plated onto Transwell filters. After 4 days, the filters were incubated for 4 h in media (left) or in media with 2 mM AICAR (right). Filters were then incubated with concanavalin A coupled to CY3 (red), fixed, and immunofluorescently labeled using anti-FLAG antibody (green) and TO-PRO-3 nuclear stain (blue). Scale bar = 10 μM. B: quantification of V-ATPase-associated MPI ratio of apical region of interest (ROI-1; here the A subunit colocalizes with concanavalin A) and cytoplasmic ROI-2 (A subunit alone). This ROI-1/ROI-2 ratio under the different conditions reveals a significant AICAR-mediated inhibition of apical V-ATPase accumulation in cells expressing the WT A subunit compared with cells expressing the S384A mutant. Values are means ± SE. *P < 0.05 vs. WT-A Control. #P < 0.05 vs. WT-A AICAR; n = 20–45 cells analyzed for both conditions. C: representative immunoblots of pThr-172-AMPKα and β-actin in clone C cell lysate filters treated with media alone (left) or with media+AICAR (right). D. quantification of the ratio of pThr-172-AMPK-α to β-actin Western blot signals shown in C normalized to Control as a measure of relative AMPK activity. *P < 0.05; n = 3.
Immunofluorescence labeling of the A subunit using an anti-FLAG antibody followed by confocal fluorescence microscopy (Fig. 7A; green) revealed that cells transfected with either WT-A or with S384A-A in untreated media exhibited A-subunit-associated immunolabeling (Fig. 7A, left, green) that was largely at or near the plasma membrane (labeled with ConA-CY3, red), with less prominent cytosolic staining. In the presence of media, the apical V-ATPase distribution in clone C cells was expected as the components of the media include bicarbonate and several growth factors that may activate PKA, which induces apical V-ATPase accumulation and activity (2, 16, 41). On the other hand, when clone C monolayers expressing the WT-A subunit were incubated in the presence of the AMPK activator AICAR, the V-ATPase-associated fluorescence was mostly cytosolic (Fig. 7A, top right). The cytosolic redistribution of the A subunit in response to AICAR treatment was not observed in clone C cells transfected with the S384A-A mutant (Fig. 7A, bottom right). Quantifications of the apical-to-cytoplasmic MPI of FLAG-associated fluorescence in these confocal stack reconstructions are shown in Fig. 7B. We confirmed a significantly lower apical accumulation of the WT-A subunit in clone C cells exposed to the AMPK activator AICAR compared with vehicle control (*P < 0.05). In addition, we confirmed the unresponsiveness of the S384A-A mutant to cytosolic redistribution by AICAR, as evidenced by its more apical distribution than that of WT-A-expressing cells treated with AICAR (#P < 0.05). Taken altogether, our results suggest that A subunit phosphorylation at Ser-384 by AMPK plays a functional role in the cytosolic subcellular distribution of the V-ATPase induced by the AMPK activator AICAR.
Finally, we performed coimmunoprecipitation experiments to determine whether the exogenously expressed WT-A and S384A-A subunits incorporated into endogenous V-ATPase complexes. We used a specific antibody that recognizes the endogenous V-ATPase V0 sector a (ATP6V0A1) subunit to reprobe immunoblots of immunoprecipitated FLAG-tagged A subunits (WT-A and S384A-A). These coimmunoprecipitation blots demonstrate that both the WT-A and S384A-A subunits form complexes with the V0 sector a subunit (Fig. 8A). Moreover, these experiments suggest that there is no significant difference in apparent binding affinity between the V0 a subunit and either the WT-A subunit or the S384A-A subunit of the V1 sector (Fig. 8B).
Fig. 8.

The AMPK phosphorylation-deficient S384A V-ATPase A subunit mutant coimmunoprecipitates with the V0 sector of the a subunit. Clone C cell monolayers were transiently transfected to express either the WT-A or S384A-A subunit. A: representative set of immunoblots of an immunoprecipitation experiment using anti-FLAG antibody (IP FLAG; left) followed by immunoblotting with antibodies against FLAG (top) and the V0 sector V-ATPase a subunit (bottom). Whole cell lysate samples (4.5% of total) of the transfected monolayers were also directly immunoblotted for the A subunit (top right) and the a subunit (bottom right). B: results shown are the apparent a subunit binding (coimmunoprecipitation) relative to the WT-A subunit. Values are means ± SE. Densitometric quantitation of the relevant bands and determinations of relative binding strength were performed as described in materials and methods (n = 3).
DISCUSSION
Researchers studying the V-ATPase in the kidney and other epithelia have been interested in linking this transport protein complex with cellular metabolism. The V-ATPase is present in almost all cells in the body, where it is involved in the acidification of intracellular organelles. However, certain epithelial cells have very abundant V-ATPase, including kidney intercalated cells and epididymal clear cells (reviewed in Ref. 55). The V-ATPase consumes one ATP per two protons translocated, and this transport can generate a steep proton gradient. This pump could therefore be considered as a potential target of AMPK, which would fit well with the role of this kinase as a regulator of cellular energy balance, especially in cells with high levels of V-ATPase expression. There are now several examples of AMPK-dependent inhibition of ion transport proteins, such as CFTR, a channel gated by ATP binding and hydrolysis, the creatine transporter, and the epithelial Na+ channel (ENaC) (reviewed in Ref. 39). In addition, AMPK inhibition of downhill ion transport proteins such as ENaC should also indirectly inhibit ATP consumption by preventing the dissipation of ion gradients maintained by the Na+-K+-ATPase, limiting its activity and indirectly decreasing cellular ATP consumption (17).
Our group initially implicated AMPK in the trafficking of the V-ATPase in epithelial proton-secreting cells of the epididymis in vivo and in ex vivo kidney slices (16, 18). We also showed that the V-ATPase A subunit in the cytoplasmic V1 sector is phosphorylated in vitro and in HEK cells by both PKA and AMPK. Our results thus confirmed prior findings that the V-ATPase A subunit was a potential direct AMPK phosphorylation target in an unbiased substrate screen (52). The present study uncovers a molecular mechanism of V-ATPase regulation by AMPK in kidney intercalated cells via direct phosphorylation of the V-ATPase A subunit by this metabolic sensing kinase. To our knowledge, this study is the first to characterize the functional role of an AMPK phosphorylation site in any of the V-ATPase subunits in mammalian cells.
In the current study, we confirmed that V-ATPase activity is downregulated by AMPK in the intercalated cells of isolated, perfused collecting ducts (Fig. 1). We then identified Ser-384 as a target for AMPK phosphorylation by mass spectrometry and candidate site mutagenesis with subsequent phosphorylation studies in vitro and in HEK-293 cells (Figs. 2 and 3). Immunolocalization studies performed in clone C cells indicated that AMPK activation induces cytoplasmic localization of the wild-type A subunit, while the S384A mutant remained at the membrane despite AMPK activation (Fig. 7). In the nominal absence of extracellular bicarbonate and Na+, the proton-secreting activity of clone C cells transfected with the A subunit was largely bafilomycin-sensitive and thus V-ATPase-dependent (Fig. 6). Moreover, under these conditions AICAR inhibited extracellular acidification by the V-ATPase in cells transfected with the WT-A subunit but failed to inhibit V-ATPase-dependent proton secretion in cells transfected with the AMPK phosphorylation-deficient S384A-A mutant. Taken together, these findings suggest that phosphorylation at Ser-384 in the A subunit by AMPK plays a key role in the AMPK-dependent inhibition of V-ATPase functional expression at the apical plasma membrane in kidney intercalated cells. This inhibition of V-ATPase-mediated proton secretion by AMPK may be important for cell survival under conditions of kidney hypoperfusion or ischemia when the organism may be in an acidotic state. Moreover, AMPK inhibition of the V-ATPase would act to preserve cellular ATP at a time when proton pumping into the lumen may be compromised due to reduced luminal flow rates.
We also studied whether the phosphorylation-deficient S384A A subunit mutant induced changes in the affinity of this V1 sector subunit for the V0 sector a subunit. Research on the assembly of the V-ATPase in yeast (reviewed in Ref. 23) established that the A subunit of the V1 sector associates at a faster rate with the V0 sector a subunit than with other subunits of the V1 sector (reviewed in Ref. 35). Of note, however, our findings suggest that the association of the V0 and V1 sectors at steady state is not affected by mutations at Ser-384, as discerned by coimmunoprecipitation studies of the V1 sector A subunit and the V0 sector a subunit (Fig. 8).
Of note, the sequences surrounding the PKA and AMPK phosphorylation sites in the A subunit are highly conserved in eukaryotes, including yeast. Although the PKA site at Ser-175 in the A subunit is in the nonhomologous, glucose-sensing region of the A subunit, the Ser-384 target AMPK phosphorylation site is outside of this domain. It has been proposed that this so-called nonhomologous domain may link pump function to metabolic status (15, 45, 46). It is intriguing that the AMPK phosphorylation site lies outside of this region, which underscores the fact that the mechanisms linking V-ATPase phosphorylation status to intracellular localization and function are still unclear.
Based on the analysis of early identified AMPK substrates, a loose consensus target motif for AMPK phosphorylation was proposed [Φ-(K/R,X)-X-X-S/T-X-X-X-Φ; where a hydrophobic residue (Φ) is 5 residues upstream of the phosphorylation position (P-5) and 4 residues downstream (P+4), a basic residue (K, R, or H) is at the P-4 and/or P-3 positions, and S or T is at the phosphorylated (P = 0) position] (10). However, more recent studies have identified relevant AMPK phosphorylation sites that do not conform to this consensus motif in other proteins, such as endothelial nitric oxide synthase and phosphofructokinase-2 (8, 22, 32). Indeed, in the present study we found that AMPK phosphorylated Ser-384, a site that is preceded by the hydrophobic Leu residue at the P-6 position instead of P-5 and that lacks a hydrophobic residue at the P+4 position. Our results underscore the challenges of predicting an AMPK phosphorylation site by sequence inspection or prediction algorithms alone.
Our previous work indicated that PKA and AMPK have opposing effects on the subcellular localization and activity of the V-ATPase in epididymal clear cells and kidney intercalated cells. Specifically, PKA phosphorylation of the A subunit at Ser-175 promotes V-ATPase apical membrane localization. The accumulation of the V-ATPase at the apical membrane of intercalated cells and its increased activity in response to PKA activation can occur after a variety of stimuli, including changes in extracellular [HCO3−] and pH, and is dependent on the activity of carbonic anhydrase and sAC (16). Conversely, AMPK activation generally antagonizes the effects of PKA stimulation on V-ATPase localization and activity. The apparent mutual antagonism between AMPK and PKA on regulation of the V-ATPase are similar to the findings from our previous work in lung epithelial cells showing that AMPK phosphorylates and inhibits the PKA-dependent activation of CFTR gating (19). Moreover, preliminary data suggest that AMPK blocks the PKA-mediated stimulation of ENaC downstream of vasopressin in kidney collecting duct cells (26). The mechanisms by which AMPK regulates the PKA-dependent accumulation of the V-ATPase at the apical plasma membrane are currently unclear. However, it is notable that our earlier work suggested that AMPK knockdown accentuates PKA-dependent phosphorylation of the V-ATPase A subunit in HEK cells (2). It is unclear whether this AMPK-inhibitory effect on PKA phosphorylation is direct or indirect.
In summary, additional studies are needed to better define the mechanisms by which both PKA-dependent phosphorylation at Ser-175 and AMPK-dependent phosphorylation at Ser-384 in the A subunit modulate plasma membrane accumulation and activity of the V-ATPase in intercalated cells. For example, it is conceivable that AMPK phosphorylation of the A subunit disturbs protein-protein interactions of V-ATPase subunits with regulatory proteins established by the PKA-mediated phosphorylation of Ser-175 in the same subunit. Future studies will be required to elucidate the coregulation of the trafficking and activity of the V-ATPase via phosphorylation at the highly conserved residues Ser-175 and Ser-384 by PKA and AMPK. Specifically, it will be important to define how PKA-dependent phosphorylation at Ser-175 may couple the sensing of acid-base status to pump function while AMPK-dependent phosphorylation at Ser-384 may couple its activity to metabolic stresses.
GRANTS
This study was supported by National Institutes of Health Grants P30 DK079307 (Pittsburgh Kidney Research Center), R01 DK-075048 (to K. R. Hallows), and R01 DK-084184 (to N. M. Pastor-Soler), an American Society of Nephrology Carl W. Gottschalk Research Scholar Award (to N. M. Pastor- Soler), American Heart Association Grant AHA 09GRNT2060539 (to N. M. Pastor-Soler) and postdoctoral fellowship 0825540D (to R. Alzamora), EU FP6 contract LSHM-CT-2004-005272 (EXGENESIS), Netherlands Organization for Scientific Research VIDI Grant (864.10.007), a graduate training fellowship ETHIIRA (to R. F. Thali), and a Sanofi Research Fellowship Grant (to M. Al-bataineh).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: R.A., M.A.-b., W.L., F.G., H.L., R.F.T., Y.J.-A., R.A.B., L.M.S., D.N., K.R.H., and N.M.P.-S. provided conception and design of research; R.A., M.A.-b., W.L., F.G., H.L., R.F.T., Y.J.-A., R.A.B., D.N., and N.M.P.-S. performed experiments; R.A., M.A.-b., W.L., F.G., H.L., R.F.T., Y.J.-A., R.A.B., L.M.S., D.N., K.R.H., and N.M.P.-S. analyzed data; R.A., M.A.-b., W.L., F.G., H.L., R.F.T., Y.J.-A., R.A.B., L.M.S., D.N., K.R.H., and N.M.P.-S. interpreted results of experiments; R.A., M.A.-b., W.L., F.G., H.L., R.F.T., Y.J.-A., R.A.B., D.N., K.R.H., and N.M.P.-S. prepared figures; R.A., M.A.-b., W.L., R.A.B., L.M.S., D.N., K.R.H., and N.M.P.-S. drafted manuscript; R.A., R.A.B., L.M.S., D.N., K.R.H., and N.M.P.-S. edited and revised manuscript; R.A., M.A.-b., W.L., F.G., H.L., R.F.T., Y.J.-A., R.A.B., D.N., K.R.H., and N.M.P.-S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Qais Al-Awqati for the generous gift of clone C rabbit intercalated cells. We thank Christy Smolak and Heather Bladek for technical assistance and also gratefully acknowledge the excellent technical assistance and teaching skills of Beth Zavilowitz.
REFERENCES
- 1.Al-Awqati Q, Gao XB. Differentiation of intercalated cells in the kidney. Physiology (Bethesda) 26: 266–272, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Alzamora R, Thali RF, Gong F, Smolak C, Li H, Baty CJ, Bertrand CA, Auchli Y, Brunisholz RA, Neumann D, Hallows KR, Pastor-Soler NM. PKA regulates vacuolar H+-ATPase localization and activity via direct phosphorylation of the A subunit in kidney cells. J Biol Chem 285: 24676–24685, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bens M, Vallet V, Cluzeaud F, Pascual-Letallec L, Kahn A, Rafestin-Oblin ME, Rossier BC, Vandewalle A. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol 10: 923–934, 1999 [DOI] [PubMed] [Google Scholar]
- 4.Bhalla V, Oyster NM, Fitch AC, Wijngaarden MA, Neumann D, Schlattner U, Pearce D, Hallows KR. AMP-activated kinase inhibits the epithelial Na+ channel through functional regulation of the ubiquitin ligase Nedd4–2. J Biol Chem 281: 26159–26169, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Bouley R, Pastor-Soler N, Cohen O, McLaughlin M, Breton S, Brown D. Stimulation of AQP2 membrane insertion in renal epithelial cells in vitro and in vivo by the cGMP phosphodiesterase inhibitor sildenafil citrate (Viagra). Am J Physiol Renal Physiol 288: F1103–F1112, 2005 [DOI] [PubMed] [Google Scholar]
- 6.Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci USA 85: 7972–7976, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carattino MD, Edinger RS, Grieser HJ, Wise R, Neumann D, Schlattner U, Johnson JP, Kleyman TR, Hallows KR. Epithelial sodium channel inhibition by AMP-activated protein kinase in oocytes and polarized renal epithelial cells. J Biol Chem 280: 17608–17616, 2005 [DOI] [PubMed] [Google Scholar]
- 8.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett 443: 285–289, 1999 [DOI] [PubMed] [Google Scholar]
- 9.Constantinescu A, Silver RB, Satlin LM. H-K-ATPase activity in PNA-binding intercalated cells of newborn rabbit cortical collecting duct. Am J Physiol Renal Physiol 272: F167–F177, 1997 [DOI] [PubMed] [Google Scholar]
- 10.Dale S, Wilson WA, Edelman AM, Hardie DG. Similar substrate recognition motifs for mammalian AMP-activated protein kinase, higher plant HMG-CoA reductase kinase-A, yeast SNF1, and mammalian calmodulin-dependent protein kinase I. FEBS Lett 361: 191–195, 1995 [DOI] [PubMed] [Google Scholar]
- 11.Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J 29: 469–481, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dobyan DC, Magill LS, Friedman PA, Hebert SC, Bulger RE. Carbonic anhydrase histochemistry in rabbit and mouse kidneys. Anat Rec 204: 185–197, 1982 [DOI] [PubMed] [Google Scholar]
- 13.Edwards JC, van Adelsberg J, Rater M, Herzlinger D, Lebowitz J, al-Awqati Q. Conditional immortalization of bicarbonate-secreting intercalated cells from rabbit. Am J Physiol Cell Physiol 263: C521–C529, 1992 [DOI] [PubMed] [Google Scholar]
- 14.Estilo G, Liu W, Pastor-Soler N, Mitchell P, Carattino MD, Kleyman TR, Satlin LM. Effect of aldosterone on BK channel expression in mammalian cortical collecting duct. Am J Physiol Renal Physiol 295: F780–F788, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forgac M. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol 8: 917–929, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Gong F, Alzamora R, Smolak C, Li H, Naveed S, Neumann D, Hallows KR, Pastor-Soler NM. Vacuolar H+-ATPase apical accumulation in kidney intercalated cells is regulated by PKA and AMP-activated protein kinase. Am J Physiol Renal Physiol 298: F1162–F1169, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hallows KR. Emerging role of AMP-activated protein kinase in coupling membrane transport to cellular metabolism. Curr Opin Nephrol Hypertens 14: 464–471, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Hallows KR, Alzamora R, Li H, Gong F, Smolak C, Neumann D, Pastor-Soler NM. AMP-activated protein kinase inhibits alkaline pH- and PKA-induced apical vacuolar H+-ATPase accumulation in epididymal clear cells. Am J Physiol Cell Physiol 296: C672–C681, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallows KR, McCane JE, Kemp BE, Witters LA, Foskett JK. Regulation of channel gating by AMP-activated protein kinase modulates cystic fibrosis transmembrane conductance regulator activity in lung submucosal cells. J Biol Chem 278: 998–1004, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Hamm LL, Hering-Smith KS. Acid-base transport in the collecting duct. Semin Nephrol 13: 246–255, 1993 [PubMed] [Google Scholar]
- 21.Hamm LL, Nakhoul NL. Renal acidification. In: Brenner and Rector's The Kidney (8th ed.), edited by Brenner BM. Philadelphia, PA: Saunders Elsevier, 2008, p. 248–279 [Google Scholar]
- 22.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett 546: 113–120, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Kane PM. The where, when, and how of organelle acidification by the yeast vacuolar H+-ATPase. Microbiol Mol Biol Rev 70: 177–191, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, Sanjad SA, Rodriguez-Soriano J, Santos F, Cremers CW, Di Pietro A, Hoffbrand BI, Winiarski J, Bakkaloglu A, Ozen S, Dusunsel R, Goodyer P, Hulton SA, Wu DK, Skvorak AB, Morton CC, Cunningham MJ, Jha V, Lifton RP. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21: 84–90, 1999 [DOI] [PubMed] [Google Scholar]
- 25.Lang K, Wagner C, Haddad G, Burnekova O, Geibel J. Intracellular pH activates membrane-bound Na+/H+ exchanger and vacuolar H+-ATPase in human embryonic kidney (HEK) cells. Cell Physiol Biochem 13: 257–262, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Li H, King JD, Alzamora R, Lee J, Pastor-Soler NM, Hallows KR. Epithelial Na+ transport regulation by AMP-activated protein kinase in kidney collecting duct and its role in ischemia (Abstract). J Am Soc Nephrol 22: 524A, 2011 [Google Scholar]
- 27.Liu W, Pastor-Soler NM, Schreck C, Zavilowitz B, Kleyman TR, Satlin LM. Luminal flow modulates H+-ATPase activity in the cortical collecting duct (CCD). Am J Physiol Renal Physiol 302: F205–F215, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu M, Ammar D, Ives H, Albrecht F, Gluck SL. Physical interaction between aldolase and vacuolar H+-ATPase is essential for the assembly and activity of the proton pump. J Biol Chem 282: 24495–24503, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Lu M, Holliday LS, Zhang L, Dunn WA, Jr, Gluck SL. Interaction between aldolase and vacuolar H+-ATPase: evidence for direct coupling of glycolysis to the ATP-hydrolyzing proton pump. J Biol Chem 276: 30407–30413, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Lu M, Sautin YY, Holliday LS, Gluck SL. The glycolytic enzyme aldolase mediates assembly, expression, and activity of vacuolar H+-ATPase. J Biol Chem 279: 8732–8739, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Marshansky V, Ausiello DA, Brown D. Physiological importance of endosomal acidification: potential role in proximal tubulopathies. Curr Opin Nephrol Hypertens 11: 527–537, 2002 [DOI] [PubMed] [Google Scholar]
- 32.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol 10: 1247–1255, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Mount PF, Hill RE, Fraser SA, Levidiotis V, Katsis F, Kemp BE, Power DA. Acute renal ischemia rapidly activates the energy sensor AMPK but does not increase phosphorylation of eNOS-Ser1177. Am J Physiol Renal Physiol 289: F1103–F1115, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Munton RP, Tweedie-Cullen R, Livingstone-Zatchej M, Weinandy F, Waidelich M, Longo D, Gehrig P, Potthast F, Rutishauser D, Gerrits B, Panse C, Schlapbach R, Mansuy IM. Qualitative and quantitative analyses of protein phosphorylation in naive and stimulated mouse synaptosomal preparations. Mol Cell Proteomics 6: 283–293, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Nelson N, Harvey WR. Vacuolar and plasma membrane proton-adenosinetriphosphatases. Physiol Rev 79: 361–385, 1999 [DOI] [PubMed] [Google Scholar]
- 36.Neumann D, Woods A, Carling D, Wallimann T, Schlattner U. Mammalian AMP-activated protein kinase: functional, heterotrimeric complexes by co-expression of subunits in Escherichia coli. Protein Expr Purif 30: 230–237, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Pastor-Soler N, Beaulieu V, Litvin TN, Da Silva N, Chen Y, Brown D, Buck J, Levin LR, Breton S. Bicarbonate-regulated adenylyl cyclase (sAC) is a sensor that regulates pH-dependent V-ATPase recycling. J Biol Chem 278: 49523–49529, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pastor-Soler NM, Alzamora R, Naveed S, Smolak C, Gong F, Hallows KR. Novel regulation of V-ATPase by PKA and AMPK in kidney intercalated cells. FASEB J 23: 602.13, 2009 [Google Scholar]
- 39.Pastor-Soler NM, Hallows KR. AMP-activated protein kinase regulation of kidney tubular transport. Curr Opin Nephrol Hypertens 21: 523–533, 2012 [DOI] [PubMed] [Google Scholar]
- 40.Pastor-Soler NM, Hallows KR, Smolak C, Gong F, Brown D, Breton S. Alkaline pH- and cAMP-induced V-ATPase membrane accumulation is mediated by protein kinase A in epididymal clear cells. Am J Physiol Cell Physiol 294: C488–C494, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paunescu TG, Ljubojevic M, Russo LM, Winter C, McLaughlin MM, Wagner CA, Breton S, Brown D. Cyclic AMP stimulates apical V-ATPase accumulation, microvillar elongation and proton extrusion in kidney collecting duct A-intercalated cells. Am J Physiol Renal Physiol 298: F643–F654, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rein J, Voss M, Blenau W, Walz B, Baumann O. Hormone-induced assembly and activation of V-ATPase in blowfly salivary glands is mediated by protein kinase A. Am J Physiol Cell Physiol 294: C56–C65, 2008 [DOI] [PubMed] [Google Scholar]
- 43.Sautin YY, Lu M, Gaugler A, Zhang L, Gluck SL. Phosphatidylinositol 3-kinase-mediated effects of glucose on vacuolar H+-ATPase assembly, translocation, and acidification of intracellular compartments in renal epithelial cells. Mol Cell Biol 25: 575–589, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartz GJ, Al-Awqati Q. Carbon dioxide causes exocytosis of vesicles containing H+ pumps in isolated perfused proximal and collecting tubules. J Clin Invest 75: 1638–1644, 1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shao E, Forgac M. Involvement of the nonhomologous region of subunit A of the yeast V-ATPase in coupling and in vivo dissociation. J Biol Chem 279: 48663–48670, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Shao E, Nishi T, Kawasaki-Nishi S, Forgac M. Mutational analysis of the non-homologous region of subunit A of the yeast V-ATPase. J Biol Chem 278: 12985–12991, 2003 [DOI] [PubMed] [Google Scholar]
- 47.Smith AN, Skaug J, Choate KA, Nayir A, Bakkaloglu A, Ozen S, Hulton SA, Sanjad SA, Al-Sabban EA, Lifton RP, Scherer SW, Karet FE. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet 26: 71–75, 2000 [DOI] [PubMed] [Google Scholar]
- 48.Steinmetz PR. Cellular organization of urinary acidification. Am J Physiol Renal Fluid Electrolyte Physiol 251: F173–F187, 1986 [DOI] [PubMed] [Google Scholar]
- 49.Su Y, Zhou A, Al-Lamki RS, Karet FE. The a-subunit of the V-type H+-ATPase interacts with phosphofructokinase-1 in humans. J Biol Chem 278: 20013–20018, 2003 [DOI] [PubMed] [Google Scholar]
- 50.Thomas JA, Buchsbaum RN, Zimniak A, Racker E. Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18: 2210–2218, 1979 [DOI] [PubMed] [Google Scholar]
- 51.Tuerk RD, Auchli Y, Thali RF, Scholz R, Wallimann T, Brunisholz RA, Neumann D. Tracking and quantification of 32P-labeled phosphopeptides in liquid chromatography matrix-assisted laser desorption/ionization mass spectrometry. Anal Biochem 390: 141–148, 2009 [DOI] [PubMed] [Google Scholar]
- 52.Tuerk RD, Thali RF, Auchli Y, Rechsteiner H, Brunisholz RA, Schlattner U, Wallimann T, Neumann D. New candidate targets of AMP-activated protein kinase in murine brain revealed by a novel multidimensional substrate-screen for protein kinases. J Proteome Res 6: 3266–3277, 2007 [DOI] [PubMed] [Google Scholar]
- 53.van Adelsberg J, Edwards JC, Takito J, Kiss B, al-Awqati Q. An induced extracellular matrix protein reverses the polarity of band 3 in intercalated epithelial cells. Cell 76: 1053–1061, 1994 [DOI] [PubMed] [Google Scholar]
- 54.Voss M, Vitavska O, Walz B, Wieczorek H, Baumann O. Stimulus-induced phosphorylation of vacuolar H+-ATPase by protein kinase A. J Biol Chem 282: 33735–33742, 2007 [DOI] [PubMed] [Google Scholar]
- 55.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Renal vacuolar-ATPase. Physiol Rev 84: 1263–1314, 2004 [DOI] [PubMed] [Google Scholar]
- 56.Weiner ID, Hamm LL. Use of fluorescent dye BCECF to measure intracellular pH in cortical collecting tubule. Am J Physiol Renal Fluid Electrolyte Physiol 256: F957–F964, 1989 [DOI] [PubMed] [Google Scholar]
- 57.Zippin JH, Levin LR, Buck J. CO2/HCO3−-responsive soluble adenylyl cyclase as a putative metabolic sensor. Trends Endocrinol Metab 12: 366–370, 2001 [DOI] [PubMed] [Google Scholar]