

Abstract
Myocardial infarction (MI) induces neural and electrical remodeling at scar border zones. The impact of focal MI on global functional neural remodeling is not well understood. Sympathetic stimulation was performed in swine with anteroapical infarcts (MI; n = 9) and control swine (n = 9). A 56-electrode sock was placed over both ventricles to record electrograms at baseline and during left, right, and bilateral stellate ganglion stimulation. Activation recovery intervals (ARIs) were measured from electrograms. Global and regional ARI shortening, dispersion of repolarization, and activation propagation were assessed before and during sympathetic stimulation. At baseline, mean ARI was shorter in MI hearts than control hearts (365 ± 8 vs. 436 ± 9 ms, P < 0.0001), dispersion of repolarization was greater in MI versus control hearts (734 ± 123 vs. 362 ± 32 ms2, P = 0.02), and the infarcted region in MI hearts showed longer ARIs than noninfarcted regions (406 ± 14 vs. 365 ± 8 ms, P = 0.027). In control animals, percent ARI shortening was greater on anterior than posterior walls during right stellate ganglion stimulation (P = 0.0001), whereas left stellate ganglion stimulation showed the reverse (P = 0.0003). In infarcted animals, this pattern was completely lost. In 50% of the animals studied, sympathetic stimulation, compared with baseline, significantly altered the direction of activation propagation emanating from the intramyocardial scar during pacing. In conclusion, focal distal anterior MI alters regional and global pattern of sympathetic innervation, resulting in shorter ARIs in infarcted hearts, greater repolarization dispersion, and altered activation propagation. These conditions may underlie the mechanisms by which arrhythmias are initiated when sympathetic tone is enhanced.
Keywords: autonomic nervous system, sympathetic nerves, cardiac innervation, neural remodeling
remodeling of the cardiac sympathetic nervous system after myocardial infarction (MI) has been linked to ventricular arrhythmias in animal models (29) and in humans (4, 12, 24). Modulation of cardiac sympathetic signaling is a major therapeutic strategy to prevent and treat ventricular arrhythmias (1, 8, 22). Despite its importance, the global electrophysiological consequences of postinfarct neural remodeling from a focal infarct remain poorly characterized.
Patchy myocardial scars with surviving islands of myocytes characterize human infarcts (10, 17). Heterogeneous intramyocardial sympathetic nerve sprouting at scar border zones leads to labile repolarization, increased peak Ca2+ current, and susceptibility to ventricular fibrillation in hypercholesterolemic rabbits (15). Increased transmural dispersion of repolarization has also been reported (13, 28), along with alterations in transient outward and inward rectifier K+ currents (21) in a postinfarct model with nerve sprouts.
However, the electrophysiological responses to sympathetic activation exhibited by myocytes at and remote to a focal infarction have not been characterized. Specifically, the functional effects on action potential duration in a global context induced by a focal infarct remain poorly understood.
We used a patchy porcine anteroapical MI model induced by microsphere delivery into the distal left anterior descending coronary artery (LAD) via a coronary artery catheter to characterize this (18). We hypothesized that focal MI in the distal anterior wall and apex induces remodeling of the entire cardiac sympathetic nervous system, resulting in altered global patterns of activation recovery interval (ARI) shortening and altered electrical propagation in scar-border zones during sympathetic activation. The distal anterior wall was chosen to avoid interrupting traveling sympathetic and parasympathetic nerves coursing from the base to the apex.
METHODS
Infarct induction.
Animal experimentation was approved by and in accordance with guidelines set by the Institutional Animal Care and Use Committee of the University of California and the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
MI was induced as previously described (18). Briefly, animals weighing 77 ± 10 lb (n = 12) were sedated with Telazol (8–10 mg/kg im) and Fentanyl (50–100 mcg iv). After intubation, general endotracheal anesthesia was maintained using inhaled isoflurane (0.8–1.5%), and analgesia was maintained by hourly boluses of fentanyl (50–100 mcg iv). Next, a balloon-tipped coronary angioplasty catheter was advanced over a guide wire to the mid-LAD. The balloon was inflated to subocclusive pressures, and a 10-ml suspension containing radioopaque contrast, sterile saline, and 5–7.5 ml polystyrene microspheres (Polybead, 90 μm, Polysciences, Warrington, PA) was slowly injected over 3–5 min via the angioplasty catheter. ST segment elevation in leads I and II confirmed immediate myocardial injury. Ventricular arrhythmias resulting in cardiac arrest occurred in seven animals, four of which could not be resuscitated. Ex vivo contrast-enhanced MRI was performed in four of the remaining nine animals at 6 wk postinfarction.
Electrical mapping and sympathetic stimulation.
Cardiac electrical mapping and sympathetic stimulation were performed in infarcted (n = 9) and control (n = 9) animals 6 wk after infarction, as previously described (3). Briefly, animals were sedated, and anesthesia was maintained as described in Infarct induction. A median sternotomy was performed to expose the heart and both stellate ganglia. Customized bipolar cuff electrodes were placed around each stellate ganglion (3). After surgery, animals were allowed to recover before experimentation. Stimulation of left, right, and bilateral stellate ganglion (LSG, RSG, and BSG, respectively) was performed for 30 s using previously described parameters; repeated square-wave pulses, 5 ms in duration, were delivered at 5-Hz frequency for 5 min at a stimulus amplitude of 5 V (3). Upon completion of the experimental protocol, animals were euthanized with a lethal dose of pentobarbital sodium (100 mg/kg). Hearts were then removed for histology. One control animal died from ventricular fibrillation before RSG stimulation, and one MI animal died before simultaneous sympathetic stimulation and scar pacing.
ARI measurements.
ARIs have been demonstrated to correlate well with local action potential duration (16). Electrograms (EGMs) were recorded with a 56-electrode sock as previously described (3) using the Prucka Cardiolab system (GE Healthcare). Using customized software to generate the first derivative of recorded unipolar electrograms, activation time (AT) and repolarization time (RT) in each EGM were measured as the times of minimal dV/dt (in the QRS complex) and maximal dV/dt (near the peak of the T wave), respectively, both relative to the global QRS onset. AT, RT, and ARIs were calculated from EGMs collected at 15 s into stimulation, as previously described (ARI = RT − AT) (3, 16).
Regional ARI responses were measured in six prespecified regions (Fig. 1): the anteroapical left ventricle (LV; region of patchy “scar-border zone” in MI animals), basal anterior right ventricle (RV), lateral LV, posterior LV, lateral RV, and posterior RV. In infarcted animals, we did not distinguish scar from the border zone due to the patchy nature of the scar. The scar-border zone region was located anteroapically and was readily recognized on gross inspection. In addition, two- and three-dimensional polar maps, contrast-enhanced MRI, and histological analyses confirmed scar locations. Mean global ARI refers to the average ARI from all 56 electrodes.
Fig. 1.

Polar map of 56-electrode sock and prespecified regions. The mesh represents the locations of all 56 electrodes (nos. 1–56) contained in the multielectrode sock and the 6 prespecified regions (with abbreviations) used in the study.
Global dispersion of repolarization (DOR) refers to the variance of ARIs measured across all 56 electrodes. To describe functional innervation patterns, the percent ARI shortening from baseline was computed from five to eight electrodes in each prespecified region during RSG, LSG, and BSG stimulation, as previously described (3, 24).
Electrical maps.
Multielectrode sock data were projected onto a two-dimensional polar map or three-dimensional geometry using publicly available Map3D software (http://www.sci.utah.edu/cibc/software/107-map3d.html, Scientific Computing and Imaging Institute, University of Utah).
Histological and immunohistochemical experiments.
Myocardial tissue was stained with Geske's modification of Verhoeff's elastic and Masson's trichrome. Sympathetic nerves were stained using anti-tyrosine hydroxylase (TH) antibody (1:200 dilution, Abcam, Cambridge, MA) detected by diaminobenzidine (Life Technologies, Green Island, NY). All slides were scanned, and digital images were electronically stored for analysis (Scan Scope, Aperio, Vista, CA). Immunohistochemical quantifications were performed by computerized morphometry (Tissue Studio, Definiens, Parsippany, NJ).
Statistical analyses.
Data are reported as means ± SE. Multigroup comparisons were performed using two-way ANOVA. If the P value using ANOVA was <0.05, post hoc comparisons were made using a Student-Neuman-Keuls test. Pre- and post-stimulation comparisons were performed using a paired two-tailed Student's t-test. For all comparison, P values of <0.05 were considered statistically significant.
RESULTS
In total, 22 pigs were studied. Nine pigs served as controls, whereas thirteen pigs underwent MI induction. Four pigs died from refractory ventricular tachycardia or ventricular fibrillation during infarct induction.
Characterization of postinfarct neural remodeling.
The infarcts induced in this model were limited to the distal anterior and apical myocardium (Fig. 2A, top). As shown in the three- and two-dimensional ARI maps (Fig. 2A, bottom), the anteroapical region in infarcted animals demonstrated longer ARIs than other regions of the epicardium (406 ± 14 vs. 365 ± 8 ms, P = 0.027; Fig. 2B); however, this was not true in control animals (454 ± 23 vs. 438 ± 10 ms, P = 0.51).
Fig. 2.

Myocardial infarct model: structure-function correlation. A, top: representative gross tissue (left) and MRI (right) images of the patchy anteroapical myocardial infarct. Bottom, three-dimensional (3-D; left) and two-dimensional (2-D; right) activation recovery interval (ARI) maps. Yellow arrows show the myocardial infarct (MI) location. B: graphic representation of ARI in the infarcted region versus all other locations in infarcted and control animals. Data are means ± SE. C: representative histological slides showing the patchy intramyocardial scar (black arrows) after trichrome/elastic von Giessen (EVG) staining and diaminobenzidine detection of anti-tyrosine hydroxylase (TH) antibody (red arrows) from the peri-infarcted (red box) and remote (blue box) myocardium. Magnification: ×20. Scale bars = 100 μm. Scant intramyocardial staining and a subepicardial nerve bundle are labeled (red arrow) in the remote myocardium compared with extensive staining in the infarct scar-border zone (BZ). D: graphic representation (means ± SE) of TH immunoreactivity expressed as the area of positive staining (in μm2) divided by the total area (in mm2) in the scar-BZ region, remote regions from the scar, and in normal control animals. The P value represents ANOVA of the three groups.
Consistent with previous studies (12, 29), scar-border zone regions showed significantly greater density of TH-positive sympathetic nerves (red arrows) compared with remote regions and normal control regions (19,983 ± 4,167 vs. 7,603 ± 1,788 and 8,020 ± 1,945 μm2/mm2, P = 0.009 by ANOVA; Fig. 2, C and D). Myocytes were, however, sparse in this area. These findings confirmed that despite greater sympathetic nerve staining in the anteroapical scar region, there was significant functional denervation and prolonged ARIs compared with other regions.
Hemodynamic and global ARI responses to sympathetic stimulation.
Hemodynamic responses to RSG, LSG, and BSG stimulation did not differ between control and infarcted animals and were similar to previous observations (3, 20, 25). Systolic blood pressures at baseline and during RSG, LSG, and BSG stimulation were 65 ± 12, 89 ± 18, 79 ± 24, and 108 ± 18 mmHg, respectively, in control animals and 65 ± 17, 90 ± 23, 83 ± 18, and 109 ± 27 mmHg, respectively, in infarcted animals. Heart rates (HRs) at baseline and during RSG, LSG, and BSG stimulation were 69 ± 5, 110 ± 21, 69 ± 8, and 112 ± 21 beats/min, respectively, in control animals and 64 ± 13, 102 ± 26, 64 ± 16, and 100 ± 22 beats/min, respectively, in infarcted animals [P < 0.05 compared with baseline (except for HR during LSG stimulation), and P > 0.25 when control and infarct animals were compared for all conditions].
Representative EGMs and calculated ARIs from the anteroapical and posterior LV regions from control and infarcted hearts are shown in Fig. 3, A and B, respectively. Slurring and diminished amplitude can be seen in the anteroapical EGMs (recorded from the infarcted region; Fig. 3B); however, the minimum dV/dt, reflecting the local AT of adjacent myocytes, can be well seen. As shown in Fig. 4A, RSG stimulation significantly decreased mean ARI in normal and infarcted hearts. Compared with normal hearts, the mean ARI response to RSG stimulation was blunted in hearts from infarcted animals (P = 0.041). Mean ARI shortening to LSG and BSG stimulation were similar in control and infarcted hearts (Fig. 4A).
Fig. 3.
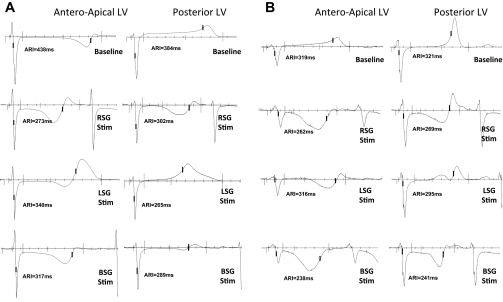
Electrogram responses to sympathetic stimulation. A and B: representative electrograms from control animals (A) and infarcted animals (B) showing two sampled regions: the Ant-Ap LV (location of the focal scar in infarcted animals) and Post LV (a location disparate from the infarct bed, with a separate coronary artery supply). Representative responses to right, left, and bilateral stellate ganglia (RSG, LSG, and BSG, respectively) stimulation are shown.
Fig. 4.
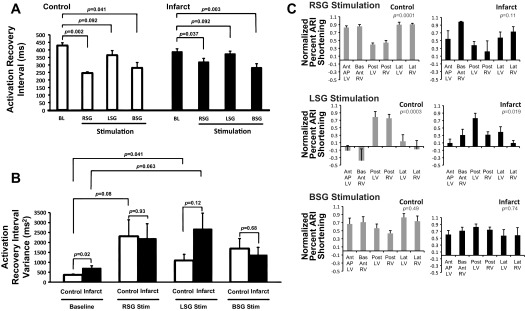
Effects of sympathetic stimulation on ARIs and repolarization heterogeneity. A: ARI shortening at baseline (BL) and during RSG, LSG, and BSG stimulation for control and infarcted animals (n = 8 for control RSG stimulation and n = 9 for all others). Note the compressed scales for RSG and BSG stimulation; the pattern of ARI shortening during BSG stimulation is more even, as emphasized in C and in Fig. 5C. B: global dispersion of repolarization (DOR) at BL and during RSG, LSG, and BSG stimulation in control and infarcted hearts. C: percent ARI shortening in each region normalized to the maximum in each animal during RSG, LSG, and BSG stimulation in control and infarcted hearts (n = 8 for control RSG stimulation and n = 9 for all others). All graphic data shown in A–C are means ± SE. P values by ANOVA are shown.
Global DOR, a marker of repolarization lability (measured by the ARI variance) was greater in infarcted hearts compared with control hearts at baseline (Fig. 4B). In control hearts, compared with baseline, LSG stimulation significantly increased DOR (Fig. 4B). In infarcted hearts, DOR during LSG stimulation was 2,677 ± 797 compared with 734 ± 123 ms2 (P = 0.063) at baseline. There were no statistically significant differences in DOR between control and infarcted hearts during RSG, LSG, or BSG stimulation, as shown in Fig. 4B. DOR during BSG stimulation was 1,690 ± 504 ms2 in control hearts and 1,378 ± 376 ms2 in infarcted hearts compared with baseline values (0.099 for both; Fig. 4B).
MI alters functional sympathetic innervation patterns.
In control animals, both RSG and LSG stimulation induced significant differences in regional ARI shortening (P = 0.0001 and P = 0.0003 for RSG and LSG stimulation, respectively, by ANOVA). Specifically, RSG stimulation resulted in significantly greater ARI shortening on the anterior and lateral ventricular walls (P < 0.000001; Fig. 4C, top left) compared with the posterior wall. LSG stimulation resulted in the reverse pattern (P < 0.000001; Fig. 4C, middle left). Stimulation of both stellate ganglia simultaneously (BSG stimation) resulted in an even pattern (P = 0.49 by ANOVA; Fig. 4C, bottom left). Representative two-dimensional polar maps displaying ARI responses (reported above) to RSG, LSG, and BSG stimulation in control animals are shown in Fig. 5A, demonstrating the anteroposterior innervation pattern seen in control animals during RSG and LSG stimulation. During BSG stimulation (Fig. 5A, bottom right), the smaller ARI scale is indicative of a more even functional innervation pattern, as emphasized by the maps shown in Fig. 5C.
Fig. 5.
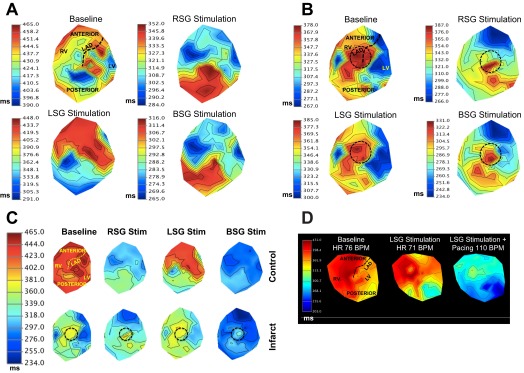
2-D ARI maps in control and infarcted hearts. A and B: representative 2-D polar maps of ARIs of control animals (A) and infarcted animals (B) at BL and during RSG, LSG, and BSG stimulation. Myocardial regions are shown in the BL control map (and in Fig. 1). The black dashed circle represents the anteroapical location of the infarct. Note the more even pattern of ARI shortening during BSG stimulation in control animals (A, bottom right), as indicated by the smaller ARI scale. C: polar maps from A and B on a synchronized scale. The top and bottom maps represent control and infarcted animals, respectively, at BL and during RSG, LSG, and BSG stimulation. Myocardial regions are shown in the BL control map. The black dashed circle represents the anteroapical location of the infarct. D: representative 2-D ARI map of a normal animal at BL, during LSG stimulation, and during LSG stimulation with simultaneous atrial pacing at 110 beats/min (BPM). The posterior wall of the heart exhibited shorter ARI during LSG stimulation; this was unchanged when the heart rate was increased. LAD, left anterior descending coronary artery.
In infarcted animals, LSG but not RSG stimulation induced significant regional differences in ARI shortening (P = 0.019 for LSG stimulation and P = 0.11 for RSG stimulation by ANOVA), although the regional pattern observed during LSG stimulation in infarcted animals was different from control animals (Fig. 4C, top right and middle right). Specifically, the posterior LV showed greater ARI shortening in response to LSG stimulation compared with the anterolateral regions (P = 0.025), whereas the posterior (or posterolateral) RV did not (P = 0.44). During BSG stimulation, an even functional innervation pattern was observed (P = 0.74 by ANOVA; Fig. 4C, bottom right), similar to BSG stimulation in control animals. Representative two-dimensional polar maps displaying ARI responses (reported above) to RSG, LSG, and BSG stimulation in infarcted animals are shown in Fig. 5B, demonstrating the loss of an anteroposterior response to RSG and LSG stimulation. The same polar maps synchronized to the same scale are shown in Fig. 5C. It can be appreciated that BSG stimulation resulted in a more even global pattern of ARI shortening; however, the anteroapical myocardium containing the infarcted myocardium exhibited diminished responses to BSG stimulation (Fig. 5C).
To determine whether the difference in anteroposterior innervation patterns seen during RSG and LSG stimulation in control animals is related to the fact that RSG stimulation induces tachycardia, whereas LSG stimulation does not, atrial pacing was performed during LSG stimulation (n = 3) to match the HR observed during RSG stimulation in the same animal. There were no differences observed in the anteroposterior pattern of innervation when HR was increased during LSG stimulation (Fig. 5D). As shown in Fig. 5, LSG stimulation alone resulted in shorter ARIs in the posterior myocardium compared with the anterior myocardium. Atrial pacing at 110 beats/min during LSG stimulation (the same HR obtained during RSG stimulation in the same animal) still demonstrated posterior dominance of LSG stimulation. This finding suggests that tachycardia plays no role in the anterior-posterior innervation pattern observed during RSG and LSG stimulation.
Regional ARI dispersion during sympathetic stimulation in normal and infarcted hearts.
Regional DOR responses to RSG, LSG, and BSG stimulation in control and infarcted hearts are presented in Table 1. In control animals, no significant differences were observed before and during RSG, LSG, and BSG stimulation in the anteroapical LV, basal anterior RV, lateral LV, and lateral RV. On the posterior wall (both posterior LV and posterior RV), BSG but not RSG or LSG stimulation increased DOR (P = 0.02 for the posterior LV and P = 0.041 for the posterior RV).
Table 1.
Regional dispersion of repolarization in control and infarcted hearts during sympathetic stimulation
| Control Hearts |
Infarcted Hearts |
|||||||
|---|---|---|---|---|---|---|---|---|
| Region | Baseline | RSG | LSG | BSG | Baseline | RSG | LSG | BSG |
| Anteroapical LV | 212 ± 41 | 687 ± 247 | 589 ± 481 | 361 ± 127 | 337 ± 130 | 3,662 ± 1,741 | 1,279 ± 780 | 1,357 ± 770 |
| Basal anterior RV | 115 ± 43 | 619 ± 231 | 84 ± 32 | 447 ± 188 | 187 ± 75 | 1,433 ± 780 | 5,188 ± 3,547 | 2,275 ± 1,612 |
| Posterior LV | 292 ± 85 | 207 ± 98 | 443 ± 152 | 1329 ± 336* | 585 ± 161† | 921 ± 411 | 1,405 ± 591 | 707 ± 245 |
| Posterior RV | 350 ± 159 | 919 ± 403 | 454 ± 193 | 983 ± 330* | 436 ± 128 | 1,927 ± 1,306 | 1,028 ± 342 | 670 ± 192 |
| Lateral LV | 209 ± 74 | 589 ± 218 | 437 ± 296 | 639 ± 190 | 449 ± 156† | 3,183 ± 1,450 | 2,392 ± 874 | 1,153 ± 458 |
| Lateral RV | 100 ± 35 | 1,842 ± 746 | 407 ± 173 | 480 ± 398 | 750 ± 244† | 1,749 ± 1,060 | 702 ± 233 | 1,166 ± 487 |
Values (in ms2) are means ± SE. Activation recovery interval dispersions in the six prespecified myocardial regions were compared across all control and infarcted animals at baseline and during right, left, and bilateral stellate ganglion (RSG, LSG, and BSG, respectively) stimulation. LV, left ventricle; RV, right ventricle.
P value for BSG stimulation compared with baseline control values (P = 0.02 and P = 0.04, respectively);
P value for baseline infarcted values compared with baseline control values (P = 0.012, 0.018, and 0.009, respectively).
At baseline, compared with control animals, regional DOR in infarcted animals was greater in the lateral LV and RV and in the posterior LV. On the anteroapical LV, DOR in infarcted versus control animals was 755 ± 298 and 179 ± 51 ms2, respectively (P = 0.09). The two remaining prespecified regions (the posterior and basal anterior RV) showed no significant differences compared with control animals (Table 1).
Although baseline regional DOR in some prespecified regions was overall greater in infarcted animals compared with control animals, regional DOR during RSG, LSG, and BSG stimulation were not further significantly increased in infarcted animals (Table 1).
Sympathetic tone modulates local activation propagation in the heterogeneous myocardium.
To determine the functional consequences of postinfarct neural remodeling described above, we evaluated the effect of sympathetic stimulation on the activation wavefront emanating from the myocardial scar. This was achieved by simulating an ectopic activation wavefront exiting scar-border zones by pacing within the scar. We performed epicardial pacing at the same site in the scar region before and during RSG, LSG, and BSG stimulation. Pacing was performed at the same rate at baseline and during sympathetic stimulation (5–10 beats/min above that achieved during RSG or BSG stimulation, whichever was higher). In control animals, pacing at the anteroapical region resulted in a centrifugal spread of activation (Fig. 6A); however, in all eight MI animals studied, a nonuniform spread of electrical activation occurred during baseline pacing (Fig. 6, B–D). In four of the eight animals, no significant changes occurred in the direction of the activation propagation from the scar region during any sympathetic stimulation; an example is shown in Fig. 6D. In the other four animals, however, significant changes were noted during LSG and RSG stimulation (2 animals) and during LSG, RSG, and BSG stimulation (2 animals), although each pattern was different (Fig. 6, B and C). In Fig. 6B, scar pacing, indicated by the white electrical pulse symbol, led to impulse propagation in the anterosuperior direction, toward the RV (white dashed arrow). Pacing at the same site and rate during RSG and LSG stimulation resulted in functional block in the same direction (white equal sign), with activation proceeding likely via the midmyocardium or endocardium. Pacing during BSG stimulation resulted in a similar activation wavefront to baseline conditions.
Fig. 6.
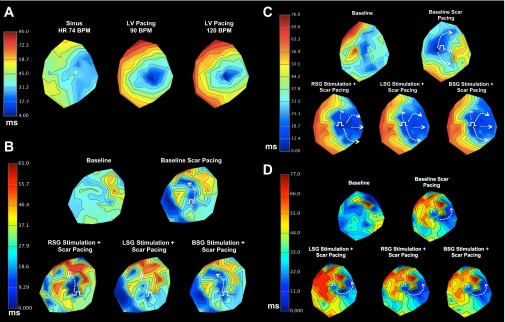
Impact of sympathetic stimulation on impulse propagation from the myocardial scar. A: 2-D activation maps showing pacing in a control animal. B and C: 2-D activation maps for two animals at baseline (sinus rhythm), during scar pacing, and during scar pacing simultaneous with RSG, LSG, and BSG stimulation. The direction of activation propagation was modulated by sympathetic stimulation in both animals. D: representative example of an animal where sympathetic stimulation did not result in modulation of activation propagation. For B–D, white dashed arrows show the region of activation propagation, whereas the white equal signs indicate regions of functional block during. The white electrical pulse symbols represent pacing sites.
Baseline pacing (Fig. 6C) resulted in activation wavefronts propagating in the antero- and posterosuperior directions (white dashed arrows), whereas there was delayed propagation in the superolateral direction (white equal sign). During RSG, LSG, and BSG, activation of the LV occurred rapidly and in a predominantly lateral direction. In the two remaining animals where sympathetic stimulation modulated activation propagation, activation progressed via a narrower isthmus during any sympathetic stimulation compared with baseline in one animal; the other animals showed more rapid activation posteriorly during RSG and LSG (but not BSG) stimulations.
DISCUSSION
The major findings of the present study are as follows: 1) the scar-border zone region shows prolonged ARIs compared with the myocardium remote from the infarct, despite having greater TH-positive sympathetic nerve density in the scar-border zone region; 2) ARIs in infarcted hearts are shorter at baseline compared with normal control hearts at the same HRs; 3) focal anteroapical MI distorts the global patterns of functional myocardial innervation during LSG and RSG stimulation; 4) regional and global DOR are worsened after MI at baseline; and 5) the direction of myocardial activation propagation out of a scar can be altered by sympathetic activation. This study represents the first comprehensive investigation at the tissue level of the functional changes induced by postinfarct neural remodeling in a model closely resembling human myocardial scars. It highlights the complex functional effects of myocardial neural remodeling after infarction, providing a spatial context to such changes.
Structure-function correlation of neural remodeling in infarcted hearts.
Heterogeneous postinfarct sprouting of sympathetic nerves has been reported in humans and animals (12, 26, 29, 30). Neurotrophins released at the infarct site are trafficked to stellate ganglia (29) and presumably the intrinsic cardiac network (ICN), where they induce neuronal remodeling (2, 4, 9), increase stellate ganglion nerve activity (9), and cause sprouting of sympathetic nerves in the heart. The porcine model of patchy myocardial scars showed nerve sprouting and increased nerve density at scar-border zone regions consistent with previous findings (13, 21).
Studies of cardiac neural remodeling at the myocardial tissue or cell level have examined models that differ from this study, including a hypercholesterolemic rabbit model (15), a partially denervated heart by phenol application (28), and a cryoablation model of MI (21) using a super-cooled rod passed through the diaphragm to the heart. Arrhythmia susceptibility was also studied in a Langendorff preparation, which excludes contributions from the higher neural centers to arrhythmogenesis (15, 21). In the present study, a nondestructive method of obtaining epicardial action potential durations (ARIs) was used, yielding global ventricular assessments of myocardial excitability and also placed these data in a spatial context. Furthermore, the entire cardiac neuraxis was unperturbed. Our findings of prolonged baseline ARI in the scar region compared with other regions in infarcted but not control animals suggest that this region is relatively denervated in infarcted but not control animals. The shorter ARIs in MI versus control animals (at the same HR range) suggest a higher resting sympathetic tone.
Postinfarct alterations in innervation patterns and DOR.
Our findings on the anteroposterior pattern of sympathetic innervation from LSG and RSG stimulation in swine are consistent with those from a canine study by Yanowitz et al. (27). The dramatic alteration of this pattern globally after an anteroapical MI suggests that, in addition to heterogeneous sprouting at the border zones, there is remodeling of sympathetic innervation of the entire heart. This is supported by results from a canine study (29) where significant nerve sprouting in the remote noninfarcted myocardium was observed, although no functional experiments were performed. While we did not observe increased nerve density in the remote region compared with the normal myocardium, the pattern, organization, or function of these nerves in the postinfarct heart may be altered, resulting in greater global DOR. Jiang et al. (13) demonstrated that the scar border zone of a rabbit LAD ligation MI model showed the greatest nerve density and a strong correlation between the density of sympathetic nerves after infarction and transmural dispersion of repolarization.
The blunted ARI shortening during RSG (but not LSG or BSG) stimulation in infarcted animals compared with control animals likely reflects the location of the infarct. The anterior and lateral walls, including the apex, are subtended by the RSG stimulation, as evidenced by the greatest ARI shortening occurring in the anterolateral wall. The loss of the anteroapical myocardium and ensuing sympathetic neural remodeling eliminates a region of ARI shortening. Mean ARI shortening during LSG and BSG stimulation was, however, less affected by this and showed no significant differences compared with baseline conditions. This highlights the potential for differential neural remodeling based on infarct location, along with a variety of potential proarrhythmic implications.
In the present study, we observed increased DOR during LSG stimulation in normal hearts. In infarcted animals, however, DOR was already elevated at baseline both globally and in half of the regions evaluated, possibly explaining the lack of significant increase to LSG stimulation in infarcted hearts. In addition, infarcted animals showed significant variability in responses to sympathetic stimulation. Consequently, no statistically significant increases were observed before and during sympathetic stimulation in MI hearts. The absence of a consistent increase in DOR in infarcted hearts (and a consistent pattern of functional innervation) suggests that the pattern of remodeling with intramyocardial nerves is variable. This may be partly due to heterogeneity in innervation, genetic differences, and differences in myocardial scar morphology. The mechanistic explanation for increased DOR on the posterior wall during BSG but not RSG or LSG stimulation is unclear but reflects unique properties of functional innervation of the posterior myocardium in the pig. Whether this contributes to arrhythmogenesis remains unclear.
The ICN may exert the most significant control of cardiac function (5–7). Multiple ganglionated plexi within fat pads distributed over the atria and ventricles form the ICN. Autonomic signaling within spatially disparate ganglionated plexi are synchronized via local circuit neurons, which allow the coordination of multiple ganglionated plexi and forms the ICN. Changes in functional responses at remote regions may be mediated by changes to the ICN. The observations in the present study are consistent with findings in humans where the remote myocardium in humans showed attenuated ARI shortening in response to reflex sympathetic stimulation induced by nitroprusside (24).
Sympathetic tone and impulse propagation.
Myocardial scars are thought to have “channels” that facilitate reentry (14) and lead to multiple exits out of the scar (23). Propagation or conduction block within a particular channel determines the direction of activation within these complex scars, and these, in turn, are determined by functional refractory periods of the myocytes that make up the channel. The functional refractory periods within the myocardial scar may be modulated by sympathetic stimulation, as evidenced by ventricular arrhythmias arising during high-frequency stimulation of the proximal pulmonary artery to activate sympathetic nerves en route to the heart (11). In the present study, 50% of the animals studied showed altered epicardial propagation during sympathetic stimulation. This suggests that during states of heightened sympathetic tone, a previously dormant channel may now conduct during sympathetic activation (or vice versa), completely altering the electrical properties of the scar and setting the stage for reentrant arrhythmias. Some of the MI animals that showed no changes in epicardial activation propagation may have had endocardial changes. These findings have significant implications for mapping and ablation of ventricular arrhythmias, as activation of cardiac sympathetic nerves with concomitant substrate mapping of the ventricle could add value to arrhythmia ablation, by identifying reentrant pathways or facilitating arrhythmia induction not otherwise possible under general anesthesia.
Limitations.
The present study is limited by the lack of endocardial recordings, although the electrophysiological effects of the sympathetic nervous system on the endocardium and epicardium are similar (19). It remains possible, despite this fact, that the four subjects lacking epicardial alteration of activation propagation may have shown endocardial alterations. Although control and infarcted animals exhibited a similar range of HRs to sympathetic stimulation, HRs were not fixed (by atrial pacing) in this study. In addition, because animals were sedated, an unquantifiable effect of anesthesia on cardiac excitability cannot be excluded. Additionally, the effects of circulating catecholamines in addition to direct innervation may explain some of our findings. Finally, control animals in our study were not sham operated due to the high risk of ventricular fibrillation induced by coronary artery spasm, dissection, or injury during placement of the 5-Fr catheter into the mid-LAD.
Conclusions.
In summary, the present study (in a patchy scar model similar to human infarcts) demonstrates that neural remodeling after focal MI dramatically alters the global pattern of functional sympathetic innervation and is associated with increased regional and global DOR in infarcted hearts. Furthermore, sympathetic stimulation altered electrical propagation from the scar, underscoring the proarrhythmic potential of sympathetic stimulation in cardiomyopathic hearts, as this altered propagation may initiate and maintain reentrant rhythms. In conclusion, both repolarization heterogeneity and altered activation propagation may underlie mechanisms of postinfarct arrhythmogenesis during states of enhanced sympathetic tone.
GRANTS
This work was supported by an A. P. Giannini Postdoctoral Award (to O. A. Ajijola), by National Institutes of Health (NIH) Grant R01-HL-084261 (to K. Shivkumar), and in part by software from NIH Center for Integrative Biomedical Computing (NIH Grant 2-P41-RR-0112553-12).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: O.A.A. and K.S. conception and design of research; O.A.A., D.Y., K.J.P., M.V., W.Z., K.Y., and E.S. performed experiments; O.A.A., K.J.P., and K.S. analyzed data; O.A.A., D.Y., M.V., W.Z., R.L.L., A.M., and K.S. interpreted results of experiments; O.A.A. prepared figures; O.A.A. and K.S. drafted manuscript; O.A.A., D.Y., K.J.P., M.V., W.Z., K.Y., E.S., R.L.L., A.M., and K.S. edited and revised manuscript; O.A.A., D.Y., K.J.P., M.V., W.Z., K.Y., E.S., R.L.L., A.M., and K.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Mariko Takemoto, Naveen K. Reddy, Stephanie Midtling, Jonathan Hoang, Joanna Shen, Chloe Su, and Brian Ngo for excellent surgical and technical assistance. The authors also thank Dr. Jeffrey Gornbein [University of California-Los Angeles (UCLA) Biostatistics Core] and Dr. Daniel Ennis (Department of Radiology, UCLA).
REFERENCES
- 1.Ajijola OA, Lellouche N, Bourke T, Tung R, Ahn S, Mahajan A, Shivkumar K. Bilateral cardiac sympathetic denervation for the management of electrical storm. J Am Coll Cardiol 59: 91–92, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ajijola OA, Shivkumar K. Neural remodeling and myocardial infarction: the stellate ganglion as a double agent. J Am Coll Cardiol 59: 962–964, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ajijola OA, Vaseghi M, Zhou W, Yamakawa K, Benharash P, Hadaya J, Lux RL, Mahajan A, Shivkumar K. Functional differences between junctional and extrajunctional adrenergic receptor activation in mammalian ventricle. Am J Physiol Heart Circ Physiol 304: H579–H588, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ajijola OA, Wisco JJ, Lambert HW, Mahajan A, Stark E, Fishbein MC, Shivkumar K. Extracardiac neural remodeling in humans with cardiomyopathy. Circ Arrhythm Electrophysiol 5: 1010–1116, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Armour JA. Functional anatomy of intrathoracic neurons innervating the atria and ventricles. Heart Rhythm 7: 994–996, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Armour JA. The little brain on the heart. Cleve Clin J Med 74, Suppl 1: S48–S51, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Armour JA. Potential clinical relevance of the ‘little brain’ on the mammalian heart. Exp Physiol 93: 165–176, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Bourke T, Vaseghi M, Michowitz Y, Sankhla V, Shah M, Swapna N, Boyle NG, Mahajan A, Narasimhan C, Lokhandwala Y, Shivkumar K. Neuraxial modulation for refractory ventricular arrhythmias: value of thoracic epidural anesthesia and surgical left cardiac sympathetic denervation. Circulation 121: 2255–2262, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han S, Kobayashi K, Joung B, Piccirillo G, Maruyama M, Vinters H, March K, Lin SF, Shen C, Fishbein MC, Chen PS, Chen LS. Electroanatomical remodeling of the left stellate ganglion after myocardial infarction. J Am Coll Cardiol 59: 954–961, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanich RF, de Langen CD, Kadish AH, Michelson EL, Levine JH, Spear JF, Moore EN. Inducible sustained ventricular tachycardia 4 years after experimental canine myocardial infarction: electrophysiologic and anatomic comparisons with early healed infarcts. Circulation 77: 445–456, 1988 [DOI] [PubMed] [Google Scholar]
- 11.Hasdemir C, Alp A, Aydin M, Can LH. Human model simulating right ventricular outflow tract tachycardia by high-frequency stimulation in the left pulmonary artery: autonomics and idiopathic ventricular arrhythmias. J Cardiovasc Electrophysiol 20: 759–763, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS, Chen LS. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation 101: 1960–1969, 2000 [DOI] [PubMed] [Google Scholar]
- 13.Jiang H, Lu Z, Yu Y, Zhao D, Yang B, Huang C. Relationship between sympathetic nerve sprouting and repolarization dispersion at peri-infarct zone after myocardial infarction. Auton Neurosci 134: 18–25, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Kocovic DZ, Harada T, Friedman PL, Stevenson WG. Characteristics of electrograms recorded at reentry circuit sites and bystanders during ventricular tachycardia after myocardial infarction. J Am Coll Cardiol 34: 381–388, 1999 [DOI] [PubMed] [Google Scholar]
- 15.Liu YB, Wu CC, Lu LS, Su MJ, Lin CW, Lin SF, Chen LS, Fishbein MC, Chen PS, Lee YT. Sympathetic nerve sprouting, electrical remodeling, and increased vulnerability to ventricular fibrillation in hypercholesterolemic rabbits. Circ Res 92: 1145–1152, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Millar CK, Kralios FA, Lux RL. Correlation between refractory periods and activation-recovery intervals from electrograms: effects of rate and adrenergic interventions. Circulation 72: 1372–1379, 1985 [DOI] [PubMed] [Google Scholar]
- 17.Nakahara S, Tung R, Ramirez RJ, Gima J, Wiener I, Mahajan A, Boyle NG, Shivkumar K. Distribution of late potentials within infarct scars assessed by ultra high-density mapping. Heart Rhythm 7: 1817–1824, 2010 [DOI] [PubMed] [Google Scholar]
- 18.Nakahara S, Vaseghi M, Ramirez RJ, Fonseca CG, Lai CK, Finn JP, Mahajan A, Boyle NG, Shivkumar K. Characterization of myocardial scars: electrophysiological imaging correlates in a porcine infarct model. Heart Rhythm 8: 1060–1067, 2011 [DOI] [PubMed] [Google Scholar]
- 19.Opthof T, Misier A, Coronel R, Vermeulen J, Verberne H, Frank R, Moulijn A, Capelle Fv Janse M. Dispersion of refractoriness in canine ventricular myocardium. Effects of sympathetic stimulation. Circ Res 68: 1204–1215, 1991 [DOI] [PubMed] [Google Scholar]
- 20.Ramirez RJ, Ajijola OA, Zhou W, Holmstrom B, Luning H, Laks MM, Shivkumar K, Mahajan A. A new electrocardiographic marker for sympathetic nerve stimulation: modulation of repolarization by stimulation of stellate ganglia. J Electrocardiol 44: 694–699, 2011 [DOI] [PubMed] [Google Scholar]
- 21.Ren C, Wang F, Li G, Jiao Q, Bai J, Yu D, Hao W, Wang R, Cao JM. Nerve sprouting suppresses myocardial Ito and IK1 channels and increases severity to ventricular fibrillation in rat. Auton Neurosci 144: 22–29, 2008 [DOI] [PubMed] [Google Scholar]
- 22.Schwartz PJ, Motolese M, Pollavini G, Lotto A, Ruberti U, Trazzi R, Bartorelli C, Zanchetti A. Prevention of sudden cardiac death after a first myocardial infarction by pharmacologic or surgical antiadrenergic interventions. J Cardiovasc Electrophysiol 3: 2–16, 1992 [Google Scholar]
- 23.Tung R, Mathuria N, Michowitz Y, Yu R, Buch E, Bradfield J, Mandapati R, Wiener I, Boyle N, Shivkumar K. Functional pace-mapping responses for identification of targets for catheter ablation of scar-mediated ventricular tachycardia. Circ Arrhythm Electrophysiol 5: 264–272, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vaseghi M, Lux RL, Mahajan A, Shivkumar K. Sympathetic stimulation increases dispersion of repolarization in humans with myocardial infarction. Am J Physiol Heart Circ Physiol 302: H1838–H1846, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaseghi M, Zhou W, Shi J, Ajijola OA, Hadaya J, Shivkumar K, Mahajan A. Sympathetic innervation of the anterior left ventricular wall by the right and left stellate ganglia. Heart Rhythm 9: 1303–1309, 2012 [DOI] [PubMed] [Google Scholar]
- 26.Vracko R, Thorning D, Frederickson RD. Nerve fibers in human myocardial scars. Hum Pathol 22: 138–146, 1991 [DOI] [PubMed] [Google Scholar]
- 27.Yanowitz F, Preston JB, Abildskov JA. Functional distribution of right and left stellate innervation to the ventricles. Production of neurogenic electrocardiographic changes by unilateral alteration of sympathetic tone. Circ Res 18: 416–428, 1966 [DOI] [PubMed] [Google Scholar]
- 28.Yoshioka K, Gao DW, Chin M, Stillson C, Penades E, Lesh M, O'Connell W, Dae M. Heterogeneous sympathetic innervation influences local myocardial repolarization in normally perfused rabbit hearts. Circulation 101: 1060–1066, 2000 [DOI] [PubMed] [Google Scholar]
- 29.Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, Fishbein MC, Sharifi B, Chen PS. Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res 95: 76–83, 2004 [DOI] [PubMed] [Google Scholar]
- 30.Zhou S, Jung BC, Tan AY, Trang VQ, Gholmieh G, Han SW, Lin SF, Fishbein MC, Chen PS, Chen LS. Spontaneous stellate ganglion nerve activity and ventricular arrhythmia in a canine model of sudden death. Heart Rhythm 5: 131–139, 2008 [DOI] [PubMed] [Google Scholar]