

Abstract
Recent studies in prehypertensive spontaneously hypertensive rats (SHR) have shown larger calcium transients and reduced norepinephrine transporter (NET) activity in cultured stellate neurons compared with Wistar-Kyoto (WKY) controls, although the functional significance of these results is unknown. We hypothesized that peripheral sympathetic responsiveness in the SHR at 4 wk of age would be exaggerated compared with the WKY. In vivo arterial pressure (under 2% isoflurane) was similar in SHRs (88 ± 2/50 ± 3 mmHg, n = 18) compared with WKYs (88 ± 3/49 ± 4 mmHg, n = 20). However, a small but significant (P < 0.05) tachycardia was observed in the young SHR despite the heart rate response to vagus stimulation (3 and 5 Hz) in vivo being similar (SHR: n = 12, WKY: n = 10). In isolated atrial preparations there was a significantly greater tachycardia during right stellate stimulation (5 and 7 Hz) in SHRs (n = 19) compared with WKYs (n = 16) but not in response to exogenous NE (0.025–5 μM, SHR: n = 10, WKY: n = 10). There was also a significantly greater release of [3H]NE to field stimulation (5 Hz) of atria in the SHR (SHR: n = 17, WKY: n = 16). Additionally, plasma levels of neuropeptide Y sampled from the right atria in vivo were also higher in the SHR (ELISA, n = 12 for both groups). The difference in [3H]NE release between SHR and WKY could be normalized by the NET inhibitor desipramine (1 μM, SHR: n = 10, WKY: n = 8) but not the α2-receptor antagonist yohimbine (1 μM, SHR: n = 7, WKY: n = 8). Increased cardiac sympathetic neurotransmission driven by larger neuronal calcium transients and reduced NE reuptake translates into enhanced cardiac sympathetic responsiveness at the end organ in prehypertensive SHRs.
Keywords: spontaneously hypertensive rat, cardiac, autonomic neurotransmission, sympathetic, vagal, hypertension
sympathetic hyperactivity is a key feature of essential hypertension (2, 5, 9, 10, 28, 30) and may arise at several distinct neuroanatomical sites. Studies have suggested that a change in afferent chemo- and baroreceptor signaling (7, 23, 26, 29) as well as brainstem control of autonomic efferent output may contribute to the condition (8, 35). More recently major changes have been reported at the end organ level in terms of peripheral cardiac sympathetic neurotransmission in the adult spontaneously hypertensive rat (SHR) compared with normotensive Wistar-Kyoto rats (WKY; Refs. 13, 18, 20, 21, 31, 36). At 16 wk of age, there is evidence of left ventricular hypertrophy and both mean arterial pressure and heart rate are significantly elevated in the SHR (13). In addition, an increase in cardiac norepinephrine (NE) release (measured by 3H-radiolabeling; Ref. 21) and a larger tachycardia on stimulation of the right stellate ganglia (13) is also observed. Cultured neurons from the stellate ganglion of adult SHRs also have larger depolarization-induced calcium transients compared with WKYs (20), and this may drive a greater calcium dependent exocytotic release of NE. Increased NE release appears to be further exacerbated by a reduction in the autoinhibtory action of the presynaptic α2-adrenoceptor (36), as well as a reduction in NE reuptake by the presynaptic NE uptake transporter (NET; Ref. 31). Conversely, the heart rate response to stimulation of the cervical vagus nerve is diminished in the adult SHR compared with age-matched WKYs (11).
A number of recent studies provide evidence that dysregulation of cardiac sympathetic neurotransmission may arise even before the onset of hypertension within the SHR. At ∼4 wk of age when the SHR is still normotensive, an increased intracellular calcium transient in response to neuronal depolarization has been described in stellate and superior cervical ganglia neurons (20). A reduction in the activity of NET has also recently been reported in cultured stellate (but not superior cervical ganglia or renal ganglia) neurons of young prehypertensive SHRs compared with age-matched WKYs (31). However, the functional significance of these observations in terms of NE release and peripheral cardiac sympathetic control of heart rate is unknown. Whether a reduction in α2-receptor autoinhibition also augments NE release at this developmental stage is also unclear.
We therefore hypothesized that stimulation of the right stellate ganglion in young prehypertensive SHRs would produce a significantly larger increase in heart rate and release of NE compared with age-matched WKY controls due to a reduction in NET or α2-receptor autoinhibtion. We also investigated whether the ability of the vagus nerve to reduce heart rate in the prehypertensive SHR was impaired.
MATERIALS AND METHODS
Animals.
Age- and weight-matched male SHR and WKY rats (4 wk of age, SHR: n = 53, WKY: n = 48) were purchased from Harlan (Bicester, UK) and housed under standard laboratory conditions. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996) and the Animals Scientific Procedures (ASP) Act 1986 (UK). Procedures were performed under British Home Office license requirements (PPL 30/2360).
Hemodynamic measurements and heart rate responses to vagus nerve stimulation in-vivo.
Rats were ventilated under anesthesia (2% isoflurane and 100% oxygen) as a terminal procedure. The left carotid artery was cannulated with a 3-F portex cannula, based on a method previously described (19). The cannula was connected to a calibrated pressure transducer, and data were acquired using a biopac M150 system connected to a Macbook Pro computer running AcqKnowledge 3.9.2 software. Heart rate and blood pressure recordings were taken as an average over 30 s after a 5-min stabilization period, recorded at 200 data points/s. In a subset of experiments, the right vagus was isolated, crushed distal to the heart, and stimulated at 3 and/or 5 Hz (20-V, 1-ms pulse duration for 30 s) and the heart rate response was recorded.
Measurement of heart rate response to right stellate stimulation and NE in vitro.
Spontaneously beating atria with intact sympathetic innervations and right stellate ganglion were isolated, and responses to sympathetic nerve stimulation were measured based on a method previously described (13, 25). In brief the double atrial preparation was transferred to a preheated (37 ± 0.2°C), water jacketed, carbogen-aerated water bath containing 100 ml Tyrode solution. A suture through the left atria was attached to a stainless steel hook at the bottom of the water bath, whilst the right atrial suture was connected with an average pretension of 5 mN to an isometric force transducer (model: 60-2997; Harvard Apparatus) linked to a signal amplifier. The right stellate ganglion was threaded through two ring stimulating electrodes. Before each protocol was started, mounted atria were allowed to equilibrate for 45 min in Tyrode solution until the beating rate stabilized (±5 beats/min). The stellate was stimulated at 1, 3, 5, and 7 Hz (20-V, 1-ms pulse duration for 30 s), or increasing cumulative doses of NE (0.025–5 μM; Sigma) were added to the organ bath. Tyrode solution was replaced approximately every 30 min throughout each protocol. Data were collected using a biopac M150 system connected to a Dell computer running AcqKnowledge 3.7.3 software.
Measurement of local [3H]NE release from isolated double atria.
Spontaneously beating double atria were isolated and transferred to a preheated (37 ± 0.2°C), water jacketed, carbogen-aerated water bath containing 3 ml Tyrode solution, where the atria were pinned flat over a silver stimulating electrode. The method for determining local NE release was based on one previously described (19). Briefly, following a 20-min equilibration period, the double atria preparation was incubated with 5 μM [3H]NE (0.185 MBq, ARC) and ascorbic acid (30 μM; Sigma). The atria were field stimulated at 5 Hz (15-V, 1-ms pulse width) for 10 s every 30 s, for 30 min to facilitate uptake of [3H]NE into the transmitter stores of the presynaptic terminal. Following [3H]NE incubation, excess radioactivity was washed from the preparation with Tyrode solution superfusion for 45 min at a rate of 3 ml/min. Bath solution was then replaced every 3 min for 60 min. A 0.5-ml sample from each solution change was added to 4.5 ml scintillation liquid (Ecoscint A; National Diagnostics), and the amount of radioactivity was measured [counts/min (CPM)] using a liquid scintillation counter (Tri-Carb 2800TR, Perkin-Elmers life science). At 16 min the atria were stimulated at 5 Hz for 1 min [stimulation 1 (S1)]. The bath solution from 27 min onwards was changed to contain either desipramine or yohimbine. A second stimulation (S2) was applied at 49 min. The [3H]NE outflow was calculated as a proportional percentage increase: [3H]NE outflow = {[CPM(y) − CPM(x)]/CPM(x)} × 100, where CPM(x) is CPM immediately before stimulation and CPM(y) is CPM immediately after stimulation. Previous control experiments have demonstrated no significant change in the magnitude of the response over two stimulations (19).
Measurement of plasma neuropeptide Y levels.
In a subset of animals on opening the thorax, a 26-gauge needle was inserted through the right ventricle to the level of the right atria and a 0.5-ml blood sample was obtained. The sample was immediately centrifuged (10 min, 13,000 rpm) to isolate plasma and snap frozen at −80°C. Neuropeptide Y (NPY) concentration was measured using a commercially available ELISA (S 1346; Peninsula Laboratories, Bachem) against serially diluted protein standards.
Solutions and drugs.
Rat Tyrode solution for the isolated atria preparation contained the following: (in mmol/l): 120 NaCl, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 2 CaCl2, and 11 glucose, constantly aerated with carbogen (5% CO2-95% O2) to maintain pH 7.4. Experiments using NE, [3H]NE, and ascorbic acid were carried out in the dark due to the light sensitivity of the drug. Desipramine (1 μM; Sigma) was used at a concentration previously shown to completely inhibit NET (31), and yohimbine (1 μM; Sigma) was used at a concentration to completely block the α2-adrenoceptor in a similar preparation (3). Drugs were made up and stored as stock solutions (1 mM) and diluted to the desired concentration on the day. All drugs under went no more than one freeze thaw cycle.
Statistics.
All statistical analysis was carried out using GraphPad Prism (GraphPad Software, San Diego, CA). Data are presented as means ± SE of the mean, and all data passed a normality test. Comparisons between SHR and WKY groups were done using an unpaired Student t-test assuming unequal variance, whereas within group analysis was performed using a one-way ANOVA with a Neuman-Keuls post hoc analysis. Statistical significance was accepted at P < 0.05.
RESULTS
Baseline hemodynamic characteristics.
At 4 wk of age, the SHR (n = 18) did not show any change in systolic, diastolic, or mean arterial blood pressure compared with WKY rats (n = 20), when measured while animals were under general anesthesia in vivo. However, the SHR had a small but significant resting tachycardia under these conditions. After equilibration in the organ bath in vitro, there was no difference in baseline atrial rate between SHRs (n = 19) and WKYs (n = 16). Animals were age matched and were of a similar weight as shown in Table 1.
Table 1.
Measurements in 4-wk-old SHRs and WKYs
| WKY | SHR | |
|---|---|---|
| In vivo systolic blood pressure, mmHg | 88 ± 3 (n = 20) | 89 ± 2 (n = 18) |
| In vivo diastolic blood pressure, mmHg | 49 ± 4 (n = 20) | 51 ± 3 (n = 18) |
| In vivo mean arterial blood pressure, mmHg | 66 ± 3 (n = 20) | 69 ± 3 (n = 18) |
| In vivo heart rate, beats/min | 298 ± 11 (n = 20) | 334 ± 9* (n = 19) |
| In vitro heart rate, beats/min | 286 ± 9 (n = 16) | 298 ± 7 (n = 19) |
| Body weight, g | 97 ± 4 (n = 48) | 96 ± 3 (n = 53) |
Values are means ± SE. No statistical difference in in vivo systolic, diastolic, or mean arterial blood pressure or in vitro atrial rate is observed between 4-wk-old spontaneously hypertensive rats (SHRs) and Wistar-Kyoto rats (WKYs), which are also of a similar body weight. However, in vivo heart rate was significantly increased in the SHR (*P < 0.05, unpaired t-test).
Heart rate response to parasympathetic and sympathetic stimulation.
There was no difference in the heart rate (beats/min) response to right vagal stimulation between the SHR and WKY at either 3 or 5 Hz (3 Hz: SHR, n = 9, WKY, n = 6; 5 Hz: SHR, n = 12, WKY, n = 10) in vivo (see Fig. 1, D and E). However, the heart rate response to stimulation of the right stellate ganglia in the isolated atrial preparation was significantly higher in the prehypertensive SHR (n = 19) compared with age-matched WKYs (n = 16) at 5 and 7 Hz (5 Hz: SHR, 91.1 ± 4.2 beats/min, WKY, 77.1 ± 3.8 beats/min; 7 Hz: SHR, 103.5 ± 4.5 beats/min, WKY, 84.4 ± 3.5 beats/min; Fig. 1B). Despite this the heart rate responses to cumulative doses of NE (0.025–5 μM) in the isolated atria were similar in the SHR and WKY (n = 10 in each group) as shown in Fig. 1C.
Fig. 1.
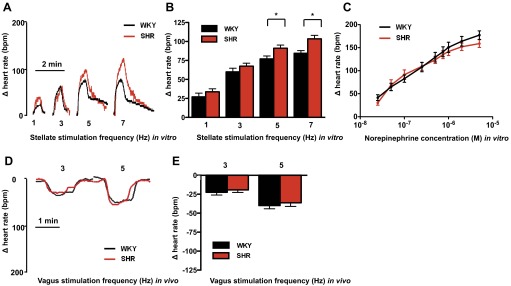
Representative raw data trace (A) and group mean data (B) demonstrating that the heart rate response to right stellate ganglion stimulation in vitro was larger in the prehypertensive spontaneously hypertensive rats (SHR; n = 19) compared with Wistar-Kyoto (WKY; n = 16), significant at 5 (*P < 0.05, unpaired t-test) and 7 Hz (*P < 0.01, unpaired t-test). However, the heart rate response to cumulative concentrations (0.025–5 μM) of norepinephrine (NE) in vitro was similar in the SHR (n = 10) and WKY (n = 10) as shown in C. In vivo the heart rate response to vagal nerve stimulation was also similar in the prehypertensive SHR (3 Hz: n = 9; 5 Hz: n = 12) compared with WKY (3 Hz: SHR, n = 6; 5 Hz: n = 10) as shown by the representative raw data trace (D) and group mean data (E).
Sympathetic neurotransmitter release.
The level of [3H]NE release in response to field stimulation of double atrial preparations was significantly higher in the prehypertensive SHR compared with age-matched WKYs (SHR: 82.14 ± 10.46%, n = 17 vs. WKY: 56.05 ± 6.98%, n = 16; Fig. 2B). Similarly, plasma levels of the sympathetic cotransmitter NPY sampled from the right atria in vivo, were also significantly elevated in the SHR (SHR: 15.70 ± 3.10 nM, n = 12; WKY: 8.461 ± 1.351 nM, n = 12) as shown in Fig. 2C.
Fig. 2.

Representative raw data (A) and group mean data (B) showing increased [3H]NE release from isolated double atrial preparations in response to electrical field stimulation (S1, 5 Hz) in the prehypertenisve SHR (n = 17) compared with age-matched WKY (n = 16, *P < 0.05, unpaired t-test). Plasma levels of the sympathetic cotransmitter neuropeptide-Y (NPY) sampled from the right atria in vivo (C) were also significantly higher within the SHR (n = 12) compared with WKY (n = 12, *P < 0.05, unpaired t-test).
Reuptake and autoinhibition of NE release.
To determine whether the rate of NE reuptake contributed to the increased NE release observed in the SHR, desipramine was used as an inhibitor of NET. Desipramine normalized the difference in release of [3H]NE to field stimulation between the SHR and WKY (SHR: 97.19 ± 7.752%, n = 10; WKY: 106.0 ± 11.53%, n = 8) as shown in Fig. 3E. To determine if reduced levels of α2-adrenoceptor autoinhibition may contribute to the increased NE release observed in the SHR, yohimbine was used a potent and selective α2-antagonist. Yohimbine did not normalize the difference in [3H]NE release between prehypertensive SHRs (n = 7) and WKYs (n = 8). In fact the difference in [3H]NE release between the SHR and WKY was significantly increased. Yohimbine also significantly increased the heart rate response to stimulation of the right stellate ganglia in vitro in the SHR at 7 Hz (99.9 ± 7.0 to 119.2 ± 8 beats/min, n = 9) with a strong trend to increasing the response at 5 Hz (85.9 ± 7.2 to 100.9 ± 7.2 beats/min, P = 0.09), but this was not observed in the WKY (7 Hz: 86.6 ± 4.9 to 96 ± 8.4 beats/min; 5 Hz: 73.4 ± 5.2 to 76.4 ± 7.4 beats/min, n = 9). Yohimbine did not significantly alter baseline heart rate in either group.
Fig. 3.

Neuronal NE reuptake transporter (NET) inhibitor desipramine (DMI; 1 μM) significantly (*P < 0.05) increased [3H]NE release in response to electrical field stimulation (S1 vs. S2, 5 Hz) of isolated atria in the WKY (n = 8, raw data in A; P < 0.01, ANOVA) and SHR (n = 10, raw data in B; P < 0.01, ANOVA). However, after desipramine pretreatment the difference in [3H]NE release between the SHR and WKY was abolished (group mean data in E, unpaired t-test). The α2-receptor antagonist yohimbine (1 μM) had no effect on [3H]NE release in the WKY (n = 8, raw data trace in C) but significantly increased the release of [3H]NE in the SHR (n = 7, raw data trace in D; P < 0.001, ANOVA), The difference in [3H]NE release in response to field stimulation between the SHR and WKY increased significantly in the presence of yohimbine (**P < 0.01, unpaired t-test, group mean data in E).
DISCUSSION
This present study reports three novel findings concerning peripheral cardiac autonomic control in the prehypertensive SHR. First, resting heart rate was significantly higher in vivo when measured while the animals were under general anesthesia in the SHR compared with age- and weight-matched WKYs, despite the intrinsic heart rate of the isolated atria and the ability of the vagus nerve to lower heart rate remaining unchanged. Secondly, the tachycardia to stimulation of the right stellate ganglia was significantly increased in the SHR although the response to exogenous NE was not different to that observed in the WKY. Finally, the release of NE and the sympathetic cotransmitter NPY was also significantly elevated in the SHR. This may be in part due to impaired reuptake of NE through NET rather than through reduced α2-adrenoceptor autoinhibition. When these results are taken together with other reports (20, 31), they indicate that impaired reuptake of NE and enhanced neuronal calcium transients in the SHR translate to an increase in NE release and tachycardia before hypertension develops (Fig. 4).
Fig. 4.

Schematic diagram of a cardiac peripheral sympathetic nerve terminal within the 4-wk prehypertensive SHR. Increased depolarization induced intracellular calcium transients (20) and reduced NE reuptake via the NET (31) facilitate greater release of NE and the cotransmitter NPY at the neuroeffector junction. The increased intracellular calcium transient is due to reduced mitochondrial buffering of intracellular calcium rather than changes in endoplasmic reticulum ryanodine receptor (RyR) function, despite a more rapid uptake into the endoplasmic reticulum by the smooth endoplasmic reticulum ATPase (SERCA; Ref. 15). An increase in the action of α2-adrenoceptor autoinhibtion may act as a compensatory mechanism within the prehypertensive SHR to limit NE release, although this is lost in the adult animal. The increase in NE release to stimulation of the right stellate ganglion leads to a greater increase in the beating rate of the sinoatrial node. Green lines represent stimulatory pathways and red lines inhibitory pathways impacting on neurotransmission.
Disrupted autonomic balance in the prehypertensive SHR.
Hypertension is associated with well-characterized physiological changes, including cardiac remodeling due to stresses in the myocardium resulting in left ventricular hypertrophy, and an increase in sympathetic nervous system activity (27). Within the 4-wk-old SHR, arterial blood pressure was not different from age- and weight-matched WKY controls when measured while the animals were under general anesthesia. In addition, we have previously reported that ventricular weight-to-body weight ratios were also not different (20), suggesting no evidence of left ventricular hypertrophy. However, there was a small but significant increase in resting heart rate in vivo in the prehypertensive SHR. Previous studies have also demonstrated no difference in arterial blood pressure between the SHR and WKY at 4–5 wk of age using the noninvasive tail-cuff method (15, 33) and also following surgical implantation of a radiotelemetry device (16). While the telemetry study reports a significant tachycardia in the SHR, reports from tail-cuff data vary from a significant tachycardia being observed in the WKY (15) to a tachycardia being observed in the SHR (33). One common feature of these studies is the degree of variability in the measurements made. The aim of our invasive blood pressure recordings while the animals were under general anesthesia was to minimize the noise and variation in the data due to recovery surgery from implantation of radiotelemeters and variations in animal activity (the SHR is often used as a model of attention deficit hyperactivity disorder), which may complicate these measurements. While the mean arterial pressures we record are lower than those observed by these other methods (≈70 vs. 90–120 mmHg), they are still within a physiological resting range. Despite our carefully controlled conditions, recordings in 38 animals (SHR: n = 20, WKY: n = 18) were required to demonstrate statistical significance in terms of a tachycardia in the SHR group, suggesting that the resting phenotype is subtle. We have previously observed this resting tachycardia to be more pronounced in the adult animal once hypertension has set in (20).
The fact that there was no difference in the beating rate of isolated atria from both the prehypertensive SHR and WKY despite the in vivo resting tachycardia suggests that the difference observed was due to a change in autonomic balance. The magnitude of vagally mediated bradycardia is small in both the SHR and WKY compared with the tachycardia mediated by sympathetic stimulation as has been previously reported in adult animals (11, 13). Unlike the adult SHR, which has a smaller heart rate response to vagus nerve stimulation than the WKY, we observe no difference in the degree of bradycardia at 4-wk of age, suggesting there is no functional cardiac parasympathetic impairment at this developmental stage. Others have reported a slight increase in the heart rate response to vagal nerve stimulation at high stimulation frequencies at ∼6 wk of age, which quickly normalizes as the animal ages (24). It is therefore unlikely that a change in vagal tone is responsible for the difference in in vivo heart rate we observe.
Conversely, we demonstrated that direct stimulation of the right stellate ganglion at 5 and 7 Hz produced a larger tachycardia in the SHR than the WKY. This was not as marked as in the adult animal where stimulation frequencies between 1 and 7 Hz produce significantly larger responses (13). However, at this age (16 wk) both [3H]NE release and β-adrenergic receptor responsiveness are also greatly increased (13). We report a greater release of NE during stimulation at 5 Hz in the 4-wk-old SHR compared with the WKY although the heart rate response to exogenous NE was no different, suggesting that it is increased neurotransmitter release that is responsible for the larger tachycardia observed both on stimulation of the right stellate in vitro and at rest in vivo.
Plasma levels of the sympathetic cotransmitter NPY are also elevated in the prehypertensive SHR compared with the WKY. Plasma catecholamine levels fluctuate dramatically making it difficult to draw accurate conclusions regarding sympathetic activity when taking measures at one time point. NPY by comparison is a slowly diffusing cotransmitter with a long half-life. Plasma levels have little diurnal variation, and so it is a more accurate indicator of sustained sympathetic hyperactivity (4, 17, 22). For these reasons we chose to measure its plasma levels sampled directly from the right atria in vivo. NPY is also able to cross talk and inhibit vagal acetylcholine release following high frequency sympathetic stimulation (12, 14). We observed no difference in the size of the vagal responses in the young SHR despite higher circulating NPY levels. This suggests the NPY levels were not high enough to impair vagal neurotransmission although both these measurements were taken in the resting state under general anesthesia, rather than following high-level sympathetic stimulation where NPY release may increase further. Nevertheless, it is conceivable that elevated cardiac NPY levels may contribute to resting vagal impairment in the adult animal when very high levels of sympathetic hyperactivity have become established (11).
A previous study has attempted to examine the changes in peripheral autonomic control of heart rate in the prehypertensive SHRs by examining the heart rate responses to field stimulation (6). Field stimulation of the entire atria will depolarize all sympathetic, parasympathetic, efferent, and afferent neurons making the subsequent atrial heart rate response to the many local neurotransmitters and cotransmitters that would be released extremely difficult to interpret. We believe that selective stimulation of the right stellate ganglion is more physiological and reliable, and we are not aware of any other previous work that has achieved this in this age group. We also demonstrate no difference in the chronotropic response to the physiological agonist NE and directly measure NE release to support our physiological heart rate data and add to their novelty.
Ideally, we would have liked to evaluate vagal responses in vitro in the SHR and WKY at this age but despite considerable effort have not been able to overcome this technical challenge. We have previously been able to perform these dissections in guinea pigs of a similar size and mice, but the mediastinal anatomy and narrow neck of the young rat makes it extremely difficult to produce a viable preparation. We therefore chose to stimulate the right vagus by exposing it at the cervical level in vivo. Conversely, it is difficult to reach the right stellate ganglion in vivo without causing a pneuomothorax (given its position posteriorly below the first rib). Measuring NPY release in vitro is also very difficult in such a small preparation when release concentrations are very low. In guinea pigs three to four times this size, measurements of NPY in atrial perfusate are on the limit of detection of ELISA assays (12). We have therefore chosen what we feel to be the most appropriate and accurate methodologies currently available.
Regulation of cardiac sympathetic neurotransmission in the prehypertensive SHR.
The adult SHR has evidence of larger calcium transients on depolarization of cultured stellate neurons (20), reduced NET activity (31), and impaired α2-adrenoceptor autoinhibition of NE release (36). The stroke-prone SHR also has increased sympathetic innervation of the left ventricle with higher tyrosine hydroxylase positive nerve fiber density (18). We observed that inhibition of NET with desipramine normalizes the difference in NE release between prehypertensive SHRs and WKYs. However, the difference in NE release between the young SHR and WKY became significantly larger following administration of the α2-adrenoceptor antagonist yohimbine suggesting that increased α2-adrenoceptor autoinhibition may be a compensatory mechanism limiting increased sympathetic neurotransmission at this developmental stage. There is strong cellular basis underpinning the enhanced functional sympathetic responses reported in this study. Specifically, NET activity is impaired in stellate neurons from 4-wk-old SHRs (31), and those neurons also have larger calcium transients on neuronal depolarization, which may drive more exocytotic release of NE (20). We have also recently reported that there appears to be heterogeneity in NET activity between neurons from the stellate ganglion and the superior cervical ganglion (which mainly innervates the vasculature of the head and neck), and the celiac/superior mesenteric ganglia (which innervate the kidneys and other abdominal organs). Only stellate neurons demonstrated impaired NET activity in the SHR at 4 wk of age compared with the WKY. We therefore do not feel that our results regarding cardiac neurotransmission can necessarily be extrapolated to other areas of the peripheral sympathetic nervous system.
Implications and future perspectives.
We present evidence that dysregulation in cardiac adrenergic signaling in the SHR develops before the onset of hypertension and functionally translates to a small but significant resting tachycardia and larger increases in heart rate on stimulation of the right stellate ganglion. Sympathetic dysfunction may therefore be an early marker of the disease and contribute to its pathogenesis rather than being an epiphenomenon that develops once hypertension has set in. The causes of increased blood pressure in the SHR are clearly multifactorial and involve changes in vascular, renal, and autonomic function at many different anatomical sites. The increase in peripheral cardiac, sympathetic control is unlikely to be the sole cause of the increased blood pressure but may be an early marker of the disease and help drive left ventricular hypertrophy (27). Increased sympathetic nerve activity within borderline hypertensive patients and normotensive individuals with first degree relatives who are hypertensive has also previously been observed (5, 10). With further research, sympathetic cardiac hyperactivity may act as an early marker for those individuals predisposed to develop hypertension, potentially providing novel therapeutic targets for treatment. Currently used treatments for hypertension target the sympatho-adrenal axis and renin-angiotensin pathways treating the end organ after manifestations of the disease have already developed. Greater identification of early hallmarks of the disease may help develop therapies that prevent the progression of hypertension if sympathetic hyperactivity can be targeted.
Whether the increase in peripheral cardiac sympathetic function we observe occurs in isolation or in response to changes in central drive or even changes in afferent brainstem input also remains unclear. For example, greater respiratory sympathetic coupling has been observed in a working heart brainstem preparation from SHRs at 3 to 5 wk of age compared with the WKY, which may reflect an increase in central sympathetic drive (32). Changes in peripheral chemoreceptor sensitivity have also been observed in the SHR compared with the WKY before the onset of hypertension (34), and carotid sinus nerve denervation in the SHR at 4 wk of age has recently been shown to reduce the development of hypertension in this model (1). How these phenomena interact and whether one component helps drive changes in the other will be an interesting area of further study.
GRANTS
N. Herring, J. Shanks, and D. J. Paterson acknowledge support from the British Heart Foundation (BHF) Centre of Research Excellence (CRE; RE/08/004), Oxford. This work was supported by a BHF project grant (PG/11/6/28660). N. Herring is a BHF CRE Intermediate Fellow at the University of Oxford and Senior Registrar in Cardiology at Oxford University Hospital National Health Service Trust.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.S., S.M.-S., C.-J.L., D.L., and N.H. performed experiments; J.S., S.M.-S., and N.H. analyzed data; J.S., S.M.-S., and N.H. interpreted results of experiments; J.S., S.M.-S., and N.H. prepared figures; J.S. and N.H. drafted manuscript; J.S., S.M.-S., C.-J.L., D.L., D.J.P., and N.H. approved final version of manuscript; D.J.P. and N.H. conception and design of research; D.J.P. and N.H. edited and revised manuscript.
REFERENCES
- 1.Abdala AP, McBryde FD, Marina N, Hendy EB, Engelman ZJ, Fudim M, Sobotka PA, Gourine AV, Paton JF. Hypertension is critically dependent on the carotid body input in the spontaneously hypertensive rat. J Physiol 590: 4269–4277, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson EA, Sinkey CA, Lawton WJ, Mark AL. Elevated sympathetic nerve activity in borderline hypertensive humans: Evidence from direct intraneural recordings. Hypertension 14: 177–183, 1989 [DOI] [PubMed] [Google Scholar]
- 3.Carlson DE, Gann DS. Alpha-adrenergic input in the locus coeruleus modulates plasma adrenocorticotropin in cats. Endocrinology 130: 2795–2803, 1992 [DOI] [PubMed] [Google Scholar]
- 4.Cuculi F, Herring N, De Caterina AR, Banning AP, Prendergast BD, Forfar JC, Choudhury RP, Channon KM, Kharbanda RK. Relationship of plasma neuropeptide Y with angiographic, electrocardiographic and coronary physiology indices of reperfusion during ST elevation myocardial infarction. Heart 99: 1198–1203, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davrath LR, Goren Y, Pinhas E, Toledo E, Akselrod S. Early automoic malfunction in normotensive individuals with a genetic predisposition to essential hypertension. Am J Physiol Heart Circ Physiol 285: H1697–H1704, 2003 [DOI] [PubMed] [Google Scholar]
- 6.Dyke AC, Angus JA, Korner PI. A functional study of the development of the cardiac sympathetic neuroeffector junction in the SHR. Hypertension 7: 345–353, 1989 [DOI] [PubMed] [Google Scholar]
- 7.Esler MD, Krum H, Sobotka PA, Schlaich MP, Schmieder RE, Bohm M. Renal sympathetic denervation in patients with treatment-resistant hypertension (The Symplicity HTN-2 Trial): a randomised controlled trial. Lancet 376: 1903–1909, 2010 [DOI] [PubMed] [Google Scholar]
- 8.Fisher JP, Paton JF. The sympathetic nervous system and blood pressure in humans: implications for hypertension. J Hum Hypertens 26: 463–475, 2012 [DOI] [PubMed] [Google Scholar]
- 9.Flaa A, Mundal HH, Eide I, Kjeldsen S, Rostrup M. Sympathetic activity and cardiovascular risk factors in young men in the low, normal, and high blood pressure ranges. Hypertension 47: 396–402, 2006 [DOI] [PubMed] [Google Scholar]
- 10.Greenwood JP, Stoker JB, Mary DASG. Single-unit sympathetic discharge: quantitative assessment in human hypertensive disease. Circ Res 100: 1305–1310, 1999 [DOI] [PubMed] [Google Scholar]
- 11.Heaton DA, Li D, Almond SC, Dawson TA, Wang L, Channon KM, Paterson DJ. Gene transfer of neuronal nitric oxide synthase into intracardiac ganglia reverses vagal impairment in hypertensive rats. Hypertension 49: 380–388, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Herring N, Cranley J, Lokale MN, Li D, Shanks J, Alston EN, Girard BM, Carter E, Parsons RL, Habecker BA, Paterson DJ. The cardiac sympathetic co-transmitter galanin reduces acetylcholine release and vagal bradycardia: implications for neural control of cardiac excitability. J Mol Cell Cardiol 52: 667–676, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herring N, Lee CW, Sunderland N, Wright K, Paterson DJ. Pravastatin normalises peripheral cardiac sympathetic hyperactivity in the spontaneously hypertensive rat. J Mol Cell Cardiol 50: 99–106, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Herring N, Lokale MN, Danson EJ, Heaton DA, Paterson DJ. Neuropeptide Y reduces acetylcholine release and vagal bradycardia via a Y2 receptor-mediated, protein kinase C-dependent pathway. J Mol Cell Cardiol 44: 477–485, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Kokubo M, Uemura A, Matsubara T, Murohara T. Noninvasive evaluation of the time course of change in cardiac function in spontaneously hypertensive rats by echocardiography. Hypertens Res 28: 601–609, 2005 [DOI] [PubMed] [Google Scholar]
- 16.Komolova M, Friberg P, Adams MA. Altered vascular resistance properties and acute pressure-natriuresis mechanism in neonatal and weaning spontaneously hypertensive rats. Hypertension 59: 979–984, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Kondo K, Matsubara T, Nakamura J, Hotta N. Characteristic patterns of circadian variation in plasma catecholamine levels, blood pressure and heart rate variability in Type 2 diabetic patients. Diabet Med 19: 359–365, 2002 [DOI] [PubMed] [Google Scholar]
- 18.Kondo M, Fujiwara T, Tabei R. Noradrenergic hyperinnervation in the heart of stroke-prone spontaneously hypertensive rats (SHRSP). Hypertens Res 19: 69–73, 1996 [DOI] [PubMed] [Google Scholar]
- 19.Lee CW, Li D, Channon KM, Paterson DJ. l-arginine supplementation reduces cardiac noradrenergic neurotransmission in spontaneously hypertensive rats. J Mol Cell Cardiol 47: 149–155, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li D, Lee CW, Buckler KJ, Parekh A, Herring N, Paterson DJ. Abnormal intracellular calcium homeostasis in sympathetic neurons from young prehypertensive rats. Hypertension 59: 642–649, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li D, Wang L, Lee CW, Dawson TA, Paterson DJ. Noradrenergic cell specific gene transfer with neuronal nitric oxide synthase reduces cardiac sympathetic neurotransmission in hypertensive rats. Hypertension 50: 1–6, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Lockinger A, Koberle D, Konig PS, Saria A, Herold M, Cornelissen G, Halberg F. Neuropeptide chronomics in clinically healthy young adults: circaoctohoran and circadian patterns. Peptides 25: 533–542, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Malliani A, Pagani M, Bergamaschi M. Positive feedback sympathetic reflexes and hypertension. Am J Cardiol 44: 860–865, 1979 [DOI] [PubMed] [Google Scholar]
- 24.Minami N, Head GA. Cardiac vagal responsiveness during development in spontaneously hypertensive rats. Auton Neurosci 82: 115–122, 2000 [DOI] [PubMed] [Google Scholar]
- 25.Mohan RM, Golding S, Paterson DJ. Intermittent hypoxia modulates nNOS expression and heart rate response to sympathetic nerve stimulation. Am J Physiol Heart Circ Physiol 281: H132–H138, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Paton JF, Sobotka PA, Fudim M, Engleman ZJ, Hart EC, McBryde FD, Abdala AP, Marina N, Gourine AV, Lobo M, Patel N, Burchell A, Ratcliffe L, Nightingale A. The carotid body as a therapeutic target for the treatment of sympathetically mediated diseases. Hypertension 61: 5–13, 2013 [DOI] [PubMed] [Google Scholar]
- 27.Schlaich MP, Kaye DM, Lambert E, Sommerville M, Socratous F, Esler MD. Relation between cardiac sympathetic activity and hypertensive left ventricular hypertrophy. Circulation 108: 560–565, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Schlaich MP, Lambert E, Kaye DM, Krozowski Z, Campbell DJ, Lambert G, Hastings JA, Aggarwal A, Esler MD. Sympathetic augmentation in hypertension: role of nerve firing, norepinephrine reuptake, and angiotensin neuromodulation. Hypertension 43: 169–175, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Schlaich MP, Sobotka PA, Krum H, Lambert E, Esler MD. Renal sympathetic-nerve ablation for uncontrolled hypertension. N Engl J Med 361: 932–934, 2009 [DOI] [PubMed] [Google Scholar]
- 30.Sendeski MM, Consolim-Colombo FM, Leite CC, Rubira MC, Lessa P, Krieger EM. Increased sympathetic nerve activity correlates with neurovascular compression at the rostral ventrolateral medulla. Hypertension 47: 988–995, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Shanks J, Mane S, Ryan R, Paterson DJ. Ganglion-specific impairment of the norepinephrine transporter in the hypertensive rat. Hypertension 61: 187–193, 2013 [DOI] [PubMed] [Google Scholar]
- 32.Simms AE, Paton JF, Pickering AE, Allen AM. Amplified respiratory-sympathetic coupling in the spontaneously hypertensive rat: does it contribute to hypertension? J Physiol 587: 597–610, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda K, Nakata T, Takesako T, Itoh H, Hirata M, Kawasaki S, Hayashi J, Oguro M, Sasaki S, Nakagawa M. Sympathetic inhibition and attenuation of spontaneous hypertension by PVN lesions in rats. Brain Res 543: 296–300, 1991 [DOI] [PubMed] [Google Scholar]
- 34.Tan ZY, Lu Y, Whiteis CA, Simms AE, Paton JF, Chapleau MW, Abboud FM. Chemoreceptor hypersensitivity, sympathetic excitation, and overexpression of ASIC and TASK channels before the onset of hypertension in SHR. Circ Res 106: 536–545, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zucker IH, Schultz HD, Li YF, Wang Y, Wang W, Patel KP. The origin of sympathetic outflow in heart failure: the roles of angiotensin II and nitric oxide. Prog Biophys Mol Biol 84: 217–232, 2004 [DOI] [PubMed] [Google Scholar]
- 36.Zugck C, Lossnitzer D, Backs J, Kristen A, Kinscherf R, Haass M. Increased cardiac norepinephrine release in spontaneously hypertensive rats: role of presynaptic alpha-2A adrenoceptors. Hypertension 21: 1363–1369, 2003 [DOI] [PubMed] [Google Scholar]