

Abstract
Mechanical ventilation may cause harm by straining lungs at a time they are particularly prone to injury from deforming stress. The objective of this study was to define the relative contributions of alveolar overdistension and cyclic recruitment and “collapse” of unstable lung units to membrane wounding of alveolar epithelial cells. We measured the interactive effects of tidal volume (VT), transpulmonary pressure (PTP), and of airspace liquid on the number of alveolar epithelial cells with plasma membrane wounds in ex vivo mechanically ventilated rat lungs. Plasma membrane integrity was assessed by propidium iodide (PI) exclusion in confocal images of subpleural alveoli. Cyclic inflations of normal lungs from zero end-expiratory pressure to 40 cmH2O produced VT values of 56.9 ± 3.1 ml/kg and were associated with 0.12 ± 0.12 PI-positive cells/alveolus. A preceding tracheal instillation of normal saline (3 ml) reduced VT to 49.1 ± 6 ml/kg but was associated with a significantly greater number of wounded alveolar epithelial cells (0.52 ± 0.16 cells/alveolus; P < 0.01). Mechanical ventilation of completely saline-filled lungs with saline (VT = 52 ml/kg) to pressures between 10 and 15 cmH2O was associated with the least number of wounded epithelial cells (0.02 ± 0.02 cells/alveolus; P < 0.01). In mechanically ventilated, partially saline-filled lungs, the number of wounded cells increased substantially with VT, but, once VT was accounted for, wounding was independent of maximal PTP. We found that interfacial stress associated with the generation and destruction of liquid bridges in airspaces is the primary biophysical cell injury mechanism in mechanically ventilated lungs.
Keywords: lung mechanics, injury, epithelial wounding
plasma membrane wounding of alveolus resident cells contributes to the pathogenesis of acute lung injury and ventilator-induced lung injury (VILI) (48). While the presence of cytopathological changes in epithelial and endothelial cells, which decorate the blood-gas barrier, has been appreciated for some time (10, 26), it should be noted that the majority of wounded alveolus resident cells repair and survive deformation-induced insults (17). This is important insofar as wounded and repaired cells activate stress response genes (23), release proinflammatory mediators, and may thereby contribute to injurious deformation responses commonly referred to as biotrauma (44). Although most students of the topic VILI agree that alveolar overdistension and cyclic recruitment and “collapse” of unstable lung units are the most prevalent biophysical lung injury mechanisms (34, 38, 43), there is less certainty how stresses associated with either mechanism are transduced to generate a specific biological response. Viewed through the lens of cellular plasma membrane stress failure, alveolar overdistension is generally thought to be associated with matrix strains that effect lytic tensions at cell-cell and cell-matrix contacts. In contrast, “recruitment and collapse” predispose airway and alveolar lining cells to injurious interfacial stresses, since liquid bridges are continually formed, displaced, and destroyed in the small airways and air spaces of edematous lungs. Experimental models of overdistension injury mechanisms typically employ cell monolayers that are stretched in liquid culture (41, 46), whereas the injury associated with recruitment and collapse is typically mimicked by advancing gas bubbles or by destroying liquid bridges in cell-coated microchannels (6, 25). While cytopathological changes in ventilator-injured lungs are widespread and involve both epithelial and endothelial lesions consisting of the loss of basement membrane anchorage, as well as loss of cell-cell contact, plasma membrane blebbing and frank basement membrane fracture (10, 16), the short time scale at which injured cells remodel and repair introduces uncertainty about the specific nature of the stresses that cause particular lesions (21).
Motivated by the hypothesis that the maintenance and/or restoration of alveolar epithelial cell integrity may improve outcomes in acute respiratory distress syndrome (ARDS), we had identified a variety of candidate compounds and tested their mechanisms of action (5, 21, 36, 39, 50). In doing so, we learned that hypertonic media protected alveolar epithelia from injury by interfacial stress and were able to attribute the cytoprotective effect to cell stiffening combined with an increase in the adhesive force between cytoskeleton and the plasma membrane (36). However, the very same mechanism, i.e., “cell stiffening,” has been shown to promote tight junction stress failure and monolayer disruptions in cultured endothelia and epithelia subjected to stretch (28, 40). Stretch-induced epithelial monolayer disruptions in liquid culture are also associated with plasma membrane wounds and have served as model systems of alveolar epithelial injury by overdistension (46). Therefore, experiments were carried out to define the relative contributions of tensile stress (associated with stretch and alveolar overdistension) and interfacial stress (associated with alveolar liquid and surfactant disruption) as principal causes of alveolar epithelial wounding in ex vivo mechanically ventilated rat lungs. To this end, we measured the interactive effects of tidal volume (VT), transpulmonary pressure (PTP), respiratory rate, and the biophysical properties of airspace liquid on the number of alveolar epithelial cells with plasma membrane wounds. Our results confirm the importance of surface forces in the genesis of epithelial injury, while revealing a remarkable tolerance of alveolar epithelial cells to alveolar distension.
METHODS
Experimental Preparation
As approved by the Institutional Animal Care and Use Committee of the Mayo Clinic, female Sprague Dawley rats (200–250 g) were killed with an overdose of pentobarbital, their trachea was intubated, and the lungs were harvested through a sternal midline incision. Rats destined for total liquid ventilation (TLV) experiments were anesthetized with an intraperitoneal injection of ketamine (90 mg/kg) and xylazine (10 mg/kg), intubated, and mechanically ventilated with 100% O2 at a VT of 6 ml/kg body wt and a positive end-expired pressure (PEEP) setting of 3 cmH2O. Five minutes later, the airway was occluded at end-expiration to promote lung collapse by gas absorption. Over the ensuing minutes during which the animals usually died, 8 ml of saline were gently instilled in the trachea. Thereafter, the thorax was opened widely, and the lungs were harvested and inflated with additional saline while residual gas and foam were aspirated.
Ventilation Protocols
Dry lung ventilation.
Eight normal rat lungs were mechanically ventilated for 20 min with room air between PTP values of zero (ZEEP) and 40 cmH2O at a rate of 40 cycles/min (Scirec, Montreal, Canada). An additional six lungs were mechanically ventilated to pressures of 40 cmH2O while PEEP was set to 3 cmH2O.
Wet lung ventilation.
A total of 54 rat lungs in which the airspaces had been flooded at the outset with 3 ml (∼11–13 ml/kg) of liquid were mechanically ventilated with room air in a bilevel pressure preset mode at settings detailed in Table 1. The initial liquid instillate consisted of either normal saline (NS) or a perfluorocarbon solution (PFC) with low surface tension. Cells with plasma membrane defects were identified by propidium iodide (PI) exclusion with label applied either from the outset as part of the initial NS instillate or post hoc, i.e., after cessation of mechanical ventilation (17).
Table 1.
| Group | n | Fluid Type | Pressure, cmH2O | Rate, min−1 | TI, s | Duration, min | Timing of PI Labeling |
|---|---|---|---|---|---|---|---|
| WLV1 | 8 | NS | 0–40 | 40 | 0.75 | 20 | Post hoc |
| WLV2 | 8 | NS | 0–40 | 40 | 0.75 | 20 | From outset |
| WLV3 | 8 | NS | 0–30 | 40 | 0.75 | 20 | From outset |
| WLV4 | 8 | NS | 0–20 | 40 | 0.75 | 20 | From outset |
| WLV5 | 8 | NS | 24–40 | 40 | 0.75 | 20 | From outset |
| WLV6 | 7 | NS | 0–40 | 1 | 6 | 60 | From outset |
| WLV7 | 7 | PFC | 0–40 | 40 | 0.75 | 20 | Post hoc |
WLV, wet lung ventilation; n, no. of observations; NS, normal saline; PFC, perfluorocarbon.; rate refers to respiratory frequency; Ddration refers to the length of the experimental exposure; TI, inspiratory time; PI, propidium iodide; timing of the PI label application identifies groups in which the initial tracheal instillate did (from the outset) or did not (post hoc) containe PI.
TLV.
Eighteen completely saline-filled lungs were suspended from the trachea in a custom-designed bioreactor and were “liquid-ventilated” for up to 1 h with a PI-containing saline solution (Applied Motion Products, Watsonville, CA). Peak PTP and VT were set to 15 cmH2O and 12 ml, respectively, whereby delivered volume was adjusted throughout to meet these targets. Using negative airway pressure during the deflation cycle as indicator, expiratory flow (time) was adjusted to minimize/avoid expiratory flow limitation. This constrained the frequency and duty cycle to 1.0/min and 0.1, respectively.
Cell Injury Assessment
Alveolar epithelial cells with plasma membrane lesions were labeled by tracheal insufflation of a fluorescein dextran (FDx)- and PI-containing solution at the end of each experimental run (4, 17). In the presence of a plasma membrane defect, PI gains access to the cytosol, intercalates with DNA, and in this form can be excited to emit red light. Twelve anatomically distinct lung regions were imaged using a Zeiss LSM 510 META laser scanning confocal microscope (Carl Zeiss, Thornwood, NY) at a depth of up to 20 μm. Two channel images were collected as follows: autofluorescence, excitation λ = 405 nm, image collected at λ between 420 and 480 nm; PI, excitation λ = 543 nm, image collected at λ >560 nm. Images were digitized at an eight-bit resolution and were stored in arrays of 512 × 512 pixels. Green light emission (on account of FDx) assured that PI-containing fluid had gained access to the regions of interest. The extent of plasma membrane injury was evaluated in a blinded fashion by two independent observers and expressed as a ratio of the number of injured (PI-positive) cells per total number of alveoli in the 12 image fields (cell injury index, CII).
In some wet lung ventilation (WLV) experiments and in all TLV experiments, the solution used to initially flood the lungs was also supplemented with PI (1 μg/ml). PI exposure during injurious ventilation labels not only mortally wounded (i.e., necrotic or apoptotic) cells but also cells with transient (successfully repaired) membrane lesions. In contrast, post hoc labeling identifies only cells with permanent membrane defects, i.e., cells that had failed to repair the plasma membrane wound.
Absolute Lung Volume at ZEEP
Lung volume at ZEEP was measured by water immersion/displacement in two groups of five partially liquid-filled lungs (WLV) after ex vivo mechanical ventilation with VT values of 10 and 30 ml/kg body wt.
Pressure/Volume Curves of Completely Saline-Filled Lungs
The saline-filled lungs were suspended from the trachea, and liquid was allowed to drain passively from the airways until the liquid pressure at midlung level reached zero. Subsequently, alveolar liquid pressure (Freescale no. MPXV7002DP) and lung weight change were measured during three successive stepwise saline inflations and deflations. To this end, a second pressure transducer (Freescale no. MPXV7002DP) was fitted with a custom highly compliant gas-tight diaphragm to allow it to function as a force transducer. A cantilever resting on ultralow friction knife edge pivot points held the lung at one end, whereas the opposing end was held captive by the force transducer. A/D conversion and acquisition was made via National Instruments USB-6215 running in house-developed Labview software. Data from the first of three inflation and deflation cycles were discarded (Fig. 1).
Fig. 1.
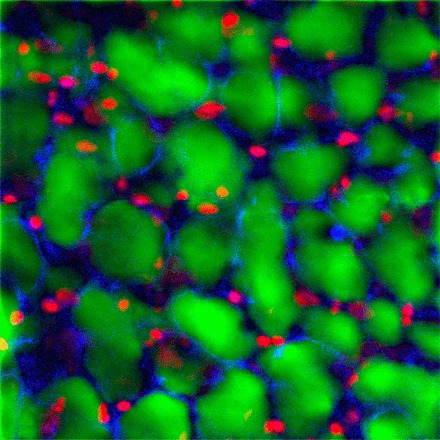
Representative optical section of an injured rat lung labeled by airspace instillation of a solution containing propidium iodide (PI) and green fluorescent dextran. The blue autofluorescence arises from the connective tissue of the lung parenchyma. The nuclei of alveolar epithelial cells with plasma membrane defects can be identified by their red nuclear fluorescence.
RESULTS
Effect of Interfacial Stress on Epithelial Injury Associated with Mechanical Hyperventilation
Cyclic inflations of normal “dry” lungs with room air from ZEEP to a target PTP of 40 cmH2O required a VT of 12.4 ± 0.1 ml (56.9 ± 3.1 ml/kg). There was no significant change in VT and by inference in lung mechanics over the course of a 20-min-long stress exposure. In contrast to dry lung ventilation (DLV), inflation of partially saline-filled lungs (WLV) to comparable peak pressures required a VT of only 9.6 ± 1.1 (44.3 ± 5.0 ml/kg; P < 0.01). Moreover, in each instance VT increased to an average of 10.7 ± 1.5 ml (49.1 ± 6.0 ml/kg) during the 20-min-long stress exposure, indicating a temporal rise in the lungs' dynamic compliance. Imaging of subpleural regions of air-ventilated dry lungs identified 0.12 ± 0.12 PI-positive cells/alveolus. The application of 3 cmH2O PEEP and associated reduction in VT to 10 ± 0.7 ml (43.1 ± 2.8 ml/kg) was associated with fewer PI-positive cells/alveolus (0.06 ± 0.03; P > 0.2), but this difference did not reach statistical significance. In contrast to DLV, partially saline-filled lungs, which had been cyclically inflated to the same peak PTP of 40 cmH2O, had significantly more PI-positive alveolus resident cells (CII = 0.52 ± 0.16; P < 0.01) when assessed by post hoc PI labeling (Fig. 2). In WLV runs, in which cells had been exposed to PI-containing liquid throughout, the cell injury estimate was even greater (0.75 ± 0.27; P < 0.01). This difference was expected given the capacity of wounded alveolar epithelial cells to repair plasma membrane lesions within tens of seconds, thus regaining the capacity to exclude PI (17, 21). Compared with saline, the tracheal instillation of a liquid with lower surface tension, i.e., PFC (3 ml or 12.9 ± 1.0 ml/kg), was associated with significantly fewer PI-positive cells/alveolus (CII = 0.52 ± 0.16 vs. 0.20 ± 0.07; P < 0.01).
Fig. 2.
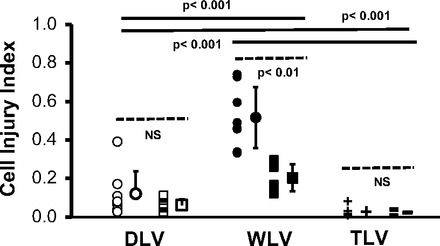
A comparison of hyperventilation-induced epithelial injury estimates between “dry” lungs (DLV), “wet” lungs, i.e., partially liquid-flooded lungs (WLV), and completely saline-filled lungs subjected to total liquid ventilation (TLV) is shown. The two DLV groups (open symbols) differ with respect to positive end-expiratory pressure and tidal volume settings. Open circles represent data from lungs ventilated between zero end-expiratory pressure and 40 cmH2O pressure. The open squares represent data from lungs ventilated between 3 and 40 cmH2O pressure. The two WLV groups differ with respect to the biophysical properties of the alveolar liquid. Closed circles represent data from partially saline-filled lungs. Closed squares represent data from partially perfluorocarbon-filled lungs. The two TLV groups differ with respect to the duration of mechanical ventilation. Crosses represent data from lungs that were ventilated for 20 min. Bars represent data from lungs that were ventilated for 60 min. Cell injury index (CII) refers to the number of wounded epithelial cells per alveolus in post hoc-labeled lungs. Individual and group mean values and their SDs are shown. Dashed and solid lines identify the groups to which statistical comparisons of data means apply. NS stands for not significant at a P value <0.05.
The elimination of surface tension in completely saline-filled and saline-ventilated lungs of groups TLV was associated with little demonstrable epithelial plasma membrane injury. There was no significant difference in cell injury estimates between lungs that were liquid ventilated with VT values between 47 and 55 ml/kg for 20 min and those ventilated for 60 min (CII = 0.03 ± 0.03 vs. 0.02 ± 0.02; P = 0.67). End-inspiratory lung volumes were consistently greater than the volume at which quasi-static pressure-volume curves of completely saline-filled rat lungs departed from linear inflation characteristics, i.e., inflation pressures exceeded the curves' upper point of maximum curvature (data not shown). Six lungs that had been repeatedly inflated to liquid pressures above 20 cmH2O showed evidence of cell injury (CII = 0.34 ± 0.31), typically in the context of profound fluid leaks (data not shown).
Because TLV imposes limits on the rate of lung emptying, TLV-associated cell injury estimates were compared with those obtained from a group of partially (3 ml) saline-filled lungs that had been ventilated with air at identical frequency (1/min) and duty cycle (0.1) settings. There was a nearly 10-fold difference in the number of cells with plasma membrane wounds, which favored TLV (CII = 0.02 ± 0.02 vs. 0.18 ± 0.07; P < 0.01).
Effects of VT on Alveolar Epithelial Injury in Flooded Lungs
Cyclic inflations of partially liquid-filled lungs from ZEEP to end-inspiratory airway and thus apparent PTP values of 40, 30, and 20 cmH2O generated VT values of 10.0 ± 1.3, 6.6 ± 0.7, and 2.3 ± 0.5 ml, i.e., 45.2 ± 3.5, 29.0 ± 4.2, and 10.1 ± 2.3 ml/kg, respectively. Only during runs with high VT values and peak pressures did we observe a minor increase in dynamic lung compliance with time. There was a strong correlation between VT and the extent of alveolar epithelial cell injury (Fig. 3). Average CII values increased with VT from 0.09 ± 0.04 to 0.27 ± 0.12 and 0.75 ± 0.27, respectively. Small VT values were less injurious than large ones, even at settings that resulted in the same maximal PTP. To demonstrate this, in a group of eight partially saline-filled lungs, the increase in PEEP to 24 cmH2O and the concomitant reduction in VT from 12 to 2.0 ml (55–9 ml/kg) was associated with the fewest number of injured alveolus resident cells (0.02 ± 0.01 PI-positive cells/alveolus). In saline-flooded lungs, the choice of VT had a significant effect on the absolute lung volume at ZEEP. Mechanical ventilation of “wet” lungs with a VT of 10 ml/kg body wt was associated with a ZEEP volume of 1.01 ± 0.1 ml/g tissue, increasing to 1.26 ± 0.1 ml/g (P < 0.01) when ventilated with 30 ml/kg. This observation suggests that the agitation of airspace liquid with large VT values promotes gas trapping and the formation of foam in airspaces and conducting small airways.
Fig. 3.
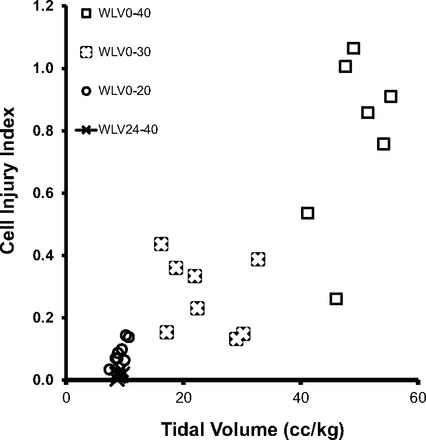
The effect of tidal volume on epithelial injury estimates in 4 groups of 8 wet lungs, i.e., saline-flooded lungs (WLV), is shown. The numbers associated with each group refer to the airway pressure limits (equal to apparent transpulmonary pressure) between which the lungs were “cycled.” CII refers to the number of wounded epithelial cells/alveolus in lungs in which the initial saline instillate contained PI.
DISCUSSION
Our study underscores the deleterious effects of mechanical hyperventilation on epithelial plasma membrane integrity in lungs with alveolar liquid. While at first glance this observation may hardly seem surprising, the remarkable tolerance of “dry lungs,” i.e., lungs without alveolar liquid, to repeated inflations of similar if not greater magnitude warrants discussion. Our study was motivated by the belief that epithelial plasma membrane wounding contributes to the innate immune and remodeling response of an injured lung and therefore represents a promising target for therapeutic interventions (48). Having identified and tested a number of cytoprotective compounds, it has become clear that achieving the desired effect depends on the specific nature of the injurious stress (28, 36). Although terms like overdistension, hyperinflation, opening/collapse, recruitment/derecruitment, interdependence, and tissue shear are commonly used to describe events on the scale of parenchymal networks, the stresses and deformations experienced by individual alveolar epithelial cells are by and large tensile and interfacial in nature. They are tensile insofar as lung inflations to volumes near total lung capacity unfold and ultimately stretch the matrix to which alveolar epithelial and endothelial cells adhere (20, 45). Such deformations have the potential of generating shear stresses between focal adhesions and matrix as well as normal stresses at tight interepithelial junctions. When similar deformations of sufficient magnitude are examined in cell culture, tight junction failure, loss of cell-matrix adherence, and plasma membrane wounds are often observed (41, 46, 49). Interestingly, we detect relatively little alveolar epithelial injury in intact unperfused dry normal lungs, in spite of having imposed huge deformations. This raises questions about the relevance of lung overdistension and tensile stress as a prevalent alveolar epithelial injury mechanism and is in stark contrast to the injurious potential of interfacial stress. Before addressing the mechanisms of interfacial stress injury, it should be stated that our research focused on only one specific manifestation of injury, namely, the wounding of epithelial plasma membranes. It neither addresses nor discounts other manifestations or mechanisms of ventilator-associated lung injury.
The term collapse is often used in the ARDS literature to describe an airless region of the lung (24). When such a region is recruited, i.e., “forced open or reaerated,” cells that line airways and air spaces are exposed to interfacial surface pressure gradients, which may be large enough to wound plasma membranes. Guided by Gaver's and Charras' computational models, Oeckler et al. proposed that the responsible cell injury mechanism involves a compressive stress gradient and apical cell deformation that causes a phase separation between the solid actin network and the incompressible cytosol, the formation of a membrane bleb, and finally its rupture (7, 19, 36). While the tissue distortion associated with alveolar collapse causes a shear deformation of the interdependent parenchymal network (33), the injurious interfacial stress is oriented normal and not tangential to the apical cell surfaces (19). As predicted by Laplace's law and confirmed experimentally, this stress varies with surface tension and the geometry of the air-liquid interface. In contrast, apical shear stresses play little to no role as a wounding mechanism of small airway and alveolar epithelia (6, 36). Consistent with these predictions and previous observations in ex vivo models, we show that an alveolar liquid with high surface tension such as NS produces significantly more cell injury than one with a lower surface tension such as PFC (Fig. 2). When air-liquid interfaces and thus surface tension were eliminated altogether by TLV, the epithelial integrity was more or less preserved even though the lungs were repeatedly inflated to near maximal volumes for up to 1 h. The cytoprotective effect of TLV cannot be attributed to the very low rates of lung inflation because partially saline-filled lungs, when ventilated at the same low-frequency settings, had substantially more injury. In contrast to TLV, in spite of its low surface tension, partial liquid ventilation (PLV) of lungs instilled with PFC was associated with moderate interfacial cell injury. Interestingly, a human trial of PLV in patients with ARDS suggested harm of this support modality (27).
Although cell injury and cell death have long been recognized as integral features of the VILI phenotype, their weight in the histological grading of lung deformation injury is marginal at best (31). It is therefore necessary to interpret our findings in the context of experimental literature in which cell injury mechanisms were inferred from surrogates, such as hemorrhage, edema, and the accumulation of inflammatory cells. In doing so, we are struck by the remarkable tolerance of the alveolar epithelial lining of nonedematous, nonperfused lungs to large tidal inflations, which amount to vital capacity maneuvers. This is in clear contrast to the epithelial and endothelial barrier lesions, which can be readily seen following high tidal ventilation in living experimental animals (12, 22, 51). While many investigators of the topic VILI have attributed the cytopathological changes of the blood-gas barrier to “overstretch” (13, 46, 48), the observations of the present study raise the possibility that injurious stresses associated with alveolar distension are in fact interfacial in nature as well.
Lung inflation can produce a protein-permeable alveolar epithelium at times allowing alveolar flooding at normal vascular pressures (15). Only at very high lung volumes and vascular pressures does the capillary hoop stress exceed lytic levels, i.e., lead to basement membrane fracture (16, 53). As long as the basement membrane remains intact, it is not clear that the epithelial lining itself undergoes stress failure at cell-cell or cell-matrix junctions (8, 14). Morphological studies in systemic capillaries exposed to high vascular pressures have demonstrated reversible endothelial openings, providing avenues for transcellular as well as paracellular fluid flux (35). However, such changes are probably manifestations of active remodeling as opposed to repair responses to structural failure. Therefore, we consider plasma membrane stress failure to be a consequence of alveolar flooding and not the primary cause of impaired epithelial barrier properties. The latter is thought to reflect Rho pathway-dependent mechanosensitive cytoskeletal remodeling events (9) and is associated with changes in the claudin tight junction protein expression profiles (54).
The importance of small VT values in lung protective mechanical ventilation is supported by compelling physiological mechanisms and by incontrovertible clinical evidence (1). Given the reduced vital capacity of injured lungs (18), the imposition of large VT values risks damaging the lungs by both overdistension and underrecruitment (“opening and collapse”). In addition, large parenchymal deformations (strains) will over time impair surfactant kinetics and function, irrespective of the absolute lung volume range over which the lungs are being “stretched” (22, 30, 47). The results of our studies in an experimental model with a narrowly focused injury endpoint, namely epithelial plasma membrane wounding, demonstrate a strain (i.e., VT) dependence of interfacial cell injury mechanisms, in agreement with a recent report in mechanically ventilated anesthetized rats (37). The effect cannot be attributed to peak parenchymal stress because neither conventional mechanical ventilation of dry normal lungs nor TLV to comparable maximal lung volumes was associated with significant cell damage. We therefore reason that large tidal oscillations promote the generation and destruction of foam in airways and airspaces, thereby injuring the epithelial lining during liquid bridge rupture (25). To test this hypothesis, we compared the volumes of the lung at ZEEP in partially liquid-filled lungs following 20 min of ex vivo mechanical ventilation with VT values of 10 ml/kg and 30 ml/kg body wt, respectively. The reason why lung volume at ZEEP (i.e., zero apparent PTP) is influenced by the amount of foam in airspaces is because foam formation promotes gas trapping. Consistent with the proposed mechanism, the end-expiratory (ZEEP) volume of lungs containing alveolar liquid was 25% greater following mechanical ventilation with 30 ml/kg than it was following mechanical ventilation with a VT of 10 ml/kg.
The application of high levels of PEEP to partially liquid-filled lungs prevented epithelial wounding even when lungs were inflated to very large end-inspiratory volumes and pressures. This finding is in line with seminal observations on PEEP-related lung protection in mechanically ventilated anesthetized rats (11, 51) and provides the rationale for the so-called “open lung mechanical ventilation” strategy (3). Although our observations with respect to PEEP-mediated lung protection may be viewed as merely confirmatory, they do offer heretofore underappreciated mechanistic insights. The many degrees of freedom in ventilator settings pose a challenge insofar as PEEP-mediated lung protection could be attributed to an increase in mean lung volumes or to the concomitant reduction in VT. We suggest that both variables contribute, insofar as both mediate reductions in interfacial stress by distinct mechanisms. An increase in end-expiratory lung volume reduces the probability of liquid bridge formation by virtue of increasing the dimensions of small airways and alveolar ducts. Moreover, an increase in mean lung volume promotes the translocation of alveolar edema to the interstitial compartment across a “leaky” epithelial barrier by interdependence mechanisms (29). Reductions of VT, in turn, not only preserve surfactant function (30, 47) but also, as already pointed out, limit foam formation in edema fluid.
Conclusions derived from reduced experimental models such as ex vivo ventilated unperfused rat lungs warrant scrutiny. As already mentioned, the probability of pulmonary capillary stress failure and by inference stress failure of the cells that decorate the capillary basement membrane depends on both lung volume and vascular pressure (16). It is therefore conceivable that, in the intact organism, epithelial stresses and microstrains could be greater than the ones we have examined in vitro. However, we consider this possibility remote because, in the absence of severe pulmonary venous hypertension, intra-alveolar capillaries are likely to collapse at alveolar pressures of 40 cmH2O and operate under zone 1 perfusion conditions (52). We can only speculate if associated cyclic “opening and closure” of intra-alveolar capillaries stresses and/or wounds endothelial cells. While in VILI models pulmonary capillary endothelial cells account for up to 50% of alveolus-resident cells with structural lesions (16), the airspace-labeling method we used in this study does not assure PI access to endothelial cells. For that reason, we can also not comment on putative effects of alveolar epithelial wounding on structure or function of adjacent endothelial cells. Finally, our conclusions are based on observations from subpleural alveoli, the deformations of which could theoretically differ from those of more centrally located lung units.
We have shown that normal dry lungs suffer only minor epithelial injury when exposed to high tidal ventilation. Because ex vivo mechanical ventilation from ZEEP is associated with cyclic alveolar collapse generating shear stresses between wetted epithelial surfaces during subsequent recruitment, we attribute epithelial wounding in dry lungs (group DLV) to interfacial stresses as well. The added cytoprotection observed during TLV (group TLV) supports this conclusion. To the extent to which epithelial injury is a significant transducer of biotrauma (44), our findings draw attention to the importance of alveolar edema in the pathogenesis of VILI. While they do not discount parenchymal remodeling and the release of matrix metalloproteases as a consequence of mechanical ventilation with high volumes and pressures, they emphasize interfacial injury mechanisms over stress failure at cell-cell and cell-matrix junctions. By inference, they support PEEP management algorithms directed at reestablishing positive end-expiratory PTP (42), suggest that surface tension and fluidity of the alveolar space filling material (e.g., edema fluid vs. hyaline membranes) are critical determinants of mechanical ventilation-associated epithelial injury (32), and are consistent with the idea that ventilation-induced surfactant dysfunction and atelectasis are causal mechanisms in the pathogenesis of the ARDS (2).
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-63178.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: O.H., B.J.W., R.W.S., P.A.V., and D.M. performed experiments; O.H., B.J.W., R.W.S., P.A.V., D.M., and R.D.H. analyzed data; O.H., B.J.W., R.W.S., P.A.V., D.M., and R.D.H. interpreted results of experiments; O.H. and R.D.H. edited and revised manuscript; O.H., B.J.W., R.W.S., P.A.V., D.M., and R.D.H. approved final version of manuscript; R.D.H. conception and design of research; R.D.H. prepared figures; R.D.H. drafted manuscript.
ACKNOWLEDGMENTS
We thank Drs. Richard Albert and Roy Brower for helpful comments and suggestions.
REFERENCES
- 1.Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes compared with traditional tidal volumes for acute lung injury, and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 342: 1301–1308, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Albert RK. The role of ventilation-induced surfactant dysfunction and atelectasis in causing acute respiratory distress syndrome. Am J Respir Crit Care Med 185: 702–708, 2012 [DOI] [PubMed] [Google Scholar]
- 3.Amato MB, Barbas CS, Medeiros DM, Schettino Gde P, Lorenzi Filho G, Kairalla RA, Deheinzelin D, Morais C, Fernandes Ede O, Takagaki TY. Beneficial effects of the “open lung approach” with low distending pressures in acute respiratory distress syndrome A prospective randomized study on mechanical ventilation. Am J Respir Crit Care Med 152: 1835–1846, 1995 [DOI] [PubMed] [Google Scholar]
- 4.Belete HA, Godin LM, Stroetz RW, Hubmayr RD. Experimental models to study cell wounding and repair. Cell Physiol Biochem 25: 71–80, 2000 [DOI] [PubMed] [Google Scholar]
- 5.Belete HA, Hubmayr RD, Wang S, Singh RD. The role of purinergic signaling on deformation induced injury and repair responses of alveolar epithelial cells. PLoS One 6: e27469, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bilek AM, Dee KC, Gaver DP., 3rd Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. J Appl Physiol 94: 770–783, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Charras GT, Coughlin M, Mitchison TJ, Mahadevan L. Life and times of a cellular bleb. Biophys J 94: 1836–1853, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costello ML, Mathieu-Costello O, West JB. Stress failure of alveolar epithelial cells studied by scanning electron microscopy. Am Rev Respir Dis 145: 1446–1455, 1992 [DOI] [PubMed] [Google Scholar]
- 9.DiPaolo BC, Margulies SS. Rho kinase signaling pathways during stretch in primary alveolar epithelia. Am J Physiol Lung Cell Mol Physiol 302: L992–L1002, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dreyfuss D, Basset G, Soler P, Saumon G. Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am Rev Respir Dis 132: 880–884, 1985 [DOI] [PubMed] [Google Scholar]
- 11.Dreyfuss D, Saumon G. Role of tidal volume, FRC, and end-inspiratory volume in the development of pulmonary edema following mechanical ventilation. Am Rev Respir Dis 148: 1194–1203, 1993 [DOI] [PubMed] [Google Scholar]
- 12.Dreyfuss D, Saumon G. Ventilator-induced lung injury: lessons from experimental studies. Am J Respir Crit Care Med 157: 294–323, 1998 [DOI] [PubMed] [Google Scholar]
- 13.Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema. Respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 137: 1159–1164, 1988 [DOI] [PubMed] [Google Scholar]
- 14.Egan EA. Lung inflation, lung solute permeability, and alveolar edema. J Appl Physiol 53: 121–125, 1982 [DOI] [PubMed] [Google Scholar]
- 15.Egan EA, Nelson RM, Olver RE. Lung inflation and alveolar permeability to non-electrolytes in the adult sheep in vivo. J Pphysiol 260: 409–424, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu Z, Costello ML, Tsukimoto K, Prediletto R, Elliott AR, Mathieu-Costello O, West JB. High lung volume increases stress failure in pulmonary capillaries. J Appl Physiol 73: 123–133, 1992 [DOI] [PubMed] [Google Scholar]
- 17.Gajic O, Lee J, Doerr CH, Berrios JC, Myers JL, Hubmayr RD. Ventilator-induced cell wounding and repair in the intact lung. Am J Respir Crit Care Med 167: 1057–1063, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Gattinoni L, Pesenti A. The concept of “baby lung.” Intensive Care Med 31: 776–784, 2005 [DOI] [PubMed] [Google Scholar]
- 19.Gaver DP, 3rd, Kute SM. A theoretical model study of the influence of fluid stresses on a cell adhering to a microchannel wall. Biophys J 75: 721–733, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gil J, Bachofen H, Gehr P, Weibel ER. Alveolar volume-surface area relation in air- and saline-filled lungs fixed by vascular perfusion. J Appl Physiol 47: 990–1001, 1979 [DOI] [PubMed] [Google Scholar]
- 21.Godin LM, Vergen J, Prakash YS, Pagano RE, Hubmayr RD. Spatiotemporal dynamics of actin remodeling and endomembrane trafficking in alveolar epithelial type I cell wound healing. Am J Physiol Lung Cell Mol Physiol 300: L615–L623, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenfield LJ, Ebert PA, Benson DW. Atelectasis and surface tension properties of lung extracts following positive pressure ventilation and overinflation. Surgical Forum 14: 239–240, 1963 [PubMed] [Google Scholar]
- 23.Grembowicz KP, Sprague D, McNeil PL. Temporary disruption of the plasma membrane is required for c-fos expression in response to mechanical stress. Mol Biol Cell 10: 1247–1257, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hubmayr RD. Perspective on lung injury and recruitment: a skeptical look at the opening and collapse story. Am J Respir Crit Care Med 165: 1647–1653, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Huh D, Fujioka H, Tung YC, Futai N, Paine R, 3rd, Grotberg JB, Takayama S. Acoustically detectable cellular-level lung injury induced by fluid mechanical stresses in microfluidic airway systems. Proc Natl Acad Sci USA 104: 18886–18891, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.John E, McDevitt M, Wilborn W, Cassady G. Ultrastructure of the lung after ventilation. Br J Exp Pathol 63: 401–407, 1982 [PMC free article] [PubMed] [Google Scholar]
- 27.Kacmarek RM, Wiedemann HP, Lavin PT, Wedel MK, Tutuncu AS, Slutsky AS. Partial liquid ventilation in adult patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 173: 882–889, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Krishnan R, Klumpers DD, Park CY, Rajendran K, Trepat X, van Bezu J, van Hinsbergh VW, Carman CV, Brain JD, Fredberg JJ, Butler JP, van Nieuw Amerongen GP. Substrate stiffening promotes endothelial monolayer disruption through enhanced physical forces. Am J Physiol Cell Physiol 300: C146–C154, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malo J, Ali J, Wood LD. How does positive end-expiratory pressure reduce intrapulmonary shunt in canine pulmonary edema? J Appl Physiol 57: 1002–1010, 1984 [DOI] [PubMed] [Google Scholar]
- 30.Mascheroni D, Kolobow T, Fumagalli R, Moretti MP, Chen V, Buckhold D. Acute respiratory failure following pharmacologically induced hyperventilation: an experimental animal study. Intensive Care Med 15: 8–14, 1988 [DOI] [PubMed] [Google Scholar]
- 31.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS, Kuebler WM. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol 44: 725–738, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McGee KP, Mariappan YK, Hubmayr RD, Carter RE, Bao Z, Levin DL, Manduca A, Ehman RL. Magnetic resonance assessment of parenchymal elasticity in normal and edematous, ventilator-injured lung. J Appl Physiol 113: 666–676, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol 28: 596–608, 1970 [DOI] [PubMed] [Google Scholar]
- 34.Muscedere JG, Mullen JB, Gan K, Slutsky AS. Tidal ventilation at low airway pressures can augment lung injury. Am J Respir Crit Care Med 149: 1327–1334, 1994 [DOI] [PubMed] [Google Scholar]
- 35.Neal CR, Michel CC. Openings in frog microvascular endothelium induced by high intravascular pressures. J Physiol 492: 39–52, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oeckler RA, Lee WY, Park MG, Kofler O, Rasmussen DL, Lee HB, Belete H, Walters BJ, Stroetz RW, Hubmayr RD. Determinants of plasma membrane wounding by deforming stress. Am J Physiol Lung Cell Mol Physiol 299: L826–L833, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pecchiari M, Monaco A, Koutsoukou A, D'Angelo E. Plasma membrane disruptions with different modes of injurious mechanical ventilation in normal rat lungs. Crit Care Med 40: 869–875, 2012 [DOI] [PubMed] [Google Scholar]
- 38.Plataki M, Hubmayr RD. The physical basis of ventilator-induced lung injury. Expert Rev Respir Med 4: 373–385, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plataki M, Lee YD, Rasmussen DL, Hubmayr RD. Poloxamer 188 facilitates the repair of alveolus resident cells in ventilator-injured lungs. Am J Respir Crit Care Med 184: 939–947, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roan E, Wilhelm K, Bada A, Makena PS, Gorantla VK, Sinclair SE, Waters CM. Hyperoxia alters the mechanical properties of alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 302: L1235–L1241, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stroetz RW, Vlahakis NE, Walters BJ, Schroeder MA, Hubmayr RD. Validation of a new live cell strain system: characterization of plasma membrane stress failure. J Appl Physiol 90: 2361–2370, 2001 [DOI] [PubMed] [Google Scholar]
- 42.Talmor D, Sarge T, Malhotra A, O'Donnell CR, Ritz R, Lisbon A, Novack V, Loring SH. Mechanical ventilation guided by esophageal pressure in acute lung injury. N Engl J Med 359: 2095–2104, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terragni PP, Rosboch G, Tealdi A, Corno E, Menaldo E, Davini O, Gandini G, Herrmann P, Mascia L, Quintel M, Slutsky AS, Gattinoni L, Ranieri VM. Tidal hyperinflation during low tidal volume ventilation in acute respiratory distress syndrome. Am J Respir Crit Care Med 175: 160–166, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Tremblay LN, Slutsky AS. Ventilator-induced injury: from barotrauma to biotrauma. Proc Assoc Am Physicians 110: 482–488, 1998 [PubMed] [Google Scholar]
- 45.Tschumperlin DJ, Margulies SS. Alveolar epithelial surface area-volume relationship in isolated rat lungs. J Appl Physiol 86: 2026–2033, 1999 [DOI] [PubMed] [Google Scholar]
- 46.Tschumperlin DJ, Margulies SS. Equibiaxial deformation-induced injury of alveolar epithelial cells in vitro. Am J Physiol Lung Cell Mol Physiol 275: L1173–L1183, 1998 [DOI] [PubMed] [Google Scholar]
- 47.Veldhuizen RA, Welk B, Harbottle R, Hearn S, Nag K, Petersen N, Possmayer F. Mechanical ventilation of isolated rat lungs changes the structure and biophysical properties of surfactant. J Appl Physiol 92: 1169–1175, 2002 [DOI] [PubMed] [Google Scholar]
- 48.Vlahakis NE, Hubmayr RD. Cellular stress failure in ventilator-injured lungs. Am J Respir Crit Care Med 171: 1328–1342, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vlahakis NE, Schroeder MA, Pagano RE, Hubmayr RD. Role of deformation-induced lipid trafficking in the prevention of plasma membrane stress failure. Am J Respir Crit Care Med 166: 1282–1289, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Wang S, Singh RD, Godin L, Pagano RE, Hubmayr RD. Endocytic response of type I alveolar epithelial cells to hypertonic stress. Am J Physiol Lung Cell Mol Physiol 300: L560–L568, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures. Protection by positive end-expiratory pressure. Am Rev Respir Dis 110: 556–565, 1974 [DOI] [PubMed] [Google Scholar]
- 52.West JB, Dollery CT, Naimark A. Distribution of blood flow in isolated lung; relation to vascular and alveolar pressures. J Appl Physiol 19: 713–724, 1964 [DOI] [PubMed] [Google Scholar]
- 53.West JB, Mathieu-Costello O. Structure, strength, failure, and remodeling of the pulmonary blood-gas barrier. Ann Rev Physiol 61: 543–572, 1999 [DOI] [PubMed] [Google Scholar]
- 54.Wray C, Mao Y, Pan J, Chandrasena A, Piasta F, Frank JA. Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am J Physiol Lung Cell Mol Physiol 297: L219–L227, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]