

Abstract
In addition to the left bundle branch block type of electrical activation, there are further remodeling aspects associated with dyssynchronous heart failure (HF) that affect the electromechanical behavior of the heart. Among the most important are altered ventricular structure (both geometry and fiber/sheet orientation), abnormal Ca2+ handling, slowed conduction, and reduced wall stiffness. In dyssynchronous HF, the electromechanical delay (EMD), the time interval between local myocyte depolarization and myofiber shortening onset, is prolonged. However, the contributions of the four major HF remodeling aspects in extending EMD in the dyssynchronous failing heart remain unknown. The goal of this study was to determine the individual and combined contributions of HF-induced remodeling aspects to EMD prolongation. We used MRI-based models of dyssynchronous nonfailing and HF canine electromechanics and constructed additional models in which varying combinations of the four remodeling aspects were represented. A left bundle branch block electrical activation sequence was simulated in all models. The simulation results revealed that deranged Ca2+ handling is the primary culprit in extending EMD in dyssynchronous HF, with the other aspects of remodeling contributing insignificantly. Mechanistically, we found that abnormal Ca2+ handling in dyssynchronous HF slows myofiber shortening velocity at the early-activated septum and depresses both myofiber shortening and stretch rate at the late-activated lateral wall. These changes in myofiber dynamics delay the onset of myofiber shortening, thus giving rise to prolonged EMD in dyssynchronous HF.
Keywords: cardiac electromechanics, electromechanical modeling, dyssynchronous heart failure
heart failure (HF) is a major cause of morbidity and mortality, contributing significantly to healthcare expenditures. A subset of HF patients exhibit contractile dyssynchrony due to electrical intraventricular delay of the left bundle branch block (LBBB) type. In addition to this altered electrical activation pattern, other detrimental remodeling aspects of HF also contribute to electromechanical dysfunction of the heart and to the dyssynchrony between its electrical and mechanical behavior. Among these are remodeled ventricular structure and fiber/sheet architecture (14), slowed electrical conduction (2), deranged Ca2+ handling (40), and reduced stiffness of the failing myocardium (18).
The electromechanical delay (EMD), the time interval between the local myocyte depolarization and the onset of myofiber shortening, and its three-dimensional (3-D) distribution are manifestations of the intricate relationship between the electrical and mechanical activity of the heart. Computational (12) and experimental (3, 27, 35) studies have previously shown that the EMD distribution in the nonfailing heart is nonuniform and is dependent on the electrical activation sequence. Recent experimental evidence has demonstrated that in the nonfailing canine heart, the EMD associated with LBBB electrical activation sequence is longer at the late-activated left ventricular (LV) lateral wall compared with that at the early-activated septum (34). Using electromechanical models of nonfailing and HF canine hearts, we demonstrated, in a previous study (9), that EMD is increased in HF, particularly at the late-activated LV lateral wall during LBBB. Furthermore, we (9) showed that pacing at the region with the longest EMD can maximize the hemodynamic benefit of cardiac resynchronization therapy (CRT), underscoring the importance of understanding the distribution of EMD in dyssynchronous HF. However, it remains unknown how each of the HF remodeling aspects contributes to the prolongation of EMD under the conditions of LBBB. Understanding the specific mechanistic contribution of the different electromechanical remodeling aspects in dyssynchronous HF has important implications for the advancement of HF therapies aiming to better recoordinate the contraction of the heart or increase cardiac contractility. Revealing the mechanisms responsible for HF-induced prolongation in EMD may help guide new strategies that aim to minimize EMD in dyssynchronous HF.
Systematically dissecting out the role of each remodeling component in determining the EMD in dyssynchronous HF cannot be currently achieved by experimentation. Computational modeling of cardiac electromechanics affords an opportunity to conduct such a mechanistic investigation (30, 38) and to reveal the role of individual HF remodeling aspects in prolonging EMD in dyssynchronous HF. In this study, we used MRI-based electromechanical models of dyssynchronous canine nonfailing and HF hearts to determine the individual and combined contributions of the major HF remodeling aspects, i.e., altered ventricular structure, slowed conduction, deranged Ca2+ handling, and decreased stiffness, in extending EMD.
METHODS
MRI-based models of canine electromechanics.
To achieve our goal, we used MRI-based electromechanical models of dyssynchronous (i.e., with LBBB) nonfailing and HF canine hearts. Using the methodology described by Gurev et al. (13), nonfailing and failing canine ventricular geometries were generated from ex vivo MRI scans of nonfailing and HF canine ventricles, and the fiber and sheet architecture were constructed from ex vivo diffusion tensor (DT) MRI data of the same nonfailing and HF canine ventricles (Fig. 1). The original MR and DT MR data are available at http://gforge.icm.jhu.edu/gf/project/dtmri_data_sets/. In HF, the ventricles become dilated and the walls are thinner; moreover, the transmural fiber gradient is increased, and the laminar sheets are oriented more vertically (14). These changes to the ventricular structure are directly incorporated into the MRI-based dyssynchronous HF canine ventricular model since individual heart MR and DT MR data were used in model reconstruction.
Fig. 1.
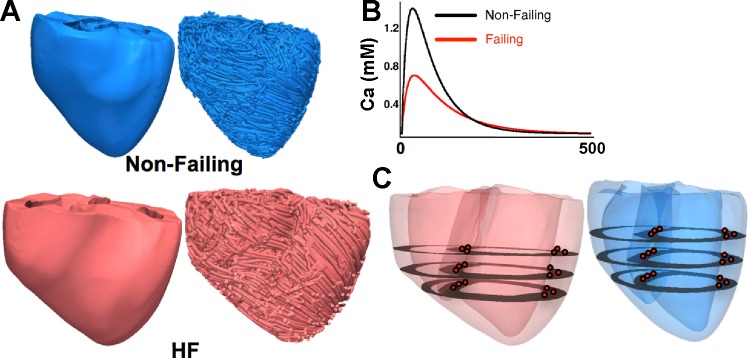
A: MRI-based electromechanical models of the canine heart. Anterior views of ventricular geometry (left) and fiber orientation (right) for the nonfailing (top) and heart failure (HF; bottom) canine heart are shown. B: Ca2+ transients for the nonfailing (black) and failing (red) myocyte as used in the mechanics component of the model (see text for details). C: locations (red dots) at which EMD and strain were calculated at the septum and lateral wall.
Briefly, the electromechanical model consisted of an electrical component and a mechanical component, which were coupled via intracellular Ca2+ cycling. The mathematical description of the electrical component of the model was governed by the monodomain representation of cardiac tissue. The Luo-Rudy dynamic model (23) was used to represent membrane dynamics. Since the electrical model was used only to calculate activation times (see detailed description below), which served as inputs (local triggers) in the mechanics model, we used a generic mammalian membrane kinetics model to avoid an unnecessary computational load.
The ventricular mechanics component was based on continuum mechanics equations, with the myocardium assumed to be an orthotropic, hyperelastic nearly incompressible material. Myofilament dynamics were based on the model of Rice et al. (32), which was parameterized for the canine heart by matching data obtained from electromechanical wave imaging, as previously described (29). Finally, the electromechanical model was coupled to a model of the systemic and pulmonic circulatory systems (20).
To minimize the electromechanical model computational load, we implemented a weak coupling scheme between electrical and mechanical components, as specified above; the local electrical activation times calculated from the electrical component determined the local time instants when the Ca2+ transient was initiated at the Gaussian points of each mechanical mesh element. In other words, the Ca2+ transient calculation was executed under the mechanics part of the model following activation time inputs from the electrical model. Calculation of the Ca2+ transient in the mechanics model used the species-specific single cell canine ionic model by Fox et al. (11). This Ca2+ transient captures changes to membrane dynamics at fast pacing rates and was thus appropriate for this study. The Ca2+ transient from the Fox et al. model was used to fit the two algebraic equations from the Rice et al. myofilament model (32), which ultimately described the Ca2+ transient in our mechanics model.
This weak electromechanical coupling scheme has significant advantages over previous weak coupling schemes (21) because it allowed us to model the Ca2+ transient at fast pacing rates and, more importantly, to directly incorporate the changes to the Ca2+ transient to reflect Ca2+ dysresgulation.
Previous studies (3, 7) have suggested that transmural heterogeneities in Ca2+ handling and mechanics do not play a significant role in altering myofiber dynamics and thus in determining the distribution of the EMD. Moreover, no differences in the Ca2+ transient and myofiber dynamics across the wall of the canine dyssynchronous failing heart have been reported (8). Therefore, transmural heterogeneity in Ca2+ handling and myofiber mechanics were not incorporated into the model.
The mechanical mesh was generated using methodology by Gurev et al. (13), and the computational mesh for the electrical component was constructed using techniques developed by Prassl et al. (26). Our framework for electromechanical modeling of the ventricles presented here has been used in a range of applications (9, 12, 17, 19, 29). Further details of our electromechanical model and the numeric approaches can be found in a previous publication (13).
Incorporating HF remodeling.
We incorporated HF remodeling of the electromechanical tissue properties in the MRI-based geometric model of failing canine ventricles, as previously described (9). Specifically, to model slowed conduction in HF (2), the longitudinal and transverse electrical conductivities in failing ventricles were reduced by 20% from the values used in nonfailing ventricles, with the latter values based on data from Ref. 33. The reduced stiffness of myocardial tissue from failing hearts (18) was incorporated into the model by decreasing the passive stress scaling constant C in the strain-energy function (39) by 50%. Finally, the peak amplitude and relaxation rate of the Ca2+ transient was reduced to 50% and increased by 30%, respectively, of the values used in the nonfailing ventricular model, to incorporate the deranged Ca2+ handling associated with HF (24). The Ca2+ transients for the nonfailing and failing models are shown in Fig. 2B. The parameters for all modified values are shown in Table 1.
Fig. 2.
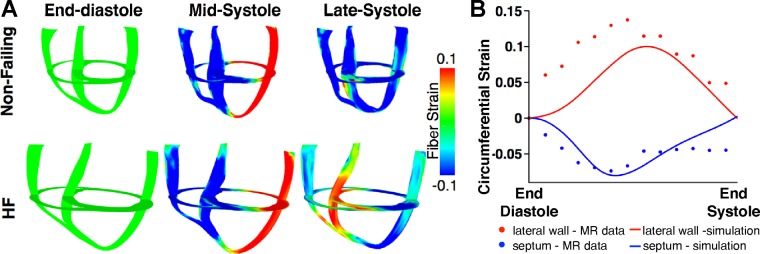
A: short- and long-axis maps of fiber strain at different time instants during the systolic phase of the cardiac cycle in the dyssynchronous nonfailing (top) and dyssynchronous HF (bottom) canine ventricular models. B: comparison of simulated (lines) and experimental (circles) circumferential strain at the midwall of the septum (blue) and lateral wall (red). See text for details.
Table 1.
Parameters of the nonfailing and failing model that were modified for the latter
| Parameter | Nonfailing Model | Failing Model |
|---|---|---|
| Parameters for electrical conductivities | ||
| σi,l, S/m | 0.74 | 0.59 |
| σi,t, S/m | 0.12 | 0.10 |
| σe,l, S/m | 0.25 | 0.20 |
| σe,t, S/m | 0.17 | 0.14 |
| Parameters for the strain energy function | ||
| C, kPA | 0.88 | 0.44 |
| Parameters for the Ca2+ transient | ||
| T1, s | 0.015 | 0.015 |
| T2, s | 0.065 | 0.097 |
| Caamp, mM | 1.4 | 0.7 |
Following the approach of Kerckhoffs et al. (21), 14 additional models with different combinations of HF remodeling aspects were constructed to dissect out the distinct role of each HF remodeling aspect as well as their combined contributions in determining the prolongation of EMD in the dyssynchronous HF canine heart. Using the dyssynchronous nonfailing model as a baseline (LBBB with normal conduction, normal Ca2+ handling, normal stiffness, and normal ventricular structure), we created four models in which only one remodeling attribute was included, six models in which a combination of two HF abnormalities was incorporated, and another four models in which a combination of three HF remodeling aspects were represented. In total, 16 simulation conditions were constructed: 1 dyssynchronous nonfailing model, 1 dyssynchronous HF model, and 14 models of dyssynchrony with varying combinations of the HF remodeling aspects. The dyssynchronous nonfailing and full dyssynchronous HF electromechanical models were first validated with experimental data (see results) before the partial HF remodeling ventricular models were assembled.
Simulating LBBB.
To represent LBBB in the nonfailing and full HF models, as well as the 14 models with various combinations of HF remodeling, the right ventricular endocardial surface was stimulated at discrete locations as if the electrical activity was emanating from the activation of the corresponding branch of the Purkinje network, as previously described (9). The locations and timings were based on experimental data (10, 31, 36). The pacing basic cycle length was 500 ms.
Data analysis.
Fiber strain was calculated throughout the ventricles, and the end-diastolic state was used as the reference state for strain calculations. As previously described (12), we determined local EMD in the following manner: the time of electrical activation was computed as the instant at which the transmembrane voltage became >0 mV, and the onset of myofiber shortening was defined as the time instant at which the myofiber shortened to 10% of its maximal value (4, 35). EMD was calculated as the time interval between the local electrical activation time and the local onset of myofiber shortening, as done in previous experiments (27, 34, 35), and 3-D distributions of EMD in dyssynchronous nonfailing and HF ventricles were determined. The onset of myofiber shortening, rather than the onset of active tension generation, was used to compute EMD to allow for comparisons between the simulation and experiment.
For all simulations, average EMDs at the septum and lateral wall were determined by calculating EMD at nine locations at the septum and lateral wall at the equatorial level (Fig. 1C), as previously described by Russel et al. (34). This allowed for comparisons between our simulation results and experimental data.
RESULTS
Model validation.
The MRI-based dyssynchronous nonfailing and HF canine electromechanical models were first validated by comparing simulation results with experimental measurements of global electrical and mechanical function. The simulation results, in terms of variables of global electrical and mechanical function, were in very good agreement with experimental results (15, 16, 22, 37) (Table 2). The mechanics components of the dyssynchronous nonfailing and HF canine models were further validated by analyzing the 3-D deformation patterns. Figure 2A shows 3-D distribution maps of fiber strain for the nonfailing and HF ventricular models at end diastole, midsystole, and late systole during LBBB. At midsystole, the septal wall contracted, whereas the LV lateral wall was prestretched, for both nonfailing and HF ventricles. The lateral wall shortened at end systole in both models. These 3-D deformation patterns during LBBB are consistent with those obtained experimentally (6, 22, 28). Finally, local circumferential strains from the simulations were compared (Fig. 2B) with MR tagging experimental data obtained by our team and used in a previously published study (15); a description of the methods can be found in the latter article. Figure 2B shows the excellent correspondence between experimental and simulation data.
Table 2.
Comparison of global metrics of electrical and mechanical function between the simulation and experiment
| Dyssynchronous Nonfailing Model |
Dyssynchronous Failing Model |
|||
|---|---|---|---|---|
| Characteristic | Simulation | Experiment | Simulation | Experiment |
| Total activation time, ms | 118 | 114.8 ± 5.6* (15) | 165 | 150.2 ± 9.8* (15) |
| Ejection fraction, % | 37 | 35.1 ± 2.7 (16) | 11 | 23 ± 12.7 (22) and 15 ± 2 (37) |
| Peak LV pressure, mmHg | 93 | 103 ± 10 (16) | 78 | 86.6 ± 7.7 (22) |
| Maximal dP/dt, mmHg/s | 2,732 | 2,487 ± 398 (16) | 1,048 | 1,048 ± 242 (22) |
LV, left ventricular
QRS duration. Numbers in parentheses are citations which the values came from.
3-D EMD distributions in nonfailing and HF ventricles.
Figure 3 shows 3-D maps of electrical activation (left) and the onset of myofiber shortening (middle) in both short- and long-axis views during LBBB for nonfailing and HF ventricles. In both nonfailing and HF dyssynchronous ventricles, depolarization began at the right ventricular septal and free wall and propagated toward the LV lateral wall. Mechanical activation followed the direction of depolarization; however, the propagation of mechanical activity was slower in the HF model. The total mechanical activation times during LBBB in nonfailing and HF models were 139 and 210 ms, respectively.
Fig. 3.

Electrical activation times (left), mechanical activation times (middle), and electromechanical delay (EMD; right) maps in the dyssynchronous nonfailing (top) and dyssynchronous HF (bottom) canine ventricular models. The short- and long-axes views are the same as those shown in Fig. 2.
Table 3.
EMD at the early-activated septal wall and late-activated LV lateral wall for all 16 simulation conditions
| Model | EMD at the Early-Activated Septum | EMD at the Late-Activated LV Lateral Wall |
|---|---|---|
| Nonfailing | 14.7 ± 3.3 | 24.1 ± 0.7 |
| CalciumHF, StiffnessN, ConductionN, StructureN | 25.1 ± 5.4* | 37.6 ± 0.9* |
| CalciumN, StiffnessHF, ConductionN, StructureN | 15.5 ± 1.6 | 25.4 ± 0.7* |
| CalciumN, StiffnessN, ConductionHF, StructureN | 14.8 ± 2.3 | 23.7 ± 0.3* |
| CalciumN, StiffnessN, ConductionN, StructureHF | 14.5 ± 2.3 | 26.0 ± 0.9* |
| CalciumHF, StiffnessHF, ConductionN, StructureN | 25.9 ± 3.7* | 40.0 ± 1.4* |
| CalciumHF, StiffnessN, ConductionHF, StructureN | 26.5 ± 4.4* | 36.8 ± 1.0* |
| CalciumHF, StiffnessN, ConductionN, StructureHF | 27.2 ± 2.3* | 39.7 ± 1.9* |
| CalciumN, StiffnessHF, ConductionHF, StructureN | 14.9 ± 2.0 | 25.1 ± 0.7* |
| CalciumN, StiffnessHF, ConductionN, StructureHF | 14.7 ± 2.1 | 27.3 ± 3.4* |
| CalciumN, StiffnessN, ConductionHF, StructureHF | 13.4 ± 2.7 | 25.7 ± 1.0* |
| CalciumHF, StiffnessHF, ConductionHF, StructureN | 25.9 ± 4.0* | 39.2 ± 1.6* |
| CalciumHF, StiffnessHF, ConductionN, StructureHF | 26.7 ± 2.0* | 42.1 ± 2.7* |
| CalciumHF, StiffnessN, ConductionHF, StructureHF | 26.5 ± 2.6* | 39.0 ± 2.1* |
| CalciumN, StiffnessHF, ConductionHF, StructureHF | 13.7 ± 2.5 | 27.0 ± 1.2* |
| HF | 26.0 ± 2.2* | 41.5 ± 2.7* |
EMD, electromechanical delay; CalciumN, normal Ca2+ handling included in the ventricular model; StiffnessN, normal stiffness included in the model; ConductionN, normal conduction included in the model; StructureN, normal ventricular structure used; CalciumHF, deranged Ca2+ handling included in the model; StiffnessHF, reduced stiffness included in the model; ConductionHF, slowed conduction included in the model; StructureHF, altered ventricular structure used; HF, full heart failure
P < 0.05 vs. nonfailing.
The 3-D distributions of EMD in nonfailing and HF dyssynchronous ventricles along the same short and long axes are shown in Fig. 3, right. In the nonfailing heart, EMD at the late-activated lateral wall was 64% longer than that at the early-activated septum (Table 2). This value is consistent with experimentally obtained EMD values in the canine heart (34). In dyssynchronous HF, EMD at the lateral wall was 88% greater compared with that at the septum (Table 2). The remodeling associated with dyssynchronous HF resulted in an overall increase in EMD compared with that in nonfailing ventricles (average EMD: 19.5 ± 5.0 vs. 33.28 ± 8.5, P < 0.05). Specifically, compared with nonfailing ventricles, local EMD in the dyssynchronous HF ventricles was 11.3 and 17.4 ms longer at the early-activated septum and late-activated LV lateral wall, respectively.
Determining the respective role of each HF remodeling component in extending EMD in dyssynchronous failing ventricles.
To understand how each HF remodeling aspect contributes to the prolongation of EMD, we computed EMD in 14 additional models in which different combinations of HF remodeling aspects were included. The simulation results revealed that Ca2+ cycling dysfunction is the primary remodeling characteristic responsible for the extension of EMD in HF. Figure 4 shows electrical (left), mechanical (middle), and EMD (right) maps for the model with only deranged Ca2+ handling, with all other electromechnical aspects being normal. Although the electrical activation sequence was identical to that in nonfailing LBBB ventricles, the mechanical activity was altered such that EMD was prolonged throughout the LV (Fig. 4, right). Similar to the full dyssynchronous HF ventricular model, EMD was 10.4 and 13.5 ms longer at the septum and lateral wall, respectively, than those values in nonfailing ventricles (Table 2). When the results from all models were compared (Table 2), EMD was significantly greater at the early-activated septum and late-activated lateral wall in all ventricular models in which deranged Ca2+ handling was incorporated, regardless of whether the other HF remodeling components were present in the model or not.
Fig. 4.

Electrical activation times (left), mechanical activation times (middle), and EMD (right) maps in the ventricular model that incorporated only deranged Ca2+ handling. Abbreviations are defined in Table 3. The short- and long-axes views are the same as those shown in Fig. 2.
In contrast, reduced stiffness, slowed conduction, and altered ventricular structure (geometry and fiber/sheet orientation) did not play a marked role in altering EMD. The electrical and mechanical activation and EMD distribution maps during LBBB for ventricular models where only reduced stiffness (top), only slowed conduction (middle), or only altered ventricular structure (bottom) were included are shown in Fig. 5. In the model with reduced stiffness only, electrical and mechanical activation patterns were almost identical to those in dyssynchronous nonfailing ventricles. Accordingly, EMD at the septum was not different from that in nonfailing ventricles and was extended only by 5.8% at the LV lateral wall (Table 2). Although incorporating slowed conduction increased the total electrical activation time by 18 ms, EMD and its distribution were not dramatically altered during LBBB; EMD was unchanged at the septum and reduced by only 1.7% at the LV lateral wall (Table 2). Finally, EMD was not markedly altered during LBBB in the model with altered ventricular structure. Since the ventricles were larger in this case, the total electrical activation time was longer than that in nonfailing ventricles (143 ms). However, the distribution of EMD was similar to that in the nonfailing case. EMD was not changed at the septum and was extended marginally at the LV lateral wall (Table 2). In all models with any combination of these three HF remodeling aspects (slowed conduction, reduced stiffness, and altered ventricular structure), EMD was not altered at the septum and was only slightly increased at the LV lateral wall. Moreover, when slowed conduction, reduced stiffness, altered ventricular structure, or any combination of these three aspects of HF remodeling were added to the model with Ca2+ handling dysregulation, EMD was altered marginally at the septum (<2.1 ms) and lateral wall (<4.5 ms) compared with the model with Ca2+ dysregulation only.
Fig. 5.

Electrical activation times (left), mechanical activation times (middle), and EMD (right) maps in the models that incorporated only reduced stiffness (top), only slowed conduction (middle), and only altered ventricular structure (bottom). Abbreviations are defined in Table 3. The short- and long-axes views are the same as those shown in Fig. 2.
These results further demonstrate that the deranged Ca2+ handling was the primary culprit in prolonging EMD in the setting of dyssynchronous HF.
Understanding the local myofiber dynamics that lead to prolonged EMD in dyssynchronous HF.
As described above, we demonstrated that changes in electrical conduction did not markedly alter EMD; thus, the prolongation of EMD in HF should be determined by the local myofiber dynamics that precede the onset of myofiber shortening. We then analyzed local myofiber dynamics at the early-activated septum and late-activated LV lateral wall to understand how remodeled Ca2+ handling in dyssynchronous HF alters the myofiber dynamics to give rise to prolonged EMD. We calculated the fiber strain at nine locations in the septum (Fig. 6A, top) and nine locations in the LV lateral wall (Fig. 6A, bottom). These locations corresponded to the points at which EMD was calculated, as shown in Table 2. We characterized local myofiber dynamics by computing the amount of local fiber stretch after electrical activation (Fig. 6A, left), maximal local fiber stretch rate (Fig. 6A, middle), and peak local myofiber shortening velocity (Fig. 6A, right). Representative fiber strain traces from the septum and lateral wall for the dyssynchronous nonfailing and dyssynchronous HF models as well as for the model in which only remodeled Ca2+ is included are shown in Fig. 6B.
Fig. 6.
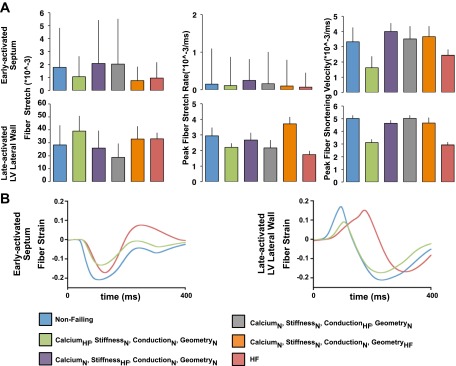
A: stretch (left), maximal stretch rate (middle), and maximal shortening velocity (right) at the early-activated septum (top) and late-activated left ventricular (LV) lateral wall (bottom) for the dyssynchronous nonfailing and dyssynchronous HF model and the models of dyssynchrony with different aspects of HF remodeling. B: representative strain traces at the early-activated septum (left) and late-activated LV lateral wall (right) for the dyssynchronous nonfailing and dyssynchronous HF models and the model of dyssynchrony that included only abnormal Ca2+ handling. Abbreviations are defined in Table 3. *P < 0.05 vs. nonfailing.
Since myofibers at the early-activated septum are not markedly prestretched during LBBB activation, the extended EMD, as observed in the dyssynchronous HF model and in the model with Ca2+ dysregulation, had to be primarily due to changes in myofiber shortening. Indeed, in the full HF model and in the model with altered Ca2+ handling, local myofiber shortening was depressed at the septum compared with the nonfailing case (Fig. 6, A, top right, and B, left). The reduced shortening velocity delays mechanical activation and therefore increases EMD. The myofibers at the septum in models with only reduced stiffness, slowed conduction, or altered ventricular structure did not exhibit depressed shortening at the septum, thus explaining why EMD was not altered at the septum by these HF remodeling aspects.
In contrast to the septum, the lateral wall is stretched during LBBB activation; thus, any changes to stretch dynamics would also affect EMD. Indeed, the local stretch rate and shortening velocity were both smaller at the late-activated LV lateral wall in dyssynchronous HF ventricles compared with nonfailing ventricles; fiber stretch, however, was not different (Fig. 6, A, bottom, and B, right). The reduced stretch rate and shortening velocity impeded the local onset of myofiber shortening and therefore extended EMD in dyssynchronous HF. When we examined local myofiber dynamics at the late-activated LV lateral wall in the models with only one HF remodeling aspect, only the model with deranged Ca2+ handling exhibited both depressed stretch rate and shortening velocity. In the models with only reduced stiffness, only slowed conduction, or only altered ventricular structure, myofibers at the late-activated lateral wall did not exhibit both depressed stretch rate and impaired shortening velocity, as in the HF model and the model with only abnormal Ca2+ handling (Fig. 6A, right and middle), explaining why EMD was not markedly increased by these remodeling attributes. These results demonstrate that the changes in myofiber mechanics, which result in markedly increased EMD in dyssynchronous HF (as shown in Fig. 3 and Table 2), are primarily caused by the remodeling of Ca2+ handling.
DISCUSSION
Dyssynchronous HF is a syndrome that is characterized by impaired pump function, partly due to the intraventricular delay in the electrical activation sequence. The diminished systolic function in dyssynchronous HF also stems from the deleterious remodeling of the dyssynchronous HF ventricles, from the organ down to the molecular level, all of which affect the electromechanical behavior of the heart. Dyssynchronous HF is associated with marked ventricular chamber dilatation and a reduction in ventricular wall thickness (14). Moreover, the laminar sheet angle is altered, and the transmural gradient in fiber angle is amplified. Due to the phosphorylation and lateralization of connexin43 (2), conduction velocity is reduced in HF. Because of changes to the ionic currents and remodeling of the Ca2+ machinery (40), the peak amplitude and relaxation rate of the intracellular Ca2+ transient are reduced. Finally, due to concomitant LV dilatation with no changes in the extracellular matrix (18), the stiffness of the chambers of the failing heart is decreased. Using our image-based electromechanical model, we (9) have demonstrated that during LBBB, the detrimental remodeling in HF increases EMD throughout the ventricular myocardium. In this study, we aimed to evaluate the individual and combined contributions of these remodeling aspects to prolonging EMD in dyssynchronous HF ventricles. Since there exists no current experimental methodology to achieve this goal, we used ex vivo MRI-based models of dyssynchronous nonfailing and HF cardiac canine ventricular electromechanics. The main findings of this study are as follows:
1. During LBBB, the prolongation of EMD in dyssynchronous HF is primarily attributed to remodeled Ca2+ handling.
2. Slowed conduction, reduced stiffness, and altered ventricular structure do not alter EMD at the septum and extend EMD at the LV lateral wall by <4 ms.
3. At the septum, remodeled Ca2+ handling slows myofiber shortening, which, in turn, delays mechanical activation, thus increasing EMD.
4. At the LV lateral wall, the reduced stretch rate and impaired myofiber shortening together result in the prolongation of EMD in dyssynchronous HF, both arising from deranged Ca2+ handling. Other remodeling aspects either alter the stretch rate or shortening velocity, but not both. Thus, they do not play a marked role in extending EMD.
Mechanisms underlying EMD prolongation.
It has been previously shown that the 3-D distribution of EMD is determined by the mechanical interactions between myofibers as well as by the loading conditions of the heart. In a computational model of the rabbit ventricles (12), we demonstrated that late-activated myofibers are prestretched by the shortening of early-activated regions, which, in turn, delays the onset of myofiber shortening and increases EMD at late-activated myofibers. Recently, Russel et al. (34) found that the rate of rise in applied load (i.e., the rate of increasing LV pressure) affects EMD. The authors demonstrated, in an experimental canine model of LBBB, that the onset of myofiber shortening occurs when the rate at which active force is generated by the myofibers is greater than the rate of rise in applied load. Since the rate of increasing LV pressure is larger at late-activated regions than at early-activated regions, it takes a longer time for late-activated regions to generate active tension at a rate faster than the rate of applied load, thus impeding the onset of myofiber shortening and increasing EMD.
In this study, we demonstrate that the changes in the inherent myocardial properties, i.e., abnormal Ca2+ handling, also play a role in extending EMD. The deranged Ca2+ handling in our model results in an altered active tension development, such that the peak active tension is reduced and the time to peak tension is longer, consistent with experimental findings in isolated myocytes (25). Therefore, compared with the nonfailing case, HF myofibers develop tension more slowly than nonfailing myofibers, which impairs myofiber shortening and delays shortening onset. This is illustrated by the myofiber dynamics at the septum. Myocytes at the septum are depolarized early in the electrical activation sequence and are contracting when the rate of increasing LV pressure (i.e., the rate of applied load) is low. Therefore, mechanical interactions between myofibers and loading conditions do not affect myofiber shortening dynamics at the septum, and myofiber shortening is only a function of active tension development. Since the rate of active tension development is slower in dyssynchronous HF compared with the nonfailing case, myofiber shortening velocity is depressed; thus, the onset of myofiber shortening at the early-activated septum is delayed in HF compared with nonfailing ventricles, giving rise to increased EMD. At the late-activated lateral wall, the loading conditions and mechanical interactions also play a role in hindering the onset of myofiber shortening; thus, EMD is further extended there.
MRI-based electromechanical model of dyssynchronous HF.
The canine model of dyssynchronous HF electromechanics presented here was created using a novel methodology that generates ventricular models directly from images (13). Therefore, it inherently incorporates the dilatation and wall thinning as well as the changes in fiber/sheet architecture associated with dyssynchronous HF. Furthermore, we used a biophysically detailed representation of myofilament dynamics, which allowed us to incorporate the effect of Ca2+-handling remodeling on active tension development. Finally, alterations in conduction and stiffness associated with HF are represented, for the first time, in a computational model of dyssynchronous HF canine electromechanics. This MRI-based model of dyssynchronous HF electromechanics was enriched with experimental data that included hemodynamic measurements, ECG metrics, and local mechanical deformation patterns. The model presented here has wide applicability; it can be used in a range of studies that aim to understand the electromechanical activity of dyssynchronous HF ventricles and can serve as a testbed for new therapies and interventions.
Clinical implications.
CRT has been shown to reduce morbidity and mortality, yet ∼30% of dyssynchronous HF patients fail to respond to the therapy (5). Further improvements in CRT efficacy will require innovative strategies that are rooted in novel insights into the electromechanical behavior of dyssynchronous HF ventricles. A new strategy to optimize CRT may be to minimize EMD by positioning the LV pacing lead near the region characterized with the longest EMD (9). Pacing near this region in CRT reverses the prolongation of EMD that is due to the loading conditions. The paced region would be activated earlier, and thus the onset of myofiber shortening would not be delayed, reducing EMD at this region and, in turn, improving electromechanical synchrony. The results of the present study suggest that there may be an additive reduction in EMD via reverse remodeling from any CRT. Aiba et al. (1) have shown that CRT partially reverses the remodeling of the Ca2+-handling machinery induced by dyssynchronous HF. Therefore, the prolongation of EMD that is due to abnormal Ca2+ handling would, to some extent, be eliminated with CRT since Ca2+ handling would be partially restored, further enhancing electromechanical resynchronization.
Limitations.
Trabeculations and papillary muscles are not represented in the model. Incorporating these structures could result in additional shortening in the longitudinal direction, which would alter the onset of myofiber shortening and thus EMD. Furthermore, the model used in this study does not incorporate heterogeneous remodeling of Ca2+ handling. Aiba et al. (11) have demonstrated that at the anterior wall in dyssynchronous failing ventricles, the peak of the Ca2+ transient is larger and the relaxation rate is slower than those at the lateral wall. Inclusion of these properties would result in a stronger contraction of the anterior wall, which, in turn, could result in even longer EMD at the lateral wall. However, these limitations are not expected to alter the mechanisms unraveled here that underlie the relation between deranged Ca2+ handling and prolonged EMD.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute (NHLBI) Grant R01-HL-103428, NHLBI Fellowship F31-HL-103090 (to J. Constantino), and National Science Foundation Grant IOS-1124804 (to N. A. Trayanova).
DISCLOSURES
N. A. Trayanova has partial ownership of CardioSolv LLC. CardioSolv LLC was not involved in this research.
AUTHOR CONTRIBUTIONS
Author contributions: J.C., Y.H., and N.A.T. conception and design of research; J.C. and A.C.L. performed experiments; J.C. analyzed data; J.C. and N.A.T. interpreted results of experiments; J.C. prepared figures; J.C. drafted manuscript; J.C. and N.A.T. edited and revised manuscript; J.C., Y.H., A.C.L., and N.A.T. approved final version of manuscript.
REFERENCES
- 1.Aiba T, Hesketh GG, Barth AS, Liu T, Daya S, Chakir K, Dimaano VL, Abraham TP, O'Rourke B, Akar FG, Kass DA, Tomaselli GF. Electrophysiological consequences of dyssynchronous heart failure and its restoration by resynchronization therapy. Circulation 119: 1220–1230, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in nonischemic dilated cardiomyopathy. Circ Res 95: 717–725, 2004 [DOI] [PubMed] [Google Scholar]
- 3.Ashikaga H, Coppola BA, Hopenfeld B, Leifer ES, McVeigh ER, Omens JH. Transmural dispersion of myofiber mechanics: implications for electrical heterogeneity in vivo. J Am Coll Cardiol 49: 909–916, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ashikaga H, Omens JH, Ingels NB, Jr, Covell JW. Transmural mechanics at left ventricular epicardial pacing site. Am J Physiol Heart Circ Physiol 286: H2401–H2407, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birnie DH, Tang AS. The problem of non-response to cardiac resynchronization therapy. Curr Opin Cardiol 21: 20–26, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Byrne MJ, Helm RH, Daya S, Osman NF, Halperin HR, Berger RD, Kass DA, Lardo AC. Diminished left ventricular dyssynchrony and impact of resynchronization in failing hearts with right versus left bundle branch block. J Am Coll Cardiol 50: 1484–1490, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Campbell SG, Howard E, Aguado-Sierra J, Coppola BA, Omens JH, Mulligan LJ, McCulloch AD, Kerckhoffs RC. Effect of transmurally heterogeneous myocyte excitation-contraction coupling on canine left ventricular electromechanics. Exp Physiol 94: 541–552, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakir K, Daya SK, Aiba T, Tunin RS, Dimaano VL, Abraham TP, Jaques-Robinson KM, Lai EW, Pacak K, Zhu WZ, Xiao RP, Tomaselli GF, Kass DA. Mechanisms of enhanced beta-adrenergic reserve from cardiac resynchronization therapy. Circulation 119: 1231–1240, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Constantino J, Hu Y, Trayanova NA. A computational approach to understanding the cardiac electromechanical activation sequence in the normal and failing heart, with translation to the clinical practice of CRT. Progr Biophys Mol Biol 110: 372–379, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durrer D, van Dam RT, Freud GE, Janse MJ, Meijler FL, Arzbaecher RC. Total excitation of the isolated human heart. Circulation 41: 899–912, 1970 [DOI] [PubMed] [Google Scholar]
- 11.Fox JJ, McHarg JL, Gilmour RF., Jr Ionic mechanism of electrical alternans. Am J Physiol Heart Circ Physiol 282: H516–H530, 2002 [DOI] [PubMed] [Google Scholar]
- 12.Gurev V, Constantino J, Rice JJ, Trayanova NA. Distribution of electromechanical delay in the heart: insights from a three-dimensional electromechanical model. Biophys J 99: 745–754, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurev V, Lee T, Constantino J, Arevalo H, Trayanova NA. Models of cardiac electromechanics based on individual hearts imaging data: image-based electromechanical models of the heart. Biomech Model Mechanobiol 10: 295–306, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Helm PA, Younes L, Beg MF, Ennis DB, Leclercq C, Faris OP, McVeigh E, Kass D, Miller MI, Winslow RL. Evidence of structural remodeling in the dyssynchronous failing heart. Circ Res 98: 125–132, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Helm RH, Byrne M, Helm PA, Daya SK, Osman NF, Tunin R, Halperin HR, Berger RD, Kass DA, Lardo AC. Three-dimensional mapping of optimal left ventricular pacing site for cardiac resynchronization. Circulation 115: 953–961, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Howard EJ, Covell JW, Mulligan LJ, McCulloch AD, Omens JH, Kerckhoffs RC. Improvement in pump function with endocardial biventricular pacing increases with activation time at the left ventricular pacing site in failing canine hearts. Am J Physiol Heart Circ Physiol 301: H1447–H1455, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 17.Hu Y, Gurev V, Constantino J, Bayer JD, Trayanova N. Effects of mechano-electric feedback on scroll wave stability in human ventricular fibrillation. PLos One 8: e60287, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaber WA, Maniu C, Krysiak J, Shapiro BP, Meyer DM, Linke WA, Redfield MM. Titin isoforms, extracellular matrix, and global chamber remodeling in experimental dilated cardiomyopathy: functional implications and mechanistic insight. Circ Heart Fail 1: 192–199, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jie X, Gurev V, Trayanova N. Mechanisms of mechanically induced spontaneous arrhythmias in acute regional ischemia. Circ Res 106: 185–192, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerckhoffs RC, Neal ML, Gu Q, Bassingthwaighte JB, Omens JH, McCulloch AD. Coupling of a 3D finite element model of cardiac ventricular mechanics to lumped systems models of the systemic and pulmonic circulation. Ann Biomed Eng 35: 1–18, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kerckhoffs RC, Omens JH, McCulloch AD, Mulligan LJ. Ventricular dilation and electrical dyssynchrony synergistically increase regional mechanical nonuniformity but not mechanical dyssynchrony: a computational model. Circ Heart Fail 3: 528–536, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leclercq C, Faris O, Tunin R, Johnson J, Kato R, Evans F, Spinelli J, Halperin H, McVeigh E, Kass DA. Systolic improvement and mechanical resynchronization does not require electrical synchrony in the dilated failing heart with left bundle-branch block. Circulation 106: 1760–1763, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Luo CH, Rudy Y. A dynamic model of the cardiac ventricular action potential. I. Simulations of ionic currents and concentration changes. Circ Res 74: 1071–1096, 1994 [DOI] [PubMed] [Google Scholar]
- 24.O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res 84: 562–570, 1999 [DOI] [PubMed] [Google Scholar]
- 25.Perreault CL, Shannon RP, Komamura K, Vatner SF, Morgan JP. Abnormalities in intracellular calcium regulation and contractile function in myocardium from dogs with pacing-induced heart failure. J Clin Invest 89: 932–938, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prassl AJ, Kickinger F, Ahammer H, Grau V, Schneider JE, Hofer E, Vigmond EJ, Trayanova NA, Plank G. Automatically generated, anatomically accurate meshes for cardiac electrophysiology problems. IEEE Trans Biomed Eng 56: 1318–1330, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prinzen FW, Augustijn CH, Allessie MA, Arts T, Delhaas T, Reneman RS. The time sequence of electrical and mechanical activation during spontaneous beating and ectopic stimulation. Eur Heart J 13: 535–543, 1992 [DOI] [PubMed] [Google Scholar]
- 28.Prinzen FW, Hunter WC, Wyman BT, McVeigh ER. Mapping of regional myocardial strain and work during ventricular pacing: experimental study using magnetic resonance imaging tagging. J Am Coll Cardiol 33: 1735–1742, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Provost J, Gurev V, Trayanova N, Konofagou EE. Mapping of cardiac electrical activation with electromechanical wave imaging: an in silico-in vivo reciprocity study. Heart Rhythm 8: 752–759, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quinn TA, Kohl P. Combining wet and dry research: experience with model development for cardiac mechano-electric structure-function studies. Cardiovasc Res 97: 601–611, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramanathan C, Jia P, Ghanem R, Ryu K, Rudy Y. Activation and repolarization of the normal human heart under complete physiological conditions. Proc Natl Acad Sci USA 103: 6309–6314, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rice JJ, Wang F, Bers DM, de Tombe PP. Approximate model of cooperative activation and crossbridge cycling in cardiac muscle using ordinary differential equations. Biophys J 95: 2368–2390, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roberts DE, Scher AM. Effect of tissue anisotropy on extracellular potential fields in canine myocardium in situ. Circ Res 50: 342–351, 1982 [DOI] [PubMed] [Google Scholar]
- 34.Russell K, Smiseth OA, Gjesdal O, Qvigstad E, Norseng PA, Sjaastad I, Opdahl A, Skulstad H, Edvardsen T, Remme EW. Mechanism of prolonged electromechanical delay in late activated myocardium during left bundle branch block. Am J Physiol Heart Circ Physiol 301: H2334–H2343, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Sengupta PP, Khandheria BK, Korinek J, Wang J, Jahangir A, Seward JB, Belohlavek M. Apex-to-base dispersion in regional timing of left ventricular shortening and lengthening. J Am Coll Cardiol 47: 163–172, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Strauss DG, Selvester RH, Wagner GS. Defining left bundle branch block in the era of cardiac resynchronization therapy. Am J Cardiol 107: 927–934, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Strik M, Rademakers LM, van Deursen CJ, van Hunnik A, Kuiper M, Klersy C, Auricchio A, Prinzen FW. Endocardial left ventricular pacing improves cardiac resynchronization therapy in chronic asynchronous infarction and heart failure models. Circ Arrhythm Electrophysiol 5: 191–200, 2012 [DOI] [PubMed] [Google Scholar]
- 38.Trayanova NA. Whole-heart modeling: applications to cardiac electrophysiology and electromechanics. Circ Res 108: 113–128, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Usyk TP, Mazhari R, McCulloch AD. Effect of laminar orthotropic myofiber architecture on regional stress and strain in the canine left ventricle. J Elasticity 61: 143–164, 2000 [Google Scholar]
- 40.Winslow RL, Rice J, Jafri S, Marban E, O'Rourke B. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, II: model studies. Circ Res 84: 571–586, 1999 [DOI] [PubMed] [Google Scholar]