

Abstract
Endothelial dysfunction is prevalent in chronic kidney disease. This study tested the hypothesis that transfusion of rat aortic endothelial cells (ECs) ameliorates endothelial dysfunction in a rat model of chronic kidney disease. Male Sprague-Dawley rats underwent sham surgery or 5/6 nephrectomy (Nx). Five weeks after Nx, EC (1.5 × 106 cells/rat) or vehicle were transfused intravenously. One week later, vascular reactivity of mesenteric artery was assessed on a wire myograph. Sensitivity of endothelium-dependent relaxation to acetylcholine and maximum vasodilation were impaired by Nx and improved by EC transfusion. Using selective pharmacological nitric oxide synthase isoform inhibitors, we demonstrated that the negative effect of Nx on endothelial function and rescue by EC transfusion are, at least in part, endothelial nitric oxide synthase mediated. Plasma asymmetric dimethylarginine was increased by Nx and decreased by EC transfusion, whereas mRNA expression of dimethylarginine dimethylaminohydrolases 1 (DDAH1) was decreased by Nx and restored by EC transfusion. Immunohistochemical staining confirmed that local expression of DDAH1 is decreased by Nx and increased by EC transfusion. In conclusion, EC transfusion attenuates Nx-induced endothelium-dependent vascular dysfunction by regulating DDAH1 expression and enhancing endothelial nitric oxide synthase activity. These results suggest that EC-based therapy could provide a novel therapeutic strategy to improve vascular function in chronic kidney disease.
Keywords: endothelial cell, chronic kidney disease, vascular reactivity, 5/6 nephrectomy, nitric oxide synthase
approximately 14% of the general population of the United States has chronic kidney disease (CKD), and the prevalence of CKD increases with increasing age (35% in those older than 60 yr) (30a). CKD increases risk for cardiovascular morbidity and mortality; e.g., among Medicare enrollees aged 66 and older, acute myocardial infarction (9.1 vs. 2.2%), heart failure (31.8 vs. 7.3%), and peripheral arterial disease (26.3 vs. 9.3%) are more prevalent among CKD than non-CKD persons, and mortality associated with cardiovascular disease is higher in those with CKD (15, 30a). Despite these worrisome statistics and although current guidelines recommend that patients with CKD be considered in the highest-risk group for subsequent cardiovascular events, few interventions have been shown to reduce cardiovascular disease risk in this patient population (24). There is, therefore, an urgent need to develop novel therapeutic strategies to improve cardiovascular health in CKD.
The relationship between CKD and cardiovascular pathology is bi-directional and likely involves alterations in multiple pathways (e.g., traditional cardiovascular risk factors, inflammation, oxidative stress, calcium-phosphorus balance, anemia, and accumulation of uremic toxins) (15). It has been suggested that endothelial dysfunction is the central link that leads to cardiovascular disease in patients with CKD (28, 32). Endothelial dysfunction is a precursor of increased arterial stiffness, atherosclerosis, restenosis after percutaneous coronary intervention, and stenosis of venous bypasses of coronary arteries and of arteriovenous grafts used for dialysis. Studies examining flow-mediated dilation of brachial arteries (35) and vascular reactivity of ex vivo resistance arteries mounted on a wire myograph (26, 29) have demonstrated impaired endothelial function in arteries of patients with CKD.
Endothelial dysfunction in CKD is thought to be related to reduced nitric oxide release secondary to endothelial nitric oxide synthase (eNOS) inhibition by asymmetric dimethylarginine (ADMA) (41). Levels of ADMA, an endogenous low-molecular weight product of protein hydrolysis, are elevated in CKD because of a combination of decreased renal clearance and decreased degradation by dimethylarginine dimethylaminohydrolases (DDAH) (6). An additional contributor to endothelial dysfunction in the setting of CKD is impairment of endothelial repair due to decrease in endothelial progenitor cell count and function (19). We have recently reported the successful use of endothelial cell (EC) transfusion to inhibit inflammation and neointima formation in balloon-injured rat carotid arteries (38). In the present study we tested the hypothesis that systemic transfusion of rat aortic ECs improves endothelial function by increasing eNOS activity in a rat model of CKD.
MATERIALS AND METHODS
Animals and procedures.
Male Sprague-Dawley rats were obtained from Charles River Breeding Laboratories, maintained at constant humidity (60 ± 5%), temperature (24 ± 1°C), and light cycle (6 am to 6 pm), and fed a standard rat pellet diet ad libitum. All protocols were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham and were consistent with the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health.
CKD was induced via renal mass reduction by performing 5/6 nephrectomy (Nx) in two stages. Eleven-week-old rats were anesthetized with 80 mg/kg ketamine and 5 mg/kg xylazine and the upper and lower thirds of the left kidney were surgically resected without renal artery ligation (17). One week later, the right kidney was completely resected. Sham-operated rats served as controls (Sham). Pilot data showed that serum creatinine progressively increases for 3 wk after Nx and then plateaus at approximately twofold baseline values. Five weeks after Nx, rats were transfused intravenously with normal saline (Nx + Veh) versus 1.5 × 106 rat aortic ECs (Nx + EC).
ECs used in the present study were previously characterized (38). Rat aortic ECs and the EC growth medium MCDB-131C were purchased from VEC Technologies (catalog No. MCDB-131C). No other growth factors were added to the medium. Passage 5 to 6 ECs were transduced with adenoviral vector containing green fluorescent protein (GFP) using the AdEasy Adenoviral Vector System (Strategene) and cultured in 100-mm culture dishes until >80% of cells expressed GFP (1.5 × 106 cells per dish). Cells were washed with 0.9% saline, collected with a cell scraper, dispersed by gentle pipetting, and then concentrated with centrifugation at 100 g and resuspended in 1.5 ml normal saline. The ECs used in this study were shown to express eNOS and DDAH by Western blot analysis. Rats assigned to Nx + EC were transfused through a femoral venous catheter with a total of 1.5 × 106 cells/1.5 ml divided into three doses (0. 5 × 106 cells/dose), each separated by 2 h.
Immunohistochemistry.
To determine the presence and location of transfused ECs, fresh frozen optimal cutting temperature (OCT)-embedded sections of mesenteric arteries were analyzed for detection of GFP and von Willebrand factor (vWF) using a VECTASTAIN ABC kit (Vector, Burlingame, CA). Sections were treated according to the manufacturer's instructions using specific antibody against GFP or vWF (Abcam, Cambridge, MA), diluted 1:300 and 1:200, respectively, in 5% normal goat serum/PBS and incubated overnight at 4°C. Sections were then incubated with biotinylated secondary antibody for 30 min followed by incubation with the enzyme conjugate for 30 min, both at room temperature. Immunodetection was determined using a VECTOR NovaRED peroxidase substrate, and the development of reaction product was monitored under a microscope. After color development, sections were washed, counterstained with hematoxylin, and mounted. The chromogen produces a red/brown reaction product at immunopositive sites, whereas cell nuclei stain blue. To quantitate the contribution of transfused ECs versus native ECs to the mesenteric artery endothelium, random sections from mesenteric arteries of seven Nx + EC rats were stained for GFP, and the number of GFP-positive and -negative ECs was counted (total number of counted cells = 999). To evaluate the effect of Nx and EC transfusion on the local expression of DDAH1 in the endothelium of the mesenteric artery, sections were stained using an anti-DDAH1 antibody (Abcam), diluted 1:300 in conjunction with the VECTASTAIN ABC kit with no counterstaining. Three readers blinded to treatment independently graded sections of arteries from Sham, Nx + Veh, and Nx + EC rats with respect to DDAH1 staining of the endothelium. The grades were averaged for all readers and presented on an arbitrary scale.
Fluorescent imaging.
Tissue fresh frozen OCT-embedded sections from mesenteric arteries, liver, lung, spleen, kidney, and heart were examined using a fluorescent microscope imaging system (Nikon TE2000U) with filters set for GFP emission. To detect whether GFP colocalizes with ECs in the mesenteric artery, indirect immunofluorescence staining was carried out, as previously described (12). The sections were blocked with 10% normal goat serum and then incubated with anti-GFP, anti-CD31 (EC marker), or normal mouse/rabbit IgG at 4°C overnight. The slides were incubated with a fluorescein-conjugated anti-mouse secondary antibody (1:100, catalog No. FI-2000; Vector) and a Texas-red-conjugated anti-rabbit secondary antibody (1:100, catalog No. TI-1000; Vector Laboratories) for 1 h at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (50 ng/ml) in PBS for 15 min. Coverslips were washed, mounted with 90% glycerol, and visualized by fluorescence microscopy (×400).
Vascular reactivity.
Seven days after EC transfusion, blood pressure was measured in the carotid artery of conscious rats using a polyethylene cannula. Rats were then anesthetized and terminated by open thoractomy. Blood samples were obtained by cardiac puncture, and the mesentery was collected and placed immediately in ice-cold Krebs-Ringer buffer (KRB) consisting of 118.5 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25.0 NaHCO3, and 5.5 d-glucose. Segments of first-order mesenteric artery were collected by pinning down the mesentery on a 90-mm glass petri dish coated with black Slygard presoaked in cold KRB. Under a dissecting microscope, the mesenteric arteries were gently cleaned of adipose and connective tissue and cut into segments of 2 mm in length. The mesenteric artery segments were mounted in a wire myograph (model 610M, Danish Myotechnology, Aarhus, Denmark) and placed in myograph chamber filled with 5 ml KRB, maintained at 37°C, and continuously aerated with 95% O2-5% CO2 to measure vascular reactivity as previously described (18). With the use of LabChart7 software (ADInstruments, Colorado Springs, CO), each segment was progressively stretched to the diameter at which the largest contractile response to 10 μM norepinephrine could be obtained (referred to as optimal diameter and set to 100% contraction). After a 20-min washout period, a cumulative concentration-response curve (CCRC) to phenylephrine (0.03–30 μM) was obtained. After a second 20-min washout period, the segments were precontracted with phenylephrine (10 μM) until a stable contraction was obtained, followed by a CCRC to the endothelium-dependent muscarinic vasodilator acetylcholine (ACh, 0.001–10 μM). The relaxation responses were recorded in the absence of any pharmacological inhibitors.
To assess the contribution of nitric oxide synthase (NOS) isoforms to endothelial relaxation, mesenteric artery segments were incubated for 30 min with NOS isoform inhibitors, with 30-min washout and resting periods, followed by relaxation responses to ACh (0.001–10 μM) as described above. First, segments were incubated with neuronal NOS (nNOS, Nω-propyl-l-arginine, l-NPA, 2 μM) (40) and inducible NOS {iNOS, N-{[3-(aminomethyl)phenyl]methyl}ethanimidamide dihydrochloride, 1400W, 10 μM} (10) inhibitors. Second, segments were incubated with eNOS [N(5)-(1-iminoethyl)-l-ornithine HCl, l-nitro, l-NIO, 10 μM] (31) and iNOS (1400W) inhibitors. Finally, segments were incubated with the nonselective NOS inhibitor (Nω-nitro-l-arginine methyl ester, l-NAME, 100 μM) (31), the soluble guanylate cyclase inhibitor (1H[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one, ODQ, 10 μM), and the cyclooxygenase inhibitor (indomethacin, 10 μM).
To study the effect of exogenous ADMA on vascular reactivity, ACh-induced relaxation (0.001–10 μM) of mesenteric arteries from unoperated male Sprague-Dawley rats was assessed in the presence of 100 μM ADMA (Sigma-Aldrich, St. Louis, MO) as previously described (20). Endothelium-independent relaxation was then tested with the nitric oxide donor sodium nitroprusside (0.001–10 μM).
ACh, phenylephrine, norepinephrine, l-NPA, l-NIO, 1400W, and l-NAME (Sigma-Aldrich) were dissolved in double distilled water. ODQ (Calbiochem, EMD Millipore, Billerica, MA) was dissolved in DMSO. Indomethacin (Sigma-Aldrich) was dissolved in ethanol. The concentrations of DMSO and ethanol did not exceed 0.1% in the organ bath.
Quantitative real-time polymerase chain reaction.
To test the hypothesis that EC transfusion ameliorates CKD-induced endothelial dysfunction via the DDAH-ADMA-eNOS axis, we measured mesenteric artery DDAH1 and eNOS mRNA expression and serum ADMA levels in Sham, Nx + Veh, and Nx + EC rats.
Mesenteric arteries were cleaned of adipose and connective tissue. RNA was extracted using an Ambion PureLink RNA kit (Life Technologies, Grand Island, NY) and quantitated by Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific). Good quality RNA (1 μg) having a ratio of A260/280 > 1.9 was reverse transcribed to cDNA using an Invitrogen SuperScript III kit (Life Technologies), amplified by polymerase chain reaction using Sybr Green master mix (Applied Biosystems) with specific primers for rat DDAH1 (5′-ACGGTGACCCAGCCAGGCAT-3′ and 5′-ACTTCCGGCCACTGAGGGGG-3′, Invitrogen) and eNOS (5′GCTGCGGGATCAGCAACG-3′ and 5′-GCGGGTCAAAGGACCAGG-3′) and quantified with the iCycler (Applied Biosystems). Levels of specific mRNAs were normalized by use of reference genes ribosomal protein S9 and 18S, as previously described (5, 14).
Serum creatinine and ADMA and plasma advanced oxidative protein products.
Serum creatinine concentration was measured using an liquid chromatography-tandem mass spectrometry (LC-MS/MS) [Waters 2795 separation module (LC)/Micromass Quatro micro API (MS/MS), Analytical Direct, Durham, CT] instrument according to the method described by Young et al. (39). ADMA concentrations were measured using an ELISA kit (MyBioSource, San Diego, CA). Since Nx has been associated with increased oxidative stress (22, 34) and since oxidative stress and eNOS uncoupling are known to induce vascular dysfunction (30), we measured the plasma concentrations of advanced oxidative protein products (AOPPs) that are generated during oxidative stress using the OxiSelect AOPP Assay Kit (Cell Biolab, San Diego, CA).
Data analysis.
Contractile responses are expressed as a percentage of the maximal contractile response to 10 μM norepinephrine, whereas relaxation responses are expressed as a percentage relative to maximal contractile response to 10 μM phenylephrine. Individual CCRCs were fitted to a nonlinear sigmoid regression curve (GraphPad Prism 5.0, GraphPad Software, San Diego, CA), and sensitivity (pEC50) and maximal effect (Emax) were calculated. Results are expressed as means ± SE without transformation. Comparisons among experimental groups were performed with one-way ANOVA, followed by Newman-Keuls multiple comparison test when the overall F-test result of ANOVA was significant. Statistical significance was set at P < 0.05.
RESULTS
Renal mass reduction by Nx in male Sprague-Dawley rats resulted in renal dysfunction as assessed by serum creatinine rise. At 6 wk after surgery, serum creatinine increased almost twofold in both Nx groups but remained unchanged from baseline in the sham-operated group (Fig. 1A, P < 0.05). Mean arterial pressure, measured with an invasive carotid artery catheter in conscious rats, was slightly but significantly increased following Nx (118 ± 3 vs. 108 ± 2 mmHg for Nx + Veh vs. Sham, Fig. 1B, P < 0.05). EC transfusion, at 5 wk after Nx, had no significant effect on serum creatinine or blood pressure measured 1 wk later.
Fig. 1.

5/6 Nephrectomy (Nx) causes a twofold increase in serum creatinine and a mild elevation in systemic blood pressure. Serum creatinine (A) and blood pressure (B) in sham-operated rats (Sham), 5/6 nephrectomized rats (Nx + Veh) and 5/6 nephrectomized rats transfused with endothelial cells (Nx + EC). The numbers above the bars indicate the number of rats analyzed in each group. P < 0.05 by ANOVA for both graphs. *P < 0.05 vs. Sham.
Transfused ECs attach to the endoluminal surface of mesenteric arteries and are integrated into the arterial endothelium.
At 7 days after intravenous transfusion of GFP-labeled ECs or saline, tissues were harvested, embedded fresh frozen in OCT, and sectioned. On fluorescent microscopy, GFP was not detected in sections from the liver, lung, or heart, but small numbers of GFP-positive cells were seen in the spleen and kidney of Nx + EC rats. GFP was detected on the endoluminal surface of mesenteric arteries of Nx + EC rats and colocalized with CD31 (Fig. 2A). No GFP was detected in sections from Sham or Nx + Veh rats.
Fig. 2.

ECs are incorporated in the mesenteric artery endothelium of Nx rats at 7 days after systemic transfusion. A: representative micrograph of cross-section of mesenteric artery from a 5/6 Nx rat transfused with ECs labeled with gren fluoresecnt protein (GFP) 7 days before harvest. Double immunofluorescent staining for GFP (green) and CD31 (red) indicates the presence of transfused cells in the endothelium and demonstrates that the GFP-positive cells are ECs. B: representative GFP immunohistochemically stained micrographs (×1000) of mesenteric artery from 5/6 Nx rats transfused with ECs. GFP-labeled ECs are seen in the endothelium of Nx + EC rats at 7 days after EC transfusion. C: staining for the EC marker, von Willebrand factor (vWF), confirms that these cells are ECs.
Immunostaining for GFP was negative in Sham and Nx + Veh rats. Nx + EC rats showed positive GFP staining in the endothelium of the mesenteric artery (Fig. 2B). vWF staining confirmed that these cells are ECs (Fig. 2C). Sections from large caliber vessels (carotid arteries) showed similar staining (data not shown). Quantitative analysis showed that GFP-positive cells constitute ∼13% of ECs of the mesenteric arteries.
EC transfusion ameliorates CKD-induced endothelial dysfunction.
Vascular function of segments of first order mesenteric arteries from Sham, Nx + Veh, and Nx + EC rats was assessed using wire myography. There was no significant effect of renal mass reduction or EC transfusion on the contractile reactivity to phenylephrine. The maximal contraction (Emax) to 30 μM phenylephrine [111 ± 3, 117 ± 7, and 112 ± 5% for Sham, Nx + Veh, and Nx + EC, respectively; P = not significant (NS)] and the sensitivity (pEC50) to phenylephrine (0.03–30 μM) (5.20 ± 0.13, 5.26 ± 0.21, and 5.23 ± 0.12, for Sham, Nx + Veh, and Nx + EC, respectively; P = NS) were comparable in the three groups (Fig. 3).
Fig. 3.

Nx does not alter vascular contractility of mesenteric arteries. Vascular contractility of mesenteric arteries from Sham, Nx + Veh, and Nx + EC rats. Left: cumulative concentration-response curves to phenylephrine (Phe; 0.03–30 μM) for the 3 groups. pEC50 (top, right) and Emax (bottom, right) are shown. The numbers above the bars indicate the number of mesenteric arteries analyzed in each group. There was no statistical difference between the groups via ANOVA.
Mesenteric arteries from Nx + Veh rats had severe impairment in endothelium-dependent relaxation in response to ACh compared with Sham controls, with a rightward shift of the CCRC curve (Fig. 4). Sensitivity to 0.001–10 μM ACh (pEC50) averaged 7.21 ± 0.10 in Nx + Veh rats and 8.02 ± 0.06 in Sham controls (Fig. 4, top, right, P < 0.05). Furthermore, mesenteric artery maximal relaxation (Emax) to 10 μM ACh was significantly lower in Nx + Veh rats (79 ± 7%) than in Sham controls (96 ± 2%, Fig. 4, bottom, right, P < 0.05). EC transfusion significantly increased ACh-induced endothelium-dependent vasorelaxation. ACh pEC50 in the Nx + EC group averaged 7.56 ± 0.05, significantly higher than 7.21 ± 0.10 in Nx + Veh group (Fig. 4, top, right, P < 0.05). Emax to 10 μM ACh was also significantly increased in Nx + EC (93 ± 3%, P < 0.05, Fig. 4, bottom, right) versus Nx + Veh (79 ± 7%). Importantly, EC transfusion in Nx rats completely normalized Emax to 10 μM ACh, with no significant difference between Nx + EC and Sham control. Sensitivity to ACh (pEC50) in Nx + EC was significantly lower than in Sham control.
Fig. 4.

Nx impairs and EC transfusion rescues vascular relaxation of mesenteric arteries. Vascular relaxation of mesenteric arteries from Sham, Nx + Veh, and Nx + EC rats. Left: cumulative concentration-response curves to acetylcholine (ACh; 0.001–10 μM) for the 3 groups. pEC50 (top, right) and Emax (bottom, right). The numbers above the bars indicate the number of mesenteric arteries analyzed in each group. P < 0.05 by ANOVA for both graphs. *P < 0.05 vs. Sham; #P < 0.05 vs. Nx + Veh.
The effect of EC transfusion on CKD-induced endothelial dysfunction is mediated by eNOS.
To elucidate the mechanism by which EC transfusion ameliorates CKD-induced endothelial dysfunction, we examined the contributions of the three NOS isoforms to endothelium-dependent ACh-induced relaxation (0.001–10 μM) of mesenteric arteries (Fig. 5). In the Sham control group (Fig. 5, A and B), inhibition of nNOS (with 2 μM l-NPA) and iNOS (with 10 μM 1400W) did not significantly change Emax to 10 μM ACh. In contrast, inhibition of eNOS (with 10 μM l-NIO) and iNOS (with 10 μM 1400W) resulted in a significant decrease in Emax compared with 10 μM ACh alone (65 ± 7 vs. 95 ± 2%, P < 0.05, Fig. 5B). Inhibition of nitric oxide-dependent ACh-induced relaxation using the nonselective NOS inhibitor l-NAME (100 μM) and the soluble guanylate cyclase inhibitor ODQ (10 μM) together with prostacyclin inhibition using the cyclooxygenase inhibitor indomethacin (10 μM) further blunted maximal relaxing responses to ACh (41 ± 7%, P < 0.05 vs. ACh alone and vs. l-NIO + 1400W).
Fig. 5.
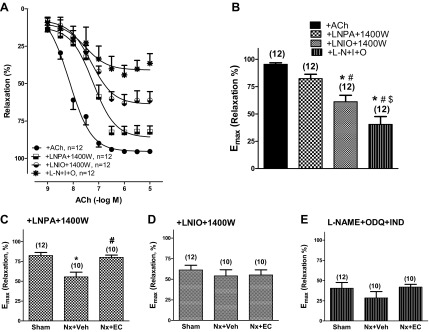
Nx induced vascular dysfunction is mediated by endothelial nitric oxide synthase. Vascular relaxation of mesenteric arteries from Sham, Nx + Veh, and Nx + EC rats in the presence of nitric oxide synthase inhibitors. Cumulative concentration-response curves to ACh (0.001–10 μM; A) and Emax (B) for mesenteric arteries from sham-operated animals that were preincubated with vehicle, Nω-propyl-l-arginine (l-NPA; 2 μM) + N-{[3-(aminomethyl)phenyl]methyl}ethanimidamide dihydrochloride (1400W; 10 μM), N(5)-(1-iminoethyl)-l-ornithine HCl (l-NIO; 10 μM) + 1400W (10 μM), or Nω-nitro-l-arginine methyl ester (l-NAME; 100 μM) + 1H[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one (ODQ; 10 μM) + indomethacin (Ind; 10 μM) for 30 min. P < 0.05 by ANOVA. *P < 0.05 vs. Veh; #P < 0.05 vs. l-NPA + 1400W; $P < 0.05 vs. l-NIO + 1400W. ACh Emax for mesenteric arteries from Sham, Nx + Veh, and Nx + EC rats preincubated with l-NPA + 1400W (C), l-NIO + 1400W (D), and l-NAME + ODQ + Ind (E) for 30 min. The numbers above the bars indicate the number of mesenteric arteries analyzed in each group. P < 0.05 by ANOVA for C. *P < 0.05 vs. Sham; #P < 0.05 vs. Nx + Veh.
In arteries from Nx + Veh animals, the maximal relaxation response to 10 μM ACh was significantly reduced compared with those from Sham control animals in the presence of nNOS and iNOS inhibition (l-NPA + 1400W) (46 ± 5 vs. 82 ± 4%, P < 0.05, Fig. 5C). EC transfusion abrogated the effect of Nx on the maximal relaxation response to 10 μM ACh (Emax, 78 ± 3% for Nx + EC vs. 46 ± 5 for Nx + Veh, P < 0.05, and P = NS vs. Sham, 82 ± 4%). In contrast, when eNOS and iNOS were inhibited (l-NIO + 1400W), Nx and EC transfusion had no effect on Emax (65 ± 7% for Sham, 58 ± 12% for Nx + Veh, and 54 ± 6% for Nx + EC, P = NS, Fig. 5D). Similarly, in the presence of l-NAME + ODQ + indomethecin, Nx, and EC transfusion did not significantly change Emax to 10 μM ACh (Fig. 5E). Thus Nx induced and EC transfusion ameliorated endothelial dysfunction only when eNOS was not inhibited, suggesting that the negative effect of Nx on endothelial function and the rescue by EC transfusion are eNOS mediated.
EC transfusion reverses the effects of CKD on the DDAH-ADMA axis and on oxidative stress.
Nx and EC transfusion did not significantly alter mRNA expression of eNOS in mesenteric arteries (1.00 ± 0.08, 0.98 ± 0.14, and 0.90 ± 0.09 for Sham, Nx + Veh, and Nx + EC, respectively; ANOVA, P = NS). Serum ADMA concentration was significantly increased at 6 wk after Nx (0.40 ± 0.06 vs. 0.25 ± 0.03 μM for Nx + Veh vs. Sham, respectively; Fig. 6A, P < 0.05). EC transfusion in Nx rats decreased serum ADMA significantly to a level not different from Sham control (0.21 ± 0.01 μM, P < 0.05 vs. Nx + Veh, P = NS vs. Sham). Mesenteric artery DDAH1 mRNA expression was significantly decreased by Nx and restored to normal levels by EC transfusion (1.00 ± 0.24, 0.41 ± 0.06, and 1.24 ± 0.15 for Sham, Nx + Veh, and Nx + EC, respectively; P < 0.05 for Nx + Veh vs. Sham and for Nx + EC vs. Nx + Veh and NS for Nx + EC vs. Sham). Immunohistochemical staining for DDAH1 confirmed that local expression of DDAH1 protein is decreased in Nx + Veh versus Sham (Fig. 6B). EC transfusion increased DDAH1 protein expression compared with Nx + Veh, but this level remained lower than that seen in sham-operated rats. To confirm that ADMA impairs endothelial function, we studied the effect of exogenous ADMA administration on ACh-induced mesenteric artery relaxation (Fig. 6C). Endothelium-dependent relaxation was inhibited by pretreatment with 100 μM ADMA, whereas endothelium-independent relaxation remained intact.
Fig. 6.

EC transfusion increases local dimethylarginine dimethylaminohydrolases 1 (DDAH1) expression in vasculature of Nx rats leading to decrease in asymmetric dimethylarginine (ADMA) levels in circulation. Serum ADMA level (A) and local expression of DDAH1 by immunohistochemistry (IHC, protein) (B) in Sham, Nx + Veh, and Nx + EC are shown. C: exogenous ADMA (100 μM) inhibits ACh-induced relaxation (0.001–10 μM) of mesenteric arteries but not endothelium-independent relaxation tested with the nitric oxide donor sodium nitroprusside (SNP; 0.001–10 μM). D: oxidative stress, measured as serum level of advanced oxidative protein products (AOPPs) in rats at 7 days after transfusion of ECs or vehicle indicates that Nx is associated with increased oxidative stress and that EC transfusion lowers AOPP levels. The numbers above the bars indicate the number of mesenteric arteries analyzed in each group. P < 0.05 by ANOVA for all graphs. *P < 0.05 vs. Sham; #P < 0.05 vs. Nx + Veh. AU, arbitrary units.
AOPP levels, a reflection of oxidative stress, were significantly elevated in Nx + Veh versus Sham (333 ± 48 vs. 211 ± 23 μM, P < 0.05, Fig. 6D). In Nx rats, EC transfusion significantly reduced AOPP levels compared with vehicle (231 ± 12 vs. 333 ± 48 μM for Nx + EC vs. Nx + Veh, respectively; P < 0.05). Importantly, EC transfusion normalized AOPP levels, resulting in no significant difference in levels between Nx + EC versus Sham control rats (Fig. 6D, P = NS for Nx + EC vs. Sham).
DISCUSSION
This study showed for the first time that rat aortic ECs transfused intravenously incorporate into the arterial endothelium of Nx rats and improve small vessel endothelial function. The findings reported here support our hypothesis that transfusion of ECs replenishes the damaged endothelium in Nx rats and normalizes elevated systemic ADMA levels, likely by delivering DDAH-1 to the endothelium, releasing inhibition of eNOS, and therefore reversing CKD-induced endothelial dysfunction. This study demonstrates the potential of EC-based therapy as a novel therapeutic strategy to improve vascular function in CKD.
The association between CKD and cardiovascular disease is well established, but the mechanism for this association is not fully understood. Since cardiovascular risk is evident even with mild reductions in renal function (11), the pathophysiological link is presumed to occur at an early stage in the disease process. The endothelium plays a crucial role in maintaining vascular health by regulating vascular tone, hemostasis, inflammatory cell adhesion, and migration into the vessel wall, platelet activity, and angiogenesis (37). Studies have demonstrated endothelial dysfunction in arteries of patients with both early and advanced-stage CKD, but these studies were often confounded by increases in traditional risk factors, which are independently associated with abnormalities in endothelial function (e.g., diabetes mellitus and hypertension) (28). Studies that have demonstrated endothelial dysfunction in animal models of CKD have also been confounded by the development of severe hypertension in the setting of CKD (36). We used a CKD renal mass reduction model that avoids renal infarction by directly resecting renal tissue. Consistent with previous studies that used this model (1), we observed only slight elevations in systemic blood pressure (mean, 10 mmHg), and the blood pressure of Nx animals remained in the normotensive range. In the absence of hypertension, CKD was associated with endothelial dysfunction with severe impairment in ACh-induced endothelium-dependent relaxation. Furthermore, we have demonstrated that CKD-induced endothelial dysfunction is dependent on eNOS (Fig. 5). In sham-operated control rats, pharmacological inhibition of nNOS and iNOS did not inhibit endothelium-dependent maximum relaxation, whereas inhibition of eNOS did. Moreover, EC transfusion rescued the endothelial dysfunction caused by Nx when nNOS and iNOS were inhibited (but eNOS was not) but had no effect on endothelial function when eNOS was inhibited. Interpretation of these data is limited by the incomplete selectivity of the inhibitors of the NOS isoforms. For example, although l-NIO is considered a selective inhibitor of eNOS, it inhibits to a lesser degree both nNOS and iNOS (3). Furthermore, there was a statistically nonsignificant trend toward inhibition of ACh-induced vasodilation with l-NPA, and l-NIO + 1400W did not completely inhibit ACh-induced vasodilation. This suggests that nNOS may also be involved in this process, as recently shown (33, 34).
Patients with CKD have markedly decreased numbers of circulating endothelial progenitor cells compared with healthy volunteers (7, 8, 23). Endothelial progenitor cells contribute to vascular integrity by homing to sites of vascular injury and incorporating into the endothelium (9). In addition to their decreased number, progenitor cells derived from CKD patients have functional impairment, including impaired migratory function in response to vascular endothelial growth factor, decreased adherence to fibronectin-coated diureido-pyrimidinone-polycaprolacton, decreased incorporation into EC vascular structures in culture (Matrigel tube formation assay), reduced endothelial outgrowth potential, and reduced antithrombogenic function (7, 23). A recent study by Lorenzen et al. (25) of 265 patients with end-stage CKD demonstrated that reduction in functionally active endothelial progenitor cells is an independent predictor of incident cardiovascular events over a median follow-up period of 36 mo. Taken together, these studies suggest that CKD is associated with impairment of endogenous capacity to repair the vasculature. When combined with increased endothelial injury in CKD, this results in an imbalance that favors endothelial dysfunction (19). Local delivery of endothelial progenitor cells to balloon-injured arteries with denuded endothelium has been shown to accelerate endothelialization and improve endothelium-dependent vasoreactivity (13, 16). Our current data show that systemically transfused, functionally active ECs integrate into the endothelium of CKD rats (Fig. 2) and improve endothelial function of their mesenteric arteries 7 days after their introduction (Fig. 4). The effect of cell therapy on vascular function in human CKD has not been evaluated. Furthermore, the mode of delivery is a critical factor in clinical cell therapy, and systemic administration, the strategy used in these experiments, is attractive for future clinical applications (38).
ADMA, a natural inhibitor of eNOS, is a metabolic by-product of the hydrolysis of methylated proteins that has been implicated in the pathogenesis of cardiovascular disease in CKD (41). Elevated levels of ADMA in CKD were originally attributed to decreased renal clearance of the protein, but it is now evident that the majority of ADMA is catabolized by DDAH into inactive metabolites. Animal experiments have demonstrated that genetic manipulation of DDAH can modulate circulating ADMA levels, which in turn can alter cardiovascular physiology (6). For example, DDAH overexpression using adenovirus-mediated gene transfer in Nx rats lowered circulating ADMA levels and prevented the elevation of blood pressure seen with Nx (27). Recently, Kajimoto et al. (20) showed that in transgenic mice overexpressing DDAH1, circulating ADMA levels did not increase with Nx and were 50% lower than in wild-type sham-operated mice. Importantly, unlike the aortic rings from wild-type Nx mice, those from transgenic Nx mice did not exhibit impaired endothelium-dependent relaxation, suggesting that an increase in DDAH1 can reverse Nx-induced endothelial dysfunction. Our current data show that the administration of EC to Nx rats replenishes DDAH1 levels in the endothelium. The increased local expression of DDAH1 was associated with systemic decreases in ADMA levels and oxidative stress and improvement in endothelial function despite a stable level of expression of eNOS. It has been shown that ADMA can uncouple eNOS in the vascular endothelium, resulting in endothelial dysfunction due to decreased bioavailability of nitric oxide and increased vascular oxidative stress (2, 21, 30). These observations offer an explanation for the elevated levels of AOPPs and the decreased activity of eNOS in our Nx + Veh rats as well as the restoration of vascular function and decreased AOPPs associated with decreased ADMA levels after the administration of ECs.
In conclusion, the studies described here provide the framework for novel therapeutic interventions that use cell therapy that homes to the endoluminal surface of the vasculature to deliver DDAH1 locally to the endothelium to reverse CKD-induced endothelial dysfunction.
GRANTS
This work was supported, in part, by an National Heart, Lung, and Blood Institute Grant T32-HL-007457 (to M. Pacurari); Veterans Affairs Biomedical Laboratory Research & Development Service Merit Award (to F. G. Hage); American Heart Association NCRP Scientist Development Grant 0930098N (to F. G. Hage); and a Greater Southeast Affiliate Grant 09BGIA2250367 (to D. Xing); and National Institutes of Health 1P30-DK-079337 (University of Alabama at Birmingham-University of California, San Diego O'Brien Core Center).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.P., D.X., R.H.H., and F.G.H. conception and design of research; M.P., D.X., R.H.H., Y.G., Z.Y., and F.G.H. performed experiments; M.P., D.X., R.H.H., Y.G., Z.Y., and F.G.H. analyzed data; M.P., D.X., R.H.H., Y.G., Z.Y., and F.G.H. interpreted results of experiments; M.P., R.H.H., and F.G.H. prepared figures; M.P. drafted manuscript; M.P., D.X., R.H.H., and F.G.H. edited and revised manuscript; M.P., D.X., R.H.H., Y.G., Z.Y., and F.G.H. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of M. Pacurari: College of Science, Engineering, and Technology, Jackson State University, Jackson, MS.
REFERENCES
- 1.Amann K, Rump LC, Simonaviciene A, Oberhauser V, Wessels S, Orth SR, Gross ML, Koch A, Bielenberg GW, Van Kats JP, Ehmke H, Mall G, Ritz E. Effects of low dose sympathetic inhibition on glomerulosclerosis and albuminuria in subtotally nephrectomized rats. J Am Soc Nephrol 11: 1469–1478, 2000 [DOI] [PubMed] [Google Scholar]
- 2.Antoniades C, Shirodaria C, Leeson P, Antonopoulos A, Warrick N, Van-Assche T, Cunnington C, Tousoulis D, Pillai R, Ratnatunga C, Stefanadis C, Channon KM. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J 30: 1142–1150, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Bardell AL, MacLeod KM. Evidence for inducible nitric-oxide synthase expression and activity in vascular smooth muscle of streptozotocin-diabetic rats. J Pharmacol Exp Ther 296: 252–259, 2001 [PubMed] [Google Scholar]
- 5.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55: 611–622, 2009 [DOI] [PubMed] [Google Scholar]
- 6.Caplin B, Leiper J. Endogenous nitric oxide synthase inhibitors in the biology of disease: markers, mediators, and regulators? Arterioscler Thromb Vasc Biol 32: 1343–1353, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi JH, Kim KL, Huh W, Kim B, Byun J, Suh W, Sung J, Jeon ES, Oh HY, Kim DK. Decreased number and impaired angiogenic function of endothelial progenitor cells in patients with chronic renal failure. Arterioscler Thromb Vasc Biol 24: 1246–1252, 2004 [DOI] [PubMed] [Google Scholar]
- 8.de Groot K, Bahlmann FH, Sowa J, Koenig J, Menne J, Haller H, Fliser D. Uremia causes endothelial progenitor cell deficiency. Kidney Int 66: 641–646, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Dzau VJ, Gnecchi M, Pachori AS, Morello F, Melo LG. Therapeutic potential of endothelial progenitor cells in cardiovascular diseases. Hypertension 46: 7–18, 2005 [DOI] [PubMed] [Google Scholar]
- 10.Garvey EP, Oplinger JA, Furfine ES, Kiff RJ, Laszlo F, Whittle BJ, Knowles RG. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J Biol Chem 272: 4959–4963, 1997 [DOI] [PubMed] [Google Scholar]
- 11.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 351: 1296–1305, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Gong K, Xing D, Li P, Hilgers RH, Hage FG, Oparil S, Chen YF. cGMP inhibits TGF-beta signaling by sequestering Smad3 with cytosolic beta2-tubulin in pulmonary artery smooth muscle cells. Mol Endocrinol 25: 1794–1803, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gulati R, Jevremovic D, Peterson TE, Witt TA, Kleppe LS, Mueske CS, Lerman A, Vile RG, Simari RD. Autologous culture-modified mononuclear cells confer vascular protection after arterial injury. Circulation 108: 1520–1526, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Hage FG, Oparil S, Xing D, Chen YF, McCrory MA, Szalai AJ. C-reactive protein-mediated vascular injury requires complement. Arterioscler Thromb Vasc Biol 30: 1189–1195, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hage FG, Venkataraman R, Zoghbi GJ, Perry GJ, DeMattos AM, Iskandrian AE. The scope of coronary heart disease in patients with chronic kidney disease. J Am Coll Cardiol 53: 2129–2140, 2009 [DOI] [PubMed] [Google Scholar]
- 16.He T, Smith LA, Harrington S, Nath KA, Caplice NM, Katusic ZS. Transplantation of circulating endothelial progenitor cells restores endothelial function of denuded rabbit carotid arteries. Stroke 35: 2378–2384, 2004 [DOI] [PubMed] [Google Scholar]
- 17.Hewitson TD, Ono T, Becker GJ. Small animal models of kidney disease: a review. Methods Mol Biol 466: 41–57, 2009 [DOI] [PubMed] [Google Scholar]
- 18.Hilgers RH, Todd J, Webb RC. Regional heterogeneity in acetylcholine-induced relaxation in rat vascular bed: role of calcium-activated K+ channels. Am J Physiol Heart Circ Physiol 291: H216–H222, 2006 [DOI] [PubMed] [Google Scholar]
- 19.Jourde-Chiche N, Dou L, Cerini C, Dignat-George F, Brunet P. Vascular incompetence in dialysis patients–protein-bound uremic toxins and endothelial dysfunction. Seminars in dialysis 24: 327–337, 2011 [DOI] [PubMed] [Google Scholar]
- 20.Kajimoto H, Kai H, Aoki H, Yasuoka S, Anegawa T, Aoki Y, Ueda S, Okuda S, Imaizumi T. Inhibition of eNOS phosphorylation mediates endothelial dysfunction in renal failure: new effect of asymmetric dimethylarginine. Kidney Int 81: 762–768, 2012 [DOI] [PubMed] [Google Scholar]
- 21.Kietadisorn R, Juni RP, Moens AL. Tackling endothelial dysfunction by modulating NOS uncoupling: new insights into its pathogenesis and therapeutic possibilities. Am J Physiol Endocrinol Metab 302: E481–E495, 2012 [DOI] [PubMed] [Google Scholar]
- 22.Kim HJ, Vaziri ND. Contribution of impaired Nrf2-Keap1 pathway to oxidative stress and inflammation in chronic renal failure. Am J Physiol Renal Physiol 298: F662–F671, 2010 [DOI] [PubMed] [Google Scholar]
- 23.Krenning G, Dankers PY, Drouven JW, Waanders F, Franssen CF, van Luyn MJ, Harmsen MC, Popa ER. Endothelial progenitor cell dysfunction in patients with progressive chronic kidney disease. Am J Physiol Renal Physiol 296: F1314–F1322, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levey AS, Coresh J. Chronic kidney disease. Lancet 379: 165–180, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Lorenzen J, David S, Bahlmann FH, de Groot K, Bahlmann E, Kielstein JT, Haller H, Fliser D. Endothelial progenitor cells and cardiovascular events in patients with chronic kidney disease—a prospective follow-up study. PLoS One 5: e11477, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luksha L, Stenvinkel P, Hammarqvist F, Carrero JJ, Davidge ST, Kublickiene K. Mechanisms of endothelial dysfunction in resistance arteries from patients with end-stage renal disease. PLoS One 7: e36056, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuguma K, Ueda S, Yamagishi S, Matsumoto Y, Kaneyuki U, Shibata R, Fujimura T, Matsuoka H, Kimoto M, Kato S, Imaizumi T, Okuda S. Molecular mechanism for elevation of asymmetric dimethylarginine and its role for hypertension in chronic kidney disease. J Am Soc Nephrol 17: 2176–2183, 2006 [DOI] [PubMed] [Google Scholar]
- 28.Moody WE, Edwards NC, Madhani M, Chue CD, Steeds RP, Ferro CJ, Townend JN. Endothelial dysfunction and cardiovascular disease in early-stage chronic kidney disease: cause or association? Atherosclerosis 223: 86–94, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Morris ST, McMurray JJ, Spiers A, Jardine AG. Impaired endothelial function in isolated human uremic resistance arteries. Kidney Int 60: 1077–1082, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Munzel T, Daiber A, Ullrich V, Mulsch A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler Thromb Vasc Biol 25: 1551–1557, 2005 [DOI] [PubMed] [Google Scholar]
- 30a.National Institutes of Health US Renal Data System, USRDS 2012 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2012 [Google Scholar]
- 31.Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo. Br J Pharmacol 101: 746–752, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schiffrin EL, Lipman ML, Mann JF. Chronic kidney disease: effects on the cardiovascular system. Circulation 116: 85–97, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Seddon M, Melikian N, Dworakowski R, Shabeeh H, Jiang B, Byrne J, Casadei B, Chowienczyk P, Shah AM. Effects of neuronal nitric oxide synthase on human coronary artery diameter and blood flow in vivo. Circulation 119: 2656–2662, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Tain YL, Freshour G, Dikalova A, Griendling K, Baylis C. Vitamin E reduces glomerulosclerosis, restores renal neuronal NOS, and suppresses oxidative stress in the 5/6 nephrectomized rat. Am J Physiol Renal Physiol 292: F1404–F1410, 2007 [DOI] [PubMed] [Google Scholar]
- 35.van Guldener C, Janssen MJ, Lambert J, Steyn M, Donker AJ, Stehouwer CD. Endothelium-dependent vasodilatation is impaired in peritoneal dialysis patients. Nephrol Dial Transplant 13: 1782–1786, 1998 [DOI] [PubMed] [Google Scholar]
- 36.Vettoretti S, Ochodnicky P, Buikema H, Henning RH, Kluppel CA, de Zeeuw D, van Dokkum RP. Altered myogenic constriction and endothelium-derived hyperpolarizing factor-mediated relaxation in small mesenteric arteries of hypertensive subtotally nephrectomized rats. J Hypertens 24: 2215–2223, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Vita JA. Endothelial function. Circulation 124: e906–e912, 2011 [DOI] [PubMed] [Google Scholar]
- 38.Xing D, Li P, Gong K, Yang Z, Yu H, Hage FG, Oparil S, Chen YF. Endothelial cells overexpressing interleukin-8 receptors reduce inflammatory and neointimal responses to arterial injury. Circulation 125: 1533–1541, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Young S, Struys E, Wood T. Quantification of creatine and guanidinoacetate using GC-MS and LC-MS/MS for the detection of cerebral creatine deficiency syndromes. Curr Protoc Hum Genet Chapt. 17: Unit 17.13, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Zhang HQ, Fast W, Marletta MA, Martasek P, Silverman RB. Potent and selective inhibition of neuronal nitric oxide synthase by N omega-propyl-l-arginine. J Med Chem 40: 3869–3870, 1997 [DOI] [PubMed] [Google Scholar]
- 41.Zoccali C. Asymmetric dimethylarginine (ADMA): a cardiovascular and renal risk factor on the move. J Hypertens 24: 611–619, 2006 [DOI] [PubMed] [Google Scholar]