

Abstract
Superoxide (O2·−) production by the NADPH oxidases is implicated in the pathogenesis of many cardiovascular diseases, including hypertension. We have previously shown that activation of NADPH oxidases increases mitochondrial O2·− which is inhibited by the ATP-sensitive K+ channel (mitoKATP) inhibitor 5-hydroxydecanoic acid and that scavenging of mitochondrial or cytoplasmic O2·− inhibits hypertension. We hypothesized that mitoKATP-mediated mitochondrial O2·− potentiates cytoplasmic O2·− by stimulation of NADPH oxidases. In this work we studied Nox isoforms as a potential target of mitochondrial O2·−. We tested contribution of reverse electron transfer (RET) from complex II to complex I in mitochondrial O2·− production and NADPH oxidase activation in human aortic endothelial cells. Activation of mitoKATP with low dose of diazoxide (100 nM) decreased mitochondrial membrane potential (tetramethylrhodamine methyl ester probe) and increased production of mitochondrial and cytoplasmic O2·− measured by site-specific probes and mitoSOX. Inhibition of RET with complex II inhibitor (malonate) or complex I inhibitor (rotenone) attenuated the production of mitochondrial and cytoplasmic O2·−. Supplementation with a mitochondria-targeted SOD mimetic (mitoTEMPO) or a mitochondria-targeted glutathione peroxidase mimetic (mitoEbselen) inhibited production of mitochondrial and cytoplasmic O2·−. Inhibition of Nox2 (gp91ds) or Nox2 depletion with small interfering RNA but not Nox1, Nox4, or Nox5 abolished diazoxide-induced O2·− production in the cytoplasm. Treatment of angiotensin II-infused mice with RET inhibitor dihydroethidium (malate) significantly reduced blood pressure. Our study suggests that mitoKATP-mediated mitochondrial O2·− stimulates cytoplasmic Nox2, contributing to the development of endothelial oxidative stress and hypertension.
Keywords: endothelial cells, hypertension, mitochondria, NADPH oxidase, superoxide
in the past decade it has become clear that production of superoxide (O2·−) plays an important role in the development of cardiovascular diseases (18). It has been suggested that NADPH oxidase, one of the main sources of vascular O2·−, can be a potential target for therapeutic intervention (19). Human endothelial cells express Nox1, Nox2, Nox4, and Nox5 isoforms (16, 22, 26, 47), but Nox2 represents a major source of O2·− in endothelial cells (31). CYBA polymorphism in human subjects result in altered Nox2 activity and affects cardiovascular diseases (3, 28). The other major source of the vascular O2·− is mitochondria. We have recently reported that scavenging of mitochondrial O2·− improves endothelial function and significantly attenuates hypertension (12); however, exact targets of mitochondrial O2·− are not clear.
Activation of vascular NADPH oxidases by angiotensin II (ANG II) increases mitochondrial O2·−, and depletion of p22phox, a docking subunit of NADPH oxidase complex, abolishes this effect (14). Interestingly, treatment of mitochondria or ANG II-stimulated endothelial cells with the ATP-sensitive K+ channel (mitoKATP) blocker 5-hydroxydecanoic acid inhibits production of mitochondrial and cytoplasmic O2·− (14). These data suggest that mitochondrial O2·− regulates activity of NADPH oxidases. Indeed, we have recently reported that overexpression of mitochondrial superoxide dismutase (SOD2) attenuates ANG II-mediated activation of NADPH oxidases (12). Moreover, acute treatment of ANG II-stimulated cells with mitochondria-targeted SOD mimetic mitoTEMPO reduces NADPH oxidase activity in intact cells (12). It is important to note that endothelial mitochondria do not express p22phox (14), which is essential for Nox1, Nox2, or Nox4 activity (1), suggesting translation of the mitochondrial signal to cytoplasm to regulate activity of the cytoplasmic NADPH oxidases; however, the exact target of mitochondrial O2·− is not known.
The activation of mitoKATP has been associated with increased production of mitochondrial O2·− in the ischemic preconditioning (41) and ANG II-mediated signaling (14). It has been reported that mitoKATP is a redox-sensitive channel which can be opened by either O2·− or H2O2 (42, 57). Several groups have shown that activation of the mitoKATP increases production of O2·− in mitochondrial matrix (2, 52), which is prevented by the mitoKATP blocker 5-hydroxydecanoic acid. Activation of the mitoKATP increases influx of K+ into mitochondrial matrix, leading to H+/K+ antiporter activation that results in mitochondrial matrix alkalization (6). Increased matrix pH has been shown to stimulate production of mitochondrial O2·− (46); however, the precise mechanism for this is unclear. It has been suggested that opening of the mitoKATP and matrix alkalization can stimulate production of mitochondrial O2·− on complex III (46). The alternative source of mitochondrial O2·− is reverse electron transfer (RET) from complex II to complex I, which can be inhibited by malonate, malate, or rotenone (39). Understanding specific mechanisms of mitochondrial O2·− regulation may help in identification of novel pharmacological targets for treatment of oxidative stress conditions.
In this work we investigated contribution of RET in mitoKATP-regulated mitochondrial O2·− production and its role in the regulation of NADPH oxidase isoforms. We tested the role of RET as a potential target for antihypertensive treatment using diazoxide-treated human aortic endothelial cells (HAECs) and ANG II-infused mice.
MATERIALS AND METHODS
Reagents.
Dihydroethidium (DHE) and MitoSOX were supplied by Roche Molecular Biochemicals (Indianapolis, IN). NADPH oxidase antibodies were obtained from Abcam (San Francisco, CA). PKCε peptide antagonist H-Glu-Ala-Val-Ser-Leu-Lys-Pro-Thr-OH (EAVSLKPT) and c-Src antibodies were received from Millipore (Billerica, MA). mitoTEMPO was purchased from Enzo Life Sciences (San Diego, CA). The Nox2 inhibitor peptide gp91ds (CSTRIRRQL) was purchased from GenScript (Piscataway, NJ). All other reagents were obtained from Sigma (St Louis, MO).
Cell culture.
HAECs were purchased from Lonza (Chicago, IL) and cultured in EGM-2 medium supplemented with 2% FBS but without antibiotics. On the day before the study, the FBS concentration was reduced to 1%. In preliminary experiments, we examined the effect of varying doses of ANG II on cellular O2·− production. We found that 4 h of ANG II treatment increased cellular O2·− in a dose-dependent manner with maximum stimulation at 200 nM (12). This concentration was therefore used in the remainder of the experiments. It should be noted that because of degradation in culture, the steady-state concentration of ANG II is substantially lower than that initially added (21). In our current study we used low dose of diazoxide (100 nM) to treat cultured cells. The primary reason for the use of nanomolar concentrations was to avoid unspecific actions of diazoxide, such as inhibition of respiratory complex II observed at micromolar concentrations (15, 17).
Inhibition of NOX expression.
To manipulate expression of the catalytic subunits of NADPH oxidase, we inhibited expression of Nox1, Nox2, Nox4, and Nox5 using small interfering RNA (siRNA) from Applied Biosystems Ambion as previously described (9, 16, 45). These siRNAs decrease NOX isoform expression by two- to threefold compared with control nonsilencing siRNA. As a control, we used nonsilencing siRNA (Qiagen AllStars negative control).
Mitochondria isolation.
Mitochondria were isolated as previously described (50). Cells collected from 100-mm dishes were washed with PBS, resuspended in ice-cold isolation buffer (consisting of 0.3 M mannitol; 0.1% BSA; 0.2 mM EDTA; and 10 mM HEPES; pH 7.4 with KOH; protease inhibitor cocktail), and homogenized in a glass homogenizer using a Teflon pestle. To prevent mitochondria damage, the progress was monitored with light microscope. Homogenate was centrifuged at 600 g for 10 min at 4°C, and supernatant was transferred into clean centrifuge tubes and centrifuged at 7,000 g for 10 min at 4°C. Pellet was resuspended with ice-cold isolation buffer without BSA and repurified by one-step centrifugation at 600 g. Mitochondria were collected by centrifugation at 7,000 g for 10 min at 4°C.
Confocal microscopy.
Intact cells cultured on coverslips were incubated with a fluorescent probe in Krebs-HEPES buffer for 20 min at 37°C in a CO2 incubator. Production of mitochondrial O2·− was visualized in using the 2 μM fluorescent probe MitoSOX [excitation (Ex)/emission (Em), 405/480 nm]. For detection of cytoplasmic O2−, cells were stained with 10 μM DHE (Ex/Em, 405/480 nm). Mitochondria localization was ensured by mitoTracker Green (Ex/Em 488/520 nm). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Ex/Em, 364/461 nm). Mitochondrial membrane potential was monitored with fluorescent probe tetramethylrhodamine methyl ester (Ex/Em, 543/573 nm). Paraformaldehyde-fixed cells were mounted in Vectashield (Vector, Burlingame, CA) and examined in a confocal imaging system (Zeiss LSM 510 META). For double-labeling experiments, images were scanned with the multitracking mode on a Zeiss LSM 510 META confocal microscope. To distinguish random color overlap from colocalization, Imaris Coloc software was used.
Superoxide measurements using HPLC.
Cells were cultured up to 80% confluence. Stock solutions of MitoSOX (4 mM) and DHE (10 mM) in DMSO were prepared and were diluted in Krebs-HEPES buffer to a final concentration of 2 μM MitoSOX and 10 μM DHE. Cells loaded with dye were incubated in a tissue culture incubator for 20 min. Buffer was next aspirated, and scraped cells were mixed with methanol (300 μl) and homogenized with a glass pestle. The cell homogenate was passed through a 0.22-μm syringe filter, and methanol filtrates were analyzed by HPLC according to previously published protocols (8). DHE and MitoSOX oxidation products 2-hydroxyethidium and ethidium were separated using a C-18 reverse-phase column (Nucleosil 250 to 4.5 mm) and a mobile phase containing 0.1% trifluoroacetic acid and an acetonitrile gradient (from 37% to 47%) at a flow rate of 0.5 ml/min. Ethidium and 2-hydroxyethidium were detected with a fluorescence detector using an excitation of 480 nm and an emission wavelength of 580 nm.
Western blot analysis.
Cells were collected and lysed in lysis buffer containing 50 mm HEPES (pH 7.4), 50 mm NaCl, 1% Triton X-100, 5 mm EDTA, 1 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 10 mm sodium pyrophosphate, 50 mm sodium fluoride, and 1 mm sodium orthovanadate. Lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes, and incubated with specific antibodies.
Animal experiments.
Hypertension was induced by ANG II (0.7 mg·kg−1·day−1) as previously described (11) using C57Bl/6 mice. In addition, six days after saline or ANG II minipump placement, mice received a second minipump for infusion of saline as vehicle or malate, as described in the figure legends. Blood pressure was monitored using either the tail cuff method or telemetry as previously described (25, 53). All procedures were performed according to guidelines and approved by Institutional Animal Care and use Committee at Vanderbilt University.
Statistics.
Experiments were analyzed using the Student-Newman-Keuls post hoc test, and ANOVA. P levels < 0.05 were considered significant.
RESULTS
Measurements of diazoxide-induced O2·− production.
We have previously reported that treatment of endothelial cells with ANG II induces both cytoplasmic and mitochondrial O2·− production, which is prevented by the mitoKATP blocker 5-hydroxydecanoic (14). In this work we tested whether stimulation of endothelial cells with the mitoKATP agonist diazoxide enhances O2·− production in a similar manner. Indeed, confocal analysis of HAECs treated with diazoxide revealed significant increase of O2·− production both in mitochondria (Figs. 1A and 2C) and cytoplasm (Figs. 1B and 2D) measured by site-specific fluorescent probes MitoSOX and DHE, respectively. The diazoxide-induced increase in mitochondrial and cytoplasmic O2·− production was confirmed by HPLC analysis of O2·−-specific 2-hydroxyethidium products (58) of DHE and MitoSOX (Fig. 2, A and B). Interestingly, treatment of HAECs with low dose of diazoxide (100 nM) increased O2·− production similarly to ANG II (Fig. 1). These data suggest that opening mitoKATP triggers both mitochondrial and cytoplasmic O2·−. The mechanism and regulation of mitoKATP-induced O2·− production is not completely recognized. It has been reported that PKCε plays a critical role in the regulation of mitoKATP (24). We therefore tested whether PKCε-specific peptide inhibitor EAVSLKPT prevents stimulation of O2− production by dizoxide. Indeed, preincubation of HAECs with EAVSLKPT (1 μM) completely abolished diazoxide-induced O2− production in mitochondria and cytoplasm (Fig. 1). These data support the concept that diazoxide-induced cytoplasmic O2− requires PKCε activity. These data are in line with previous reports on stimulation of mitochondrial O2·− by diazoxide and suggest an important role of mitoKATP in regulation of both mitochondrial and cytoplasmic O2·− production.
Fig. 1.

Stimulation of mitochondrial ATP-sensitive K+ channel (mitoKATP) with diazoxide induces production of mitochondrial and cytoplasmic superoxide (O2·−). A: mitochondrial O2·− was measured by MitoSOX (2 μM) and confocal microscopy in human aortic endothelial cells (HAECs) following stimulation with angiotensin II (ANG II; 200 nM) or diazoxide (100 nM). PKCε peptide antagonist H-Glu-Ala-Val-Ser-Leu-Lys-Pro-Thr-OH (EAVSLKPT; 1 μM) was added before MitoSOX application. B: cytoplasmic O2− was measured by dihydroethidium (DHE; 10 μM) and confocal microscopy in HAECs stimulated by ANG II or diazoxide. PKCε peptide antagonist EAVSLKPT (1 μM) was added before DHE application. Inh, inhibitor. Data represent typical Z-stack of fluorescent images.
Fig. 2.

Superoxide measurements in mitochondria and cytoplasm. Mitochondrial O2·− was measured using fluorescent probe MitoSOX detected by HPLC (A) or confocal microscopy (B) in control-unstimulated and diazoxide (Dz; 100 nM, 10 min)-stimulated HAECs. Cytoplasm O2·− was measured by HPLC detection of O2·−-specific DHE product (C) or confocal microscopy (D) in control-unstimulated or gp91d, malonate-treated cells with or without diazoxide. AU, arbitrary units. Fluorescence signal from confocal microscopy images was quantified using ImageJ software. Results represent means ± SE; n = 3–6. *P < 0.01 vs. control; **P < 0.05 vs. diazoxide.
Mitochondrial source of diazoxide-induced O2·−.
To further investigate the mechanism of diazoxide-induced production of mitochondrial O2·−, we investigated the effect of mitochondria-targeted antioxidants on diazoxide-induced mitochondrial O2·− and alterations of mitochondrial membrane potential. Figure 3 shows that diazoxide caused partial depolarization of mitochondria, and this effect of diazoxide was inhibited by the mitochondria-targeted SOD mimetic mitoTEMPO. We have previously shown that ANG II reduces mitochondrial membrane potential and that mitoTEMPO supplementation prevents these mitochondrial alterations (12). These data show that diazoxide has similar effect on mitochondrial O2·− in endothelial cells as ANG II.
Fig. 3.
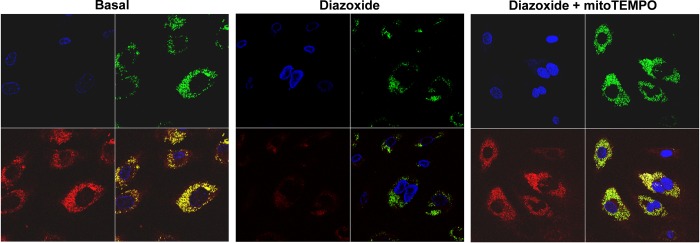
Diazoxide partially depolarize mitochondrial membrane, and mitoTEMPO supplementation abolishes effect of diazoxide. Mitochondrial membrane potential was measured by tetramethylrhodamine methyl ester (TMRM) fluorescence (red). Mitochondrial localization of TMRM signal was confirmed by colocalization with MitoTracker (green).
We tested the potential role of mitochondrial H2O2 in diazoxide-induced production of mitochondrial and cytoplasmic O2·−. To achieve this aim, HAECs were treated with the mitochondria-targeted glutathione peroxidase mimetic mitoEbselen (33). Supplementation of HAECs with low dose of mitoEbselen (1 nM) completely inhibited diazoxide-induced O2·− production in both mitochondria and cytoplasm (Fig. 4, A and B), whereas mitoEbselen did not affect the partial depolarization induced by diazoxide (Fig. 4C). Since mitoEbselen scavenges H2O2 and does not react with O2·−, these data suggest that mitochondrial and cytoplasmic O2·− production is redox dependent.
Fig. 4.

Redox sensitivity of diazoxide-induced O2·− production. Mitochondrial O2·− was measured by MitoSOX (A); cytoplasmic O2·− was detected by DHE (B), and mitochondrial potential was analyzed by TMRM fluorescence (C). HAECs were treated with 1 nM mitoEbselen 15 min before stimulation with diazoxide (100 nM, 10 min).
It is important to note that low dose of mitoEbselen selectively acts at the mitochondrial site but not in the cytoplasm. We found that a low dose of untargeted Ebselen (1 nM) did not significantly affect ANG II-induced O2·− production, whereas mitochondria-targeted H2O2 scavenger mitoEbselen completely inhibited ANG II-stimulated cytoplasm O2·− production measured by DHE and HPLC (Fig. 5). These data show that ANG II-induced cytoplasm O2·− production depends on mitochondrial H2O2.
Fig. 5.

Cytoplasm O2·− production in HAECs stimulated with ANG II (4 h, 200 nM) following acute 15-min treatment with mitochondria-targeted SOD mimetic mitoTEMPO (25 nM) or mitochondria targeted H2O2 scavenger mitoEbselen (mitoEbs, 1 nM) or untargeted analog Ebselen (Ebs, 1 nM). Cytoplasm O2·− was measured by HPLC detection of O2·−-specific DHE product using HPLC. Results represent means ± SE; n = 4–6. *P < 0.01 vs. control; **P <0.05 vs. ANG II.
RET represents of the major pathways of mitochondrial O2·− formation on complex I. We therefore investigated the potential role of RET in diazoxide-induced mitochondrial O2·− formation. We found that supplementation of HAECs with complex II inhibitor malonate (0.1 mM) or complex I inhibitor rotenone (1 μM) abolished diazoxide-induced production of mitochondrial O2− to the similar level as PKCε inhibition (Figs. 6 and 2D). Any single source of electrons from complex I or complex II is sufficient for O2− production on complex III, but diazoxide-stimulated O2·− production was completely inhibited both by malonate or rotenone, therefore, implicating RET from complex II to complex I as a source of diazoxide-induced mitochondrial O2·−. Indeed, rotenone is a well-known inhibitor of RET-mediated production of mitochondrial O2·− on complex I (39). Taken together, these data implicate RET and complex I as a major source of diazoxide-induced mitochondrial O2·−.
Fig. 6.

Production of mitochondrial O2·− was attenuated by inhibition of reverse electron transfer (RET) with rotenone, malonate, or with PKCε peptide antagonist EAVSLKPT. Mitochondrial O2·− was measured using fluorescent probe MitoSOX and confocal microscopy in unstimulated cells (basal; A) and HAECs stimulated with diazoxide (100 nM, 10 min; B) and then treatment with rotenone, malonate, or PKCε peptide antagonist EAVSLKPT before application of MitoSOX.
Cytoplasmic source of diazoxide-induced O2·−.
In the next step we aimed to determine the effect of RET on cytoplasmic O2·− production. Figure 7 shows that supplementation of HAECs with malate did not affect the basal O2·− production but completely abolished the diazoxide-induced cytoplasmic O2·− production. Interestingly, rotenone slightly increased basal cytoplasmic O2·− production but significantly inhibited the diazoxide-induced O2·− production (Fig. 7). The increase of basal O2·− in rotenone-supplemented cells is likely due to NADH-dependent stimulation of mitochondrial reactive oxygen species (ROS) on complex I (38). The PKCε antagonist EAVSLKPT inhibited diazoxide-induced O2·− production. The Nox2 inhibitor gp91ds and cSrc inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolopyrimidine (PP2) prevented stimulation of cytoplasmic O2·− by diazoxide (Fig. 7). These data confirm that inhibition of RET attenuates diazoxide-induced cytoplasmic O2·− and implicate Nox2 as a potential target of mitochondrial O2·−.
Fig. 7.
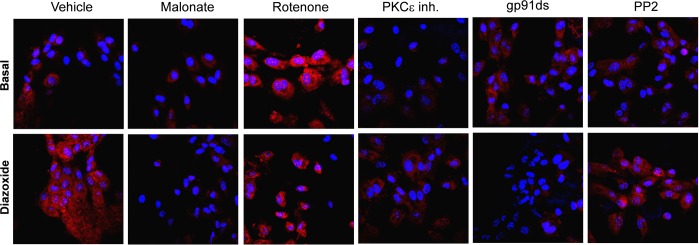
Diazoxide-induced cytoplasmic O2·− is attenuated by inhibition of RET with rotenone, malate, or PKCε peptide antagonist EAVSLKPT. Cytoplasmic O2·− was measured by DHE (10 μM) in HAECs treated with diazoxide (100 nM, 10 min), malonate (0.1 mM), rotenone (1 μM), PKCε peptide antagonist EAVSLKPT (1 μM), or Nox2 inhibitor peptide gp91ds or cSrc inhibitor 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolopyrimidine (PP2).
We next tested which Nox isoform is a target for mitochondrial O2·− in HAECs. For this purpose we downregulated Nox1, Nox2, Nox4, and Nox5 expression with specific siRNA as previously described (9, 16, 45). Depletion of Nox2 significantly attenuated stimulation of cytoplasmic O2·− production by diazoxide (Fig. 8), whereas inhibition of Nox1 expression or deletion of Nox4 and Nox5 did not significantly affect diazoxide-induced O2·− production. These data are in line with inhibition of endothelial O2·− production by Nox2 inhibitor gp91ds. The role of cSrc in activation of cytoplasmic Nox has been previously demonstrated (48). cSrc is the ROS-sensitive nonreceptor tyrosine kinase that can be activated by the cytoplasmic ROS production (51). Inhibition of endothelial O2·− production by of PP2, therefore, implicates redox-sensitive cSrc in mitochondrial ROS-mediated regulation of Nox2 activity.
Fig. 8.

Nox2 depletion diminishes diazoxide-induced cytoplasmic O2·−. Cytoplasmic O2·− was measured in unstimulated HAECs (basal), cells stimulated with ANG II (200 nM, 4 h) or diazoxide (100 nM, 10 min) using fluorescent probe DHE. NS, nonsilencing; si, small interfering.
To exclude the possibility that NOX2 is expressed in mitochondria, we tested its localization in cell fractions by Western blot analysis. As shown in Fig. 9, Nox2 is abundantly expressed in HAECs under basal and ANG II-stimulated conditions, but analysis of mitochondrial fractions did not reveal Nox2 presence (Fig. 9). Furthermore, 4-h ANG II stimulation or acute 10-min treatment with diazoxide did not change Nox2 localization. Of note, the purity of mitochondrial fraction was confirmed by enhanced presence of cytochrome-c and lack of cytoplasmic protein Akt. These data are in line with the previously reported lack of p22phox subunit in mitochondria of endothelial cells (14). Overall, these data are consistent with previously reported cytoplasmic localization of Nox2 (32) and lack of Nox2 in the mitochondria; therefore, mitochondrial ROS are likely to stimulate Nox2-dependent cytoplasmic O2·− production (Figs. 7 and 8).
Fig. 9.

Western blot analysis of cellular and mitochondrial expression of Nox2 in HAECs. Endothelial mitochondria do not contain NOX2 isoform. Molecular mass indicated on the right side of the images. Cyt C, cytochrome-c.
Inhibition of mitochondrial ROS by malate reduces blood pressure after onset of ANG II-induced hypertension.
We have previously shown that scavenging of mitochondrial O2·− attenuates development of hypertension and reduces blood pressure after mitoTEMPO treatment of hypertensive mice (12). In this work we aimed to determine if inhibition of RET attenuates hypertension. Our data show that inhibition of complex II with malonate or blocking complex I with rotenone attenuate RET-mediated ROS production (Figs. 6 and 7). Both malonate and rotenone cause severe neurodegeneration (38); therefore, their application in the setting of hypertension should not be considered as a pharmacological treatment.
Malate metabolically regulates complex II activity in the citric acid cycle and malate supplementation reduces RET-mediated production of mitochondrial ROS (39). In the present study we investigated the effect of malate infusion on ANG II-induced hypertension. Malate supplementation was started after the onset of ANG II-induced hypertension by infusion of pH-equilibrated malate solution via osmotic pump. We found that three days malate infusion had a significant antihypertensive effect, reducing blood pressure by 17 ± 6 mmHg (Fig. 10). The antihypertensive effect of malate was compared with mitoTEMPO supplementation. Treatment of hypertensive mice with mitoTEMPO caused sustained reduction of blood pressure by 25 ± 5 mmHg (Fig. 10). Although mitoTEMPO treatment had more substantial blood pressure decrease compared with malate supplementation these data support the concept that both inhibition of mitochondrial ROS production and scavenging of mitochondrial ROS may provide novel targets for design of for antihypertensive treatments.
Fig. 10.

Antihypertensive effect of malate in ANG II-infused mice. C57Bl/6J mice were infused with ANG II (0.7 mg·kg−1·day−1) and osmotic pumps with saline, malate, or mitoTEMPO (0.5 mg·kg−1·day−1) were set up after the onset of ANG II-induced hypertension. Results represent means ± SE; n = 6–8 animals per group. §P < 0.05 vs. saline; *P < 0.01 vs. saline.
DISCUSSION
The present study provides evidence that Nox2 is a potential target for mitochondrial O2·−. Furthermore, our data show an important role of mitoKATP in stimulation of mitochondrial O2·− production. Inhibitory analysis suggests that mitoKATP opening stimulates O2·− production in mitochondrial complex I due to RET-mediated electron transfer from complex II to complex I. RET has been extensively reported in mitochondria from various tissue (20, 37, 49); however, it has never been linked to the activation of NADPH oxidases. Our work shows activation of cytoplasmic Nox2 by mitoKATP and RET-mediated mitochondrial O2·− production (Fig. 11). Interestingly, attenuation of RET by malate supplementation (39) reduced blood pressure in ANG II-infused mice, which supports the role of RET-mediated mitochondrial O2·− in hypertension. It is conceivable that antihypertensive effect of systemic malate supplementation is not limited by inhibition of mitochondrial O2·− in endothelial cells and includes contribution of other tissues such as smooth muscle cells, kidney and brain, since arterial blood pressure is determined by numerous factors (36). Meanwhile, our data provide clear evidence of inhibition of mitochondrial O2·− by malate, malonate, and rotenone, implicating RET as a major source of mitochondrial ROS.
Fig. 11.
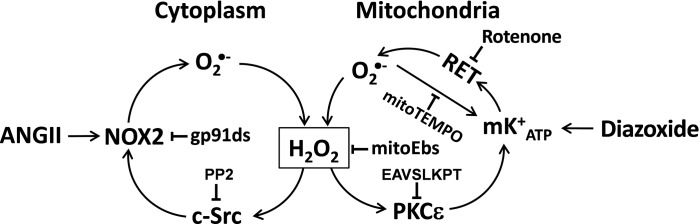
Proposed Nox2-dependent stimulation of cytoplasmic O2·− by mitochondrial reactive oxygen species produced by RET. Opening of mKATP increases O2·− production by rotenone-sensitive RET. Dismutation of O2·− produces neutral H2O2 that leaks out of mitochondria and stimulates redox-sensitive cSrc, leading to NOX2 activation.
ROS measurements in biological samples have been a challenge for investigators for many years. There are a limited number of methods that can be used for this purpose, and they all have limitations (8). Mitochondrial-targeted fluorescent probe MitoSOX has been widely used for detection of mitochondrial O2·− (30). HPLC detection of MitoSOX O2·−-specific product has been suggested as reliable and quantitative method; however, this method also has number of limitations. HPLC assay requires significantly larger amount of a sample, and it does not provide space resolution of the detected signal. For example, HPLC detection of O2·−-specific product 2OH-ethydium is unable to show precise subcellular localization of the O2·− source, unless organelles are isolated from the cells. HPLC also cannot perform live-imaging or on-line study, unable to reveal the heterogeneity of cellular responses nor account for artifacts induced by cell death (29, 43). Therefore, confocal microscopy may provide additional information that can significantly improve understanding of cell and organelle specificity of O2·− production. To minimize the contribution of nonspecific oxidation products, in this study we used optimized fluorescent parameters for MitoSOX (44) and DHE as we have recently reported (34).
Although production of mitochondrial O2·− by RET has been widely recognized, the pathophysiological significance of RET is not clear. Rotenone supplementation has been commonly used to improve mitochondrial respiration in the studies with succinate-driven respiration. Rotenone, however, inhibits succinate-stimulated ROS production by RET. It has been suggested that RET is physiologically regulated by several mechanisms, 1) by control of succinate dehydrogenase activity in the citric acid cycle (39), 2) by fatty acids (40), 3) and by mitochondrial membrane potential (35). Thus RET is not an “artifact” and rotenone supplementation may produce an artificially improved mitochondrial functions.
It has been previously shown that ROS production may represent a feed-forward regulation resulting in ROS-induced ROS release (7, 59). Our data suggest that activation of redox-sensitive RET may represent an important mechanism in this feed-forward regulation. This is in line with our previous report showing that ANG II-induced ROS production was abrogated by either inhibition of the NADPH oxidases or mitochondria (12, 14). It has been previously shown that mitoKATP-induced production of mitochondrial ROS is redox-sensitive. Activity of mitoKATP is regulated by PKCε and can be stimulated by H2O2 (5, 42). Recently, David Busija group have investigated the effects of mitoKATP openers diazoxide and BMS-191095 on endothelium-dependent relaxation (23). Interestingly, SOD mimetic MnTBAP attenuated diazoxide-induced relaxation but did not affect vascular responses to BMS-191095 (23). These data suggest potential dual actions of that mitoKATP openers by mitochondrial depolarization and ROS production. Our data suggest that even low doses of diazoxide have the potential to generate ROS in addition to depolarizing mitochondria. Activation of redox-sensitive mitoKATP and PKCε drives RET-mediated mitochondrial O2·− production; therefore, RET is a redox-dependent mechanism (Fig. 11). This suggests that RET is a part of redox-dependent ROS-induced ROS release. While there is increased O2·− production in mitochondria, it is conceivable that H2O2, the product of O2·− dismutation, is diffusing through mitochondrial membrane and affects cytosolic targets to activate NOX2.
Our in vitro data show that well-known RET inhibitor rotenone blocks mitoKATP-mediated O2·− production in mitochondria and cytoplasm. These data highlight the potential utility of rotenone in the studies of mitochondrial O2·−. Indeed, rotenone has been shown to inhibit flow-induced dilation in human coronary resistance arteries (27) and attenuated DOCA-salt induced hypertension (56). Since rotenone supplementation causes neurodegeneration (38), in our work we used malate, the citric acid-cycle substrate, which regulates complex II activity and attenuates RET. It is important to note that malate supplementation has been previously shown to reverse oxidative stress and improve antioxidant defenses in heart of aged rats (55). Our study demonstrates an antihypertensive effect of malate, which suggests RET as a potential novel pharmacological target for treatment of hypertension.
Endothelial dysfunction impairs smooth muscle relaxation, and smooth muscle cells contribute to development of hypertension. Our in vitro study, however, was limited to endothelial cells and does not include smooth muscle cells. Future studies should investigate the role of smooth muscle mitochondrial ROS to determine their contribution in endothelial and vascular dysfunction (11, 13).
Mitochondria have been previously implicated in regulation of NADPH oxidases. For example, mitochondria depletion in ethidium-treated rabbit aortic smooth muscle cells attenuates Nox1 expression (54). We have shown that decreased mitochondrial membrane potential enhances calcium-mediated activation of phagocytic NADPH oxidase, and mitochondrial ROS contributes to activation of NADPH oxidase in phagocytic cells (10). In this work we show evidence that Nox2 could be a specific target of mitochondrial ROS and demonstrate acute activation of Nox2 by mitochondrial O2·−.
Taken together, our data demonstrate an important role of mitochondrial ROS in the development of endothelial oxidative stress and hypertension. We suggest that mitoKATP opening and RET play an important role in the interaction between mitochondrial and NADPH oxidase-derived O2·− (Fig. 11). This feed-forward cycle between Nox2 and RET can be responsible for overproduction of cellular O2·−, inactivation of nitric oxide, and uncoupling endothelial nitric oxide synthase previously found in hypertension (4). The interplay between mitochondrial and NADPH oxidase-derived O2·− is a redox sensitive due to redox-dependent regulation of PKCε, mitoKATP, and c-Src (Fig. 11). Our study suggests that both Nox2 and mitochondrial RET can be important targets for the development of antioxidant strategies.
GRANTS
This work was supported by funding from National Heart, Lung, and Blood Institute Grant HL-094469.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
R.R.N. and S.I.D. conception and design of research; R.R.N., A.E.D., and A.B. performed experiments; R.R.N., A.E.D., A.B., and S.I.D. analyzed data; R.R.N., A.E.D., and S.I.D. interpreted results of experiments; R.R.N., A.E.D., A.B., and S.I.D. prepared figures; R.R.N., A.E.D., and S.I.D. drafted manuscript; R.R.N., A.E.D., A.B., and S.I.D. approved final version of manuscript; S.I.D. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Dr. Alexander Panov and Vladimir Mayorov for fruitful discussion and advice.
REFERENCES
- 1.Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP. Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J Biol Chem 279: 45935–45941, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Andrukhiv A, Costa AD, West IC, Garlid KD. Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am J Physiol Heart Circ Physiol 291: H2067–H2074, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Bedard K, Attar H, Bonnefont J, Jaquet V, Borel C, Plastre O, Stasia MJ, Antonarakis SE, Krause KH. Three common polymorphisms in the CYBA gene form a haplotype associated with decreased ROS generation. Hum Mutat 30: 1123–1133, 2009 [DOI] [PubMed] [Google Scholar]
- 4.Boulden BM, Widder JD, Allen JC, Smith DA, Al-Baldawi RN, Harrison DG, Dikalov SI, Jo H, Dudley SC., Jr Early determinants of H2O2-induced endothelial dysfunction. Free Radic Biol Med 41: 810–817, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Costa AD, Garlid KD. Intramitochondrial signaling: interactions among mitoKATP, PKCε, ROS, and MPT. Am J Physiol Heart Circ Physiol 295: H874–H882, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa AD, Quinlan CL, Andrukhiv A, West IC, Jaburek M, Garlid KD. The direct physiological effects of mitoKATP opening on heart mitochondria. Am J Physiol Heart Circ Physiol 290: H406–H415, 2006 [DOI] [PubMed] [Google Scholar]
- 7.Dikalov S. Cross talk between mitochondria and NADPH oxidases. Free Radic Biol Med 51: 1289–1301, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 49: 717–727, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dikalov SI, Dikalova AE, Bikineyeva AT, Schmidt HH, Harrison DG, Griendling KK. Distinct roles of Nox1 and Nox4 in basal and angiotensin II-stimulated superoxide and hydrogen peroxide production. Free Radic Biol Med 45: 1340–1351, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dikalov SI, Li W, Doughan AK, Blanco RR, Zafari AM. Mitochondrial reactive oxygen species and calcium uptake regulate activation of phagocytic NADPH oxidase. Am J Physiol Regul Integr Comp Physiol 302: R1134–R1142, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112: 2668–2676, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 107: 106–116, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dikalova AE, Gongora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Heart Circ Physiol 299: H673–H679, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II mediated mitochondrial dysfunction. Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 102: 488–496, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Drose S, Brandt U, Hanley PJ. K+-independent actions of diazoxide question the role of inner membrane KATP channels in mitochondrial cytoprotective signaling. J Biol Chem 281: 23733–23739, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Guzik TJ, Chen W, Gongora MC, Guzik B, Lob HE, Mangalat D, Hoch N, Dikalov S, Rudzinski P, Kapelak B, Sadowski J, Harrison DG. Calcium-dependent NOX5 NADPH oxidase contributes to vascular oxidative stress in human coronary artery disease. J Am Coll Cardiol 52: 1803–1809, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanley PJ, Mickel M, Loffler M, Brandt U, Daut J. K(ATP) channel-independent targets of diazoxide and 5-hydroxydecanoate in the heart. J Physiol 542: 735–741, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harrison D, Griendling KK, Landmesser U, Hornig B, Drexler H. Role of oxidative stress in atherosclerosis. Am J Cardiol 91: 7A–11A, 2003 [DOI] [PubMed] [Google Scholar]
- 19.Harrison DG, Cai H, Landmesser U, Griendling KK. Interactions of angiotensin II with NAD(P)H oxidase, oxidant stress and cardiovascular disease. J Renin Angiotensin Aldosterone Syst 4: 51–61, 2003 [DOI] [PubMed] [Google Scholar]
- 20.Hirst J, King MS, Pryde KR. The production of reactive oxygen species by complex I. Biochem Soc Trans 36: 976–980, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Hoch NE, Guzik TJ, Chen W, Deans T, Maalouf SA, Gratze P, Weyand C, Harrison DG. Regulation of T-cell function by endogenously produced angiotensin II. Am J Physiol Regul Integr Comp Physiol 296: R208–R216, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, Dikalov S, Giddens DP, Griendling KK, Harrison DG, Jo H. Oscillatory shear stress stimulates endothelial production of O2- from p47phox-dependent NAD(P)H oxidases, leading to monocyte adhesion. J Biol Chem 278: 47291–47298, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Katakam PV, Wappler EA, Katz PS, Rutkai I, Institoris A, Domoki F, Gaspar T, Grovenburg SM, Snipes JA, Busija DW. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol 33: 752–759, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim MY, Kim MJ, Yoon IS, Ahn JH, Lee SH, Baik EJ, Moon CH, Jung YS. Diazoxide acts more as a PKC-epsilon activator, and indirectly activates the mitochondrial K(ATP) channel conferring cardioprotection against hypoxic injury. Br J Pharmacol 149: 1059–1070, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krege JH, Hodgin JB, Hagaman JR, Smithies O. A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 25: 1111–1115, 1995 [DOI] [PubMed] [Google Scholar]
- 26.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30: 653–661, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res 93: 573–580, 2003 [DOI] [PubMed] [Google Scholar]
- 28.Mehranpour P, Wang SS, Blanco RR, Li W, Song Q, Lassegue B, Dikalov SI, Austin H, Zafari AM. The C242T CYBA polymorphism as a major determinant of NADPH oxidase activity in patients with cardiovascular disease. Cardiovasc Hematol Agents Med Chem 7: 251–259, 2009 [DOI] [PubMed] [Google Scholar]
- 29.Mukhopadhyay P, Rajesh M, Hasko G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc 2: 2295–2301, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mukhopadhyay P, Rajesh M, Yoshihiro K, Hasko G, Pacher P. Simple quantitative detection of mitochondrial superoxide production in live cells. Biochem Biophys Res Commun 358: 203–208, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murdoch CE, Alom-Ruiz SP, Wang M, Zhang M, Walker S, Yu B, Brewer A, Shah AM. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res Cardiol 106: 527–538, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murillo I, Henderson LM. Expression of gp91phox/Nox2 in COS-7 cells: cellular localization of the protein and the detection of outward proton currents. Biochem J 385: 649–657, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol 47: 629–656, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Nazarewicz RR, Bikineyeva A, Dikalov SI. Rapid and specific measurements of superoxide using fluorescence spectroscopy. J Biomol Screen 18: 498–503, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404: 787–790, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Page IH. The mosaic theory of arterial hypertension—its interpretation. Perspect Biol Med 10: 325–333, 1967 [DOI] [PubMed] [Google Scholar]
- 37.Panov A, Dikalov S, Shalbuyeva N, Hemendinger R, Greenamyre JT, Rosenfeld J. Species- and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am J Physiol Cell Physiol 292: C708–C718, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Panov A, Dikalov S, Shalbuyeva N, Taylor G, Sherer T, Greenamyre JT. Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem 280: 42026–42035, 2005 [DOI] [PubMed] [Google Scholar]
- 39.Panov A, Schonfeld P, Dikalov S, Hemendinger R, Bonkovsky HL, Brooks BR. The neuromediator glutamate, through specific substrate interactions, enhances mitochondrial ATP production and reactive oxygen species generation in nonsynaptic brain mitochondria. J Biol Chem 284: 14448–14456, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panov AV, Vavilin VA, Lyakhovich VV, Brooks BR, Bonkovsky HL. Effect of bovine serum albumin on mitochondrial respiration in the brain and liver of mice and rats. Bull Exp Biol Med 149: 187–190, 2010 [DOI] [PubMed] [Google Scholar]
- 41.Pomerantz BJ, Robinson TN, Heimbach JK, Calkins CM, Miller SA, Banerjee A, Harken AH. Selective mitochondrial KATP channel opening controls human myocardial preconditioning: too much of a good thing? Surgery 128: 368–373, 2000 [DOI] [PubMed] [Google Scholar]
- 42.Queliconi BB, Wojtovich AP, Nadtochiy SM, Kowaltowski AJ, Brookes PS. Redox regulation of the mitochondrial K(ATP) channel in cardioprotection. Biochim Biophys Acta 1813: 1309–1315, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson KM, Janes MS, Beckman JS. The selective detection of mitochondrial superoxide by live cell imaging. Nat Protoc 3: 941–947, 2008 [DOI] [PubMed] [Google Scholar]
- 44.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci USA 103: 15038–15043, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.San Martin A, Foncea R, Laurindo FR, Ebensperger R, Griendling KK, Leighton F. Nox1-based NADPH oxidase-derived superoxide is required for VSMC activation by advanced glycation end-products. Free Radic Biol Med 42: 1671–1679, 2007 [DOI] [PubMed] [Google Scholar]
- 46.Selivanov VA, Zeak JA, Roca J, Cascante M, Trucco M, Votyakova TV. The role of external and matrix pH in mitochondrial reactive oxygen species generation. J Biol Chem 283: 29292–29300, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sorescu GP, Song H, Tressel SL, Hwang J, Dikalov S, Smith DA, Boyd NL, Platt MO, Lassegue B, Griendling KK, Jo H. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based NADPH oxidase. Circ Res 95: 773–779, 2004 [DOI] [PubMed] [Google Scholar]
- 48.Touyz RM, Yao G, Schiffrin EL. c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 23: 981–987, 2003 [DOI] [PubMed] [Google Scholar]
- 49.Treberg JR, Quinlan CL, Brand MD. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I). J Biol Chem 286: 27103–27110, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol 264: 484–509, 1996 [DOI] [PubMed] [Google Scholar]
- 51.Ushio-Fukai M, Griendling KK, Becker PL, Hilenski L, Halleran S, Alexander RW. Epidermal growth factor receptor transactivation by angiotensin II requires reactive oxygen species in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 21: 489–495, 2001 [DOI] [PubMed] [Google Scholar]
- 52.Vadziuk OB. [Effects of diazoxide on the mitochondrial membrane potential and ROS generation in rat uterus cells]. Fiziol Zh 58: 86–92, 2012 [PubMed] [Google Scholar]
- 53.Widder JD, Guzik TJ, Mueller CF, Clempus RE, Schmidt HH, Dikalov SI, Griendling KK, Jones DP, Harrison DG. Role of the multidrug resistance protein-1 in hypertension and vascular dysfunction caused by angiotensin II. Arterioscler Thromb Vasc Biol 27: 762–768, 2007 [DOI] [PubMed] [Google Scholar]
- 54.Wosniak J, Jr, Santos CX, Kowaltowski AJ, Laurindo FR. Cross-talk between mitochondria and NADPH oxidase: effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid Redox Signal 11: 1265–1278, 2009 [DOI] [PubMed] [Google Scholar]
- 55.Wu JL, Wu QP, Yang XF, Wei MK, Zhang JM, Huang Q, Zhou XY. L-malate reverses oxidative stress and antioxidative defenses in liver and heart of aged rats. Physiol Res 57: 261–268, 2008 [DOI] [PubMed] [Google Scholar]
- 56.Zhang A, Jia Z, Wang N, Tidwell TJ, Yang T. Relative contributions of mitochondria and NADPH oxidase to deoxycorticosterone acetate-salt hypertension in mice. Kidney Int 80: 51–60, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang DX, Chen YF, Campbell WB, Zou AP, Gross GJ, Li PL. Characteristics and superoxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channels. Circ Res 89: 1177–1183, 2001 [DOI] [PubMed] [Google Scholar]
- 58.Zielonka J, Hardy M, Kalyanaraman B. HPLC study of oxidation products of hydroethidine in chemical and biological systems: ramifications in superoxide measurements. Free Radic Biol Med 46: 329–338, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zinkevich NS, Gutterman DD. ROS-induced ROS release in vascular biology: redox-redox signaling. Am J Physiol Heart Circ Physiol 301: H647–H653, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]