

Abstract
Over 26,000 manuscripts have been published dealing with endothelins since their discovery 25 years ago. These peptides, and particularly endothelin-1 (ET-1), are expressed by, bind to, and act on virtually every cell type in the body, influencing multiple biological functions. Among these actions, the effects of ET-1 on arterial pressure and volume homeostasis have been most extensively studied. While ET-1 modulates arterial pressure through regulation of multiple organ systems, the peptide's actions in the kidney in general, and the collecting duct in particular, are of unique importance. The collecting duct produces large amounts of ET-1 that bind in an autocrine manner to endothelin A and B receptors, causing inhibition of Na+ and water reabsorption; absence of collecting duct ET-1 or its receptors is associated with marked salt-sensitive hypertension. Collecting duct ET-1 production is stimulated by Na+ and water loading through local mechanisms that include sensing of salt and other solute delivery as well as shear stress. Thus the collecting duct ET-1 system exists, at least in part, to detect alterations in, and maintain homeostasis for, extracellular fluid volume. Derangements in collecting duct ET-1 production may contribute to the pathogenesis of genetic hypertension. Blockade of endothelin receptors causes fluid retention due, in large part, to inhibition of the action of ET-1 in the collecting duct; this side effect has substantially limited the clinical utility of this class of drugs. Herein, the biology of the collecting duct ET-1 system is reviewed, with particular emphasis on key issues and questions that need addressing.
Keywords: nephron, epithelial sodium channel, water, arterial pressure
in honor of Ernest H. Starling, this review encompasses part of the 2013 Ernest Starling Lectureship of the American Physiological Society Water and Electrolyte Section and so it is appropriate at the outset to acknowledge how Dr. Starling, who embarked on his career in physiology 125 years ago, influenced physiology research to the extent that we continue to honor his memory. A short introduction does not begin to do justice to his accomplishments; however, hopefully these brief comments will be illustrative of his influence on all of us who pursue physiology research. Dr. Starling was a renaissance man of physiology, making key discoveries with regard to the heart, vasculature, lymphatics, endocrine system, gut, and kidney. With respect to the latter, together with his colleagues, he pioneered the isolated kidney and demonstrated that glomeruli form a filtrate largely devoid of protein and described processes of tubule cell secretion (urea) and reabsorption (water, chloride, glucose, and bicarbonate). He introduced the concept of circulating hormones and suggested that these could control renal excretion of specific electrolytes. On a more personal level, three aspects of his character struck me that were noted in a 7-page obituary in the British Medical Journal written in 1927 (57a). The first was that, while he was very interested in clinical medicine, he was “determined to try the rougher path and become demonstrator in physiology” despite the fact that “the [financial] rewards of this office were minute”! Second, although “he had not a natural gift of oratory,” he excelled at teaching research, extending help and encouragement “provided [the student] was a serious inquirer after truth.” Third, he had “extraordinary mental alertness and boyish enthusiasm; like Peter Pan, he refused to grow old.” Thus, on multiple levels, it is important and timely to acknowledge Dr. Starling; he remains an inspiration to make a difference, through both research and teaching, and to remain passionate.
Introduction to the Collecting Duct Endothelin System
The collecting duct (CD) endothelin (ET) system was initially described almost 25 years ago; it is now apparent that this system is an important physiological regulator of renal Na+ and water transport and arterial pressure (50). The CD produces and binds more ET than possibly any other cell type in the body. CD ET production is increased by Na+ and/or water loading, leading to autocrine inhibition of CD Na+ and water reabsorption, thereby facilitating maintenance of volume homeostasis. In addition, CD-derived ET may exert paracrine effects on neighboring cells. Derangements of CD ET production may contribute to the pathogenesis of hypertension, whereas blockade of ET action in the CD by ET receptor antagonists is likely important in their propensity to cause fluid retention. This review focuses exclusively on the CD ET system in the control of volume homeostasis and blood pressure; however, it should be noted the ET is involved in the control of multiple systems that affect Na+ and water balance (for a detailed review of such regulation, see Ref. 50).
Overview of Endothelin Biochemistry
The ET gene family consists of three 21-amino acid peptides: ET-1, ET-2, and ET-3. This review focuses on ET-1; it is the most abundant ET isoform and its biology has been most extensively studied. ET-1 synthesis is largely controlled at the transcription level (110); ET-1 mRNA has a short half-life (∼15 min) due to a destabilizing AUUUA sequence in the 3′-untranslated region; this brief mRNA half-life is reflected in the very tight association between ET-1 mRNA and protein levels (Fig. 1). ET-1 mRNA encodes a prepropeptide that undergoes removal of a short signal sequence by furin and other enzymes, followed by dibasic-pair-specific endopeptidase cleavage to yield the 38 amino acid Big ET-1 (58, 111). Big ET-1 is converted to ET-1 primarily by three ET-converting enzymes with varying subcellular localizations, including the plasma membrane (15, 97). ET-1 is substantially degraded by neutral endopeptidases that are abundantly expressed in the kidney (15, 98). Secreted ET-1 primarily acts in an autocrine or paracrine manner; tissue concentrations are much higher than those in plasma.
Fig. 1.
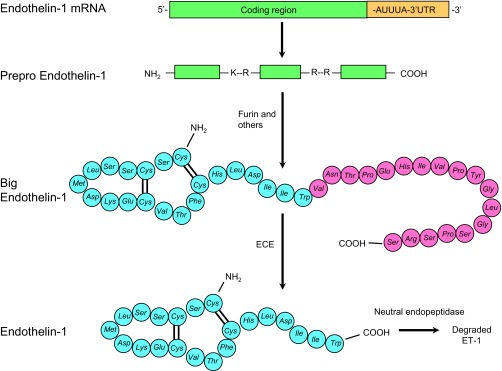
Synthesis and degradation of endothelin-1 (ET-1). ET-1 mRNA encodes prepro-ET-1. The signal peptide is cleaved to yield pro-ET-1, which is cleaved by furin and other convertases at dibasic amino acids to yield Big ET-1. Big ET-1 is cleaved by ET-converting enzymes (ECE) to mature ET-1. ET-1 is degraded by neutral endopeptidases and other enzymes.
Two receptors mediate the biological actions of ET: ETA and ETB. One or both of these receptors are found on virtually every cell type in the body. ETA binds ET-1 ≥ ET-2 >> ET-3 (34), whereas ETB binds all ET isoforms with equal affinity (2, 74). ETB, possibly by virtue of targeting to lysosomes, can function as a clearance receptor (8). Initial descriptions of ETA and ETB suggested they had opposite biological effects, i.e., ETA mediated vasoconstriction, proliferation, and fibrosis, while ETB did the opposite. However, it is now apparent that these receptors also can have similar and even interacting effects, the latter potentially through heterodimerization (20). A key feature of ET-1 binding is its very tight and prolonged association with its receptors (even after endocytosis), leading to long-lasting effects (13).
Regulation of Collecting Duct ET Production
ET-1 was originally described as an endothelium-derived factor. However, it is evident that ET-1 is made by almost all cell types and that the renal inner medulla likely has more ET-1 than any other region in the body (41). Ultimately, it was discovered that all regions of the CD produce relatively large amounts of ET-1 (43). Within the CD, the inner medullary CD (IMCD) produces the greatest amount of ET-1, followed by the outer medullary CD and then the cortical CD (CCD). Whereas intercalated cell ET-1 production has not been directly studied, it is likely that most CD ET-1 derives from principal cells (49). CD cells predominantly release ET-1 on the basolateral side (46, 92).
Under physiological conditions, CD ET-1 production is regulated by factors associated with extracellular fluid volume (ECFV) status. Water and/or Na+ loading in humans and/or experimental animals increases urinary ET-1 (1, 14, 35, 37, 39, 47, 56, 59, 73, 76, 108, 115), renal medullary ET-1 mRNA (1, 21), and acutely isolated CD ET-1 production (55). Mice with CD ET-1 knockout (KO) have markedly reduced urinary ET-1 excretion in response to salt loading (1). The mechanisms by which ECFV expansion augments CD ET-1 are now being uncovered. First, it is evident that circulating hormones, insofar as has been determined, do not account for this relationship. Vasopressin and atrial natriuretic peptide do not substantially affect CD ET-1 production, at least in vitro (44, 60, 93). Aldosterone increases CD ET-1 synthesis, an effect that is mediated by disrupter of telomeric silencing alternative splice variant (Dot1a)-Af9-induced histone hypomethylation and derepression of ET-1 gene expression (116) (Fig. 2); clearly, aldosterone cannot account for volume expansion induction of CD ET-1 production. Another possibility is medullary Na+ concentration; increased interstitial tonicity augments thick ascending limb ET-1 production (32). However, the data on this in CD are conflicting, showing that, at least in vitro, increasing media NaCl concentration increases (60, 93, 112) or decreases (47, 77, 93) CD ET-1 synthesis. Furthermore, increases in interstitial tonicity cannot explain how both Na+ and water loading increase CD ET-1 production. Nephron flow increases during Na+ and/or water loading; recent studies point to nephron flow as an important regulator of CD ET-1 production (Figs. 2 and 3). Laminar flow increased ET-1 mRNA content in a CD cell line (mpkCCDcl4) at shear stresses between 2 and 10 dyn/cm2; i.e., at flows comparable to those observed in vivo (55). The flow response was dependent on intracellular and extracellular Ca2+, phospholipase C (PLC), and protein kinase C (PKC). A subsequent study (67) found that increasing perfusate NaCl concentration increased the ET-1 flow response in mpkCCDcl4 cells and that this effect was due to Na+ but not Cl− or osmolality. The ET-1 flow response was dependent on the epithelial Na+ channel (ENaC), being inhibited by amiloride or benzamil and stimulated by aldosterone. Blockade of mitochondrial, but not plasma membrane, Na+/Ca2+ exchangers (NCX) abolished the ET-1 flow response. Taken together, these findings suggest that during ECFV expansion, increased CD Na+ delivery augments CD ET-1 synthesis via ENaC-mediated Na+ entry, mitochondrial NCX activity, and activation of PLC and PKC. One problem with this scenario is that it does not explain how water loading stimulates CD ET-1 synthesis wherein CD Na+ delivery may not necessarily be increased. To address this, mouse IMCD3 cells were studied (unpublished data; M. Pandit and D. Kohan). Interestingly, flow markedly increased IMCD3 ET-1 mRNA content, but this response was not ENaC dependent. Furthermore, increasing perfusate solute concentration, regardless of the solute used, greatly increased the IMCD3 ET-1 flow response (but in the absence of flow, did not affect ET-1 production). Thus ET-1 production in the CCD and IMCD may be differentially regulated by flow, with Na+ delivery and uptake being the prime determinants of CCD ET-1 production, whereas IMCD ET-1 synthesis is controlled by a combination of water and solute delivery.
Fig. 2.
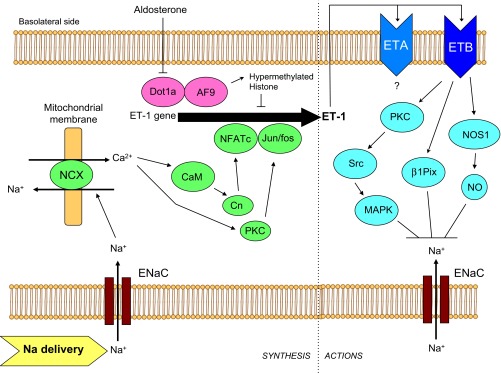
Model of ET-1 synthesis and actions in the cortical collecting duct. Increased tubule fluid Na+ delivery increases intracellular Na+ concentration via the epithelial Na+ channel (ENaC), leading to mitochondrial Na+/Ca2+ exchange (NCX), activating calmodulin (CaM), calcineurin (Cn), and protein kinase C (PKC), which lead to transactivation of the ET-1 promoter via Ca2+-dependent nuclear factor of activated T cells (NFATc) and Jun/fos. ET-1 is secreted primarily basolaterally where it can activate ETA and ETB receptors in an autocrine manner. Activation of ETB leads to increased protein kinse C (PKC) activity, increased Src tyrosine kinase, and increased mitogen-activated protein kinase (MAPK) activities, which inhibits epithelial Na+ channel (ENaC). ETB also activates β1Pix, which ultimately causes endocytosis of ENaC. In addition, ETB stimulates nitric oxide synthase 1 (NOS1), increasing NO which inhibits ENaC activity. ETA may also inhibit CD Na+ transport, but the signaling mechanisms are unknown. Finally, aldosterone can stimulate ET-1 production by repressing disrupter of telomeric silencing alternative splice variant (Dot1a), reducing interaction with AF9, and derepressing ET-1 gene transcription. Please note that the model is drawn from studies using disparate cell types under different experimental conditions, hence this should be viewed as a conceptual overview.
Fig. 3.
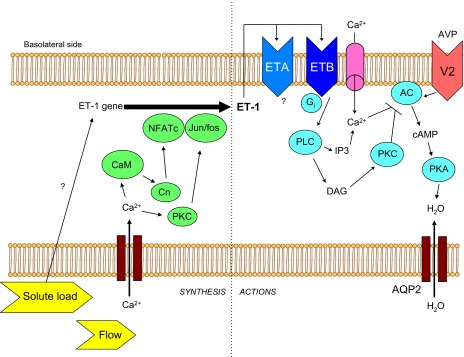
Model of ET-1 synthesis and actions in the inner medullary collecting duct. Increased tubule fluid flow increases Ca2+ entry into cells, activating calmodulin (CaM) and PKC. Ultimately, this leads to transactivation of the ET-1 promoter via NFATc and Jun/fos. Increased tubule fluid solute load also increases ET-1 synthesis, although the mechanism is unknown. ET-1 is secreted primarily basolaterally where it can activate ETA and ETB receptors in an autocrine manner. Activation of ETB leads to increased phospholipase C (PLC) activity, which increases diacylglycerol (DAG), activates PKC which inhibits vasopressin (AVP)-V2 receptor-stimulated adenylyl cyclase (AC) activity. In addition, ETB increases Ca2+ entry into cells and augments PLC production of inositol trisphosphate (IP3), leading to further increases in intracellular Ca2+ concentration; Ca2+ can inhibit AC activity as well. Inhibition of AC-dependent cAMP production leads to reduced protein kinase A (PKA)-mediated phosphorylation of aquaporin-2 (AQP2) and reduced water reabsorption. Please note that the model is drawn from studies using disparate cell types under different experimental conditions, hence this should be viewed as a conceptual overview.
The intracellular signaling pathways through which CD ET-1 synthesis is regulated are complex and may depend, as discussed above, on the region of the CD studied. Studies using primary rat IMCD cultures point to the central importance of Ca2+-regulated signaling pathways, including intracellular Ca2+, calmodulin, calmodulin kinase, and calcineurin (79) (Figs. 2 and 3). Rat ET-1 promoter-reporter analysis revealed a calmodulin-sensitive region between 0.56 and 1.9 kb 5′ to the transcription start site in IMCD but not endothelial cells (79). Two consensus nuclear factor of activated T-cells (NFAT) binding sites were identified at −1263 and −1563 5′ to the transcriptional start site which, upon mobility shift analysis, revealed that ET-1263 bound NFATc1 while ET-1563 bound NFATc3 (80). Site-directed mutagenesis of either the ET-1263 or ET-1563 sites prevented NFAT binding and reduced ET-1 promoter activity. Finally, PKC is also important in modulation of IMCD ET-1 production and promoter activity (82). Thus Ca2+, calmodulin, calmodulin kinase, calcineurin, and Ca2+-dependent NFAT regulate CD ET-1 transcription; how these pathways relate to the ET-1 flow response and whether differences exist between regions of the CD remains to be fully determined.
Expression and Regulation of Collecting Duct ET Receptors
Numerous studies involving multiple species and techniques consistently demonstrate that the CD is the major renal site of ET receptor expression (16, 101, 103, 113). As for ET-1 production, the highest binding of ET-1 is in IMCD, followed by OMCD and CCD (88). ETB is the major ET receptor isoform expressed by the CD in experimental animals and humans (3, 12, 40, 51, 88, 90, 102). Although more difficult to demonstrate, CD also express ETA (11, 18, 45, 65, 102, 113). Most recently, ETA immunoreactivity was detected in the basal infoldings of the CD, with the strongest staining in the IMCD (109). While not extensively studied, CD cells appear to bind ET-1 predominantly on the basolateral side (46); since, as described above, ET-1 is predominantly secreted basolaterally, the potential for autocrine regulation of CD function is apparent.
Relatively little is known about regulation of CD ET receptor expression. Vasopressin (AVP) reduces cultured rat CCD and IMCD ETB-dependent ET-1 binding (87, 105). Similarly, angiotensin II decreases ETA and ETB expression in rat IMCD cells (104). Notably, IMCD ETB expression is decreased in animal models of congestive heart failure (22, 106), and this downregulation is prevented by angiotensin receptor blockade (106). Thus the possibility exists that AVP and angiotensin II reduce IMCD ET receptor expression to mitigate the natriuretic effects of the ET system (see discussion in Collecting Duct ET Regulation of Na+ Transport).
Collecting Duct ET Regulation of Na+ Transport
Initial studies found that ET-1 inhibits Na+-K+-ATPase activity in rabbit IMCD (114); however, all subsequent work has focused on ET-1 regulation of ENaC. Exogenous ET-1 has consistently been demonstrated to potently inhibit ENaC-dependent Na+ reabsorption in isolated CCD or CD cell lines (10, 23, 29, 52, 94) (Fig. 2). Most importantly, CD-specific deletion of ET-1 increases systolic blood pressure (BP) by ∼15 mmHg during normal Na+ intake and by ∼35 mmHg during high Na+ intake (1). The CD ET-1 KO mice had excessive weight gain and reduced urinary Na+ excretion. All of these effects of CD ET-1 KO mice were reduced by amiloride. In addition, CD ET-1 KO mice had impaired elevations in Na+ excretion in response to controlled increases in renal perfusion pressure (76). Thus CD-derived ET-1 functions as an autocrine inhibitor of CD Na+ reabsorption. Why have an autocrine system in the CD that reduces Na+ reabsorption? One possibility is that CD ET-1, at least with respect to Na+ transport, is important in maintaining normal ECFV status. As discussed earlier, Na+ loading causes increased CD luminal flow and Na+ delivery. If unopposed, this could lead to increased CD Na+ reabsorption; nephron fluid flow increases ENaC open probability (Po) in the CCD and can augment CD Na+ reabsorption (75). Thus, by the mechanisms described above wherein ENaC-mediated Na+ uptake increases CD ET-1 production, it may be that the CD ET-1 system exists, at least in part, to sense an increase in CD Na+ delivery and mitigate against the inherent tendency of ENaC to enhance CD Na+ reabsorption. In this regard, it is likely that ET-1 works in concert with other locally derived factors to reduce ENaC activity during salt loading; for example, salt loading increases the activation of P2Y2 receptors in the CD, which in turn inhibits ENaC activity (69).
Studies in cultured cells and in isolated CCD implicate ETB in mediating ET-1 inhibition of CD Na+ transport (9, 10, 23) (Fig. 2). Mice with CD-specific ETB KO have an ∼8 mmHg elevation in systolic BP on a normal Na+ diet and an ∼20 mmHg increase in systolic BP during high-salt intake associated with an impaired natriuresis (25). Notably, the degree of hypertension in CD ETB KO mice was about one-half that observed in CD ET-1 KO mice, suggesting that CD ETB receptors do not fully account for all the antihypertensive effects of CD-derived ET-1. The data on ETA-mediated inhibition of CD Na+ transport is more complex. Studies in isolated split-open CCD did not report an effect of ETA on ENaC activity (9, 10). Similarly, mice with CD-specific ETA KO did not have altered BP or urinary Na+ excretion (28). However, mice with combined CD-specific ETA and ETB KO had systolic BP on normal and high-Na+ diet that were greater, and urinary Na+ excretion that was lower, than those seen with CD ETB KO alone and were similar to those observed in CD ET-1 KO mice (76). While interpretation of gene targeting studies is complicated by possible compensatory mechanisms, one possibility is that CD ETA and ETB receptors interact in some as yet undefined manner. In this regard, a recent study found that ET-1 inhibition of transepithelial Na+ flux in isolated perfused CCD was abolished by either ETA or ETB blockade, suggesting that both ET receptor isoforms may affect CD Na+ transport (54). In addition, nephron-wide knockout of ETA receptors caused mild fluid volume retention, although the precise cell type(s) responsible was not identified (84). As will be discussed, these considerations about the biological actions of ET receptors in the CD have taken on clinical relevance.
Several signaling pathways are involved in ET-1 modulation of CD Na+ transport (Fig. 2). First, Src and mitogen-activated protein (MAPK) kinases have been implicated. ETB-mediated inhibition of ENaC Po is Src kinase dependent as demonstrated in cultured cells and isolated rat CCD (10, 29). In addition, ETB activates MAPK in the CD (91) and blockade of MAPK abolishes ET-1 inhibition of ENaC in the CCD (10). Second, nitric oxide (NO) is likely involved. ET-1 increases NO synthase-1 (NOS1) expression and NO production in cultured IMCD cells (81, 85). CD ET-1 KO mice have reduced urinary nitrate/nitrite excretion and lower inner medullary NOS1 and NOS3 activities, while systemic NOS inhibition has a markedly blunted hypertensive effect in these mice (76). A recent study found that CD-specific KO of NOS1 caused a hypertensive and Na+-retaining phenotype virtually identical to that observed in CD ET-1 KO mice (38). Since NO has been reported to reduce Na+ flux in CCD (66) and ENaC Po in CD cells (31), these data strongly suggest that NO mediates, at least partly, ET-1 effects on ENaC. Third, ET-1 activates β1-Pix, which interacts with 14–3-3β to impair interaction with Nedd4–1, leading to increased endocytosis and degradation of ENaC (68). Finally, although ET-1 can increase CD PGE2 production (48, 114), in vivo studies do not support a role for cyclooxygenase metabolites in mediating the natriuretic effect of ET-1 in the CD (27).
Collecting Duct ET-1 Regulation of Water Transport
ET-1 dose-dependently inhibits AVP-stimulated osmotic water permeability in rat CCD and IMCD in vitro (17, 62, 64, 94) (Fig. 3). CD ET-1 KO mice have impaired excretion of an acute water load associated with enhanced sensitivity to exogenous and endogenous AVP (24). ET-1 inhibition of AVP-stimulated CD water transport is likely due, in large part, to reduced adenylyl cyclase-dependent cAMP accumulation and is independent of phosphodiesterase activity (17, 48, 60, 62, 64, 88, 94, 95). Anti-ET-1 antibodies increased AVP-stimulated cAMP accumulation by IMCD cells, suggesting that ET-1 is an autocrine inhibitor of AVP action (46). Similarly, IMCD from CD ET-1 KO mice have increased AVP-stimulated cAMP accumulation (24). These effects of ET-1 are likely mediated by ETB since ET-1, ET-3, and S6c (the latter two are relatively ETB-specific agonists) were equipotent in inhibiting AVP-induced cAMP accumulation in IMCD, while ETA blockade was without effect (17, 48, 107).
The inhibitory effect of ET-1 on AVP-dependent cAMP accumulation and water transport in the IMCD is mediated through Gi (48, 62) and PKC (48, 62, 95, 96, 107) (Fig. 3). In addition, Ca2+ is likely of central importance. ET-1 causes an initial rapid rise, followed by a sustained elevation, in CD intracellular Ca2+ concentration ([Ca2+]i) (52, 60, 63). While the data are somewhat conflicting, the bulk of evidence supports the notion that ET-1 causes a biphasic increase in [Ca2+]i wherein the initial transient peak is mainly caused by release of Ca2+ from cell stores while the sustained rise is primarily due to Ca2+ entry via dihydropyridine-sensitive channels (50, 52, 62, 63). The initial rapid rise in [Ca2+]i may be the major Ca2+ component involved in ET-1 modulation of cAMP since increased [Ca2+]i inhibits AVP-stimulated adenylyl cyclase activity in rat IMCD through PLC-mediated activation of PKC (89). In contrast, dihydropyridine treatment did not alter AVP-stimulated cAMP accumulation in rat CCD, OMCD, or IMCD (95, 96). Finally, cyclooxygenase blockade did not affect ET-1 inhibition of AVP-stimulated cAMP accumulation in rat CCD, OMCD, or IMCD (48, 95, 96), whereas neither NOS inhibition nor NO donors altered ET-1 inhibition of AVP-stimulated cAMP content in IMCD suspensions (81).
Collecting Duct ET-1 as a Paracrine Factor
Collecting duct-derived ET-1 could potentially affect neighboring blood vessels or tubules; however, direct evidence for this is lacking. An interesting finding in the CD ET-1 KO mice was that plasma renin activity and aldosterone levels were not suppressed as would be anticipated (given that the hypertension and Na+ retention are due, at least in part, to increased CD Na+ reabsorption) (1, 26). Angiotensin or mineralocorticoid receptor antagonism normalized BP in CD ET-1 KO mice during normal Na+ intake and partly corrected the hypertension during high Na+ intake, suggesting that relative overactivity of the renin-angiotensin-aldosterone axis may be of pathogenic importance in this mouse model. Since ET-1 potently inhibits juxtaglomerular renin production (19, 53, 61, 71, 72, 86), and since the CCD and connecting segment are anatomically adjacent to the juxtaglomerular apparatus and afferent arteriole (70, 99), it is tempting to speculate that CD-derived ET-1 has the potential to modulate systemic renin. Studies are needed to address this interesting possibility.
Collecting Duct ET-1 and Genetic Hypertension
Although relatively little is known, the available evidence suggests that CD ET-1 production may be reduced in genetic models of hypertension. Acutely isolated or cultured IMCD released less ET-1 from hypertensive SHR compared with normotensive Wistar-Kyoto (WKY) rats, while spontanteously hypertensive (SHR) rats had less ET-1 in the inner medulla compared with WKY rats (30, 36, 42). In addition, medullary ET-1 content is reduced in the renal papilla from Prague hypertensive rats compared with normotensive controls (100), whereas medullary ET-1 levels are lower in Dahl S compared with Dahl R rats (78). Dietary salt intake increased urinary ET-1 excretion in salt-resistant, but not salt-sensitive, hypertensive subjects (33), whereas reduced urinary ET-1 excretion has been reported in patients with essential hypertension (33, 37, 117). Given that all of the human studies have involved small cohort sizes and it is not possible to know the degree to which CD ET-1 contributes to urinary ET-1 in humans, any inferences from these studies must be drawn with caution. In addition, if renal injury is present, urinary ET-1 will be elevated (occurs in virtually every form of renal injury due to cytokine/inflammatory mediator stimulation of renal ET-1 production), hence ideally patients should be evaluated before clinically apparent renal disease is present. Ultimately, the question arises that, even if CD ET-1 is suppressed in genetic hypertension, is this a cause or effect of the hypertension? Clearly, additional studies in this area involving both animal models and larger patient cohorts are needed.
ET-1 and ET Receptor Antagonist-Induced Fluid Retention
Endothelin-1 has a multitude of physiological and pathophysiological affects. In particular, overactivity of the ET-1 system has been implicated in the pathogenesis of a wide variety of diseases, including arterial hypertension, pulmonary artery hypertension, atherosclerosis, myocardial infarction, cancer, chronic proteinuric kidney disease, and others (7). Most studies in experimental models indicate that ETA is the primary mediator of the pathological effects of ET-1, including cell proliferation, hypertrophy, fibrosis, increased vascular resistance, insulin resistance, inflammation, and others (6). Large clinical trials have been conducted using ETA antagonists in congestive heart failure, arterial hypertension, and diabetic kidney disease; however, these trials have failed to demonstrate a clear benefit of ETA antagonism (5). A major factor limiting the clinical utility of ETA blockers is their propensity to cause fluid retention; this was the reason for discontinuing a Phase III trial in patients with diabetic nephropathy (ASCEND) (57). Given that ETB mediates ET-1 inhibition of Na+ reabsorption throughout the nephron (49), it is predictable that ETB blockade might promote fluid retention. Such ETB blockade may have occurred in some of the above trials due to the use of ET blockers at doses that were not ETA selective. However, trials using highly ETA-selective antagonists still demonstrated significant fluid retention (5).
Several possibilities for the etiology of ETA antagonist-induced fluid retention exist, including reduced cardiac contractility, systemic vasodilation, and renal fluid retention (83). To dissect out these possible mechanisms, mice were generated with site-specific deletion of ETA in cardiomyocytes, smooth muscle, the entire nephron, or the CD (83). After 2 wk of ambrisentan or atrasentan (relatively ETA-selective antagonists), wild-type mice and mice with cardiomyocyte ETA knockout retained more fluid than vehicle-treated mice. Mice with smooth muscle ETA knockout had reduced fluid retention in response to either ETA antagonist, while mice with nephron or CD duct ETA KO were completely protected against ETA blocker-induced fluid retention. Thus it appears that, at least in mice, blockade of CD ETA is a major cause of ETA antagonist-induced fluid retention. These findings may have relevance to humans, suggesting that diuretics that either target the CD or that act proximal to the CD but can overwhelm enhanced CD fluid reabsorption would be a rational choice to prevent and treat ETA blocker-induced fluid retention. However, as discussed earlier, additional studies are needed to define the cellular mechanisms by which ETA modulates CD Na+ and water transport.
Perspective and Significance
This review has examined the role of the CD ET system in the control of renal function and BP (Fig. 4). The CD produces and binds more ET-1 than any other region/cell type in the body. Collecting duct ET-1 production is stimulated by Na+ and/or water loading, leading to autocrine inhibition of CD Na+ and water reabsorption, and control of BP. This system likely exists, at least in part, to mitigate luminal flow-induced increases in CD Na+ and water reabsorption. Deficiency of CD ET-1 may contribute to the pathogenesis of genetic hypertension. Antagonism of ETA receptors causes fluid retention that is likely substantially due to blockade of ET-1 action in the CD; this side effect has limited the clinical utility of these agents. Further studies on CD ET-1 and its receptors are needed to shed light on the control of renal Na+ and water handling in health and disease; it is hoped that this will lead to improved strategies for treating a variety of diseases with ET receptor antagonists.
Fig. 4.

Integrated role of collecting duct ET-1 in control of renal Na+ and water transport during salt loading. A salt load increases extracellular fluid volume (ECFV) and tends to increase arterial pressure, leading to increases in renal blood flow (RBF) and renal interstitial hydrostatic pressure (RIHP), with resultant activation of signaling pathways that reduce Na+ and water reabsorption proximal to the CD and increase CD Na+ and water delivery. If unopposed, increased CD Na+ delivery would increase CD Na+ reabsorption; however, this is mitigated by flow and Na+ delivery-induced increases in CD ET-1, which inhibits CD Na+ and water transport, facilitating a natriuresis and diuresis.
GRANTS
The research by the author's laboratory described in this manuscript was supported by P01 HL-095499 and funds from AbbVie and Gilead.
DISCLOSURES
The author is a consultant for AbbVie and Retrophin.
AUTHOR CONTRIBUTIONS
Author contributions: D.E.K. conception and design of research; D.E.K. performed experiments; D.E.K. analyzed data; D.E.K. interpreted results of experiments; D.E.K. prepared figures; D.E.K. drafted manuscript; D.E.K. edited and revised manuscript; D.E.K. approved final version of manuscript.
ACKNOWLEDGMENTS
I recognize the individuals in my laboratory without whom none of these studies would have been possible. In addition, I acknowledge my collaborators, colleagues, and friends who continue to support and inspire.
REFERENCES
- 1.Ahn D, Ge Y, Stricklett PK, Gill P, Taylor D, Hughes AK, Yanagisawa M, Miller L, Nelson RD, Kohan DE. Collecting duct-specific knockout of endothelin-1 causes hypertension and sodium retention. J Clin Invest 114: 504–511, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arai H, Hori S, Aramori I, Ohkubo H, Nakanishi S. Cloning and expression of a cDNA encoding an endothelin receptor. Nature 348: 730–732, 1990 [DOI] [PubMed] [Google Scholar]
- 3.Backer A, Bokemeyer D, Kramer HJ. Endothelin synthesis and receptors in porcine kidney. Acta Physiol Scand 171: 105–112, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Barton M, Kohan DE. Endothelin antagonists in clinical trials: lessons learned. Contribut Nephrol 172: 255–260, 2011 [DOI] [PubMed] [Google Scholar]
- 6.Barton M, Yanagisawa M. Endothelin: 20 years from discovery to therapy. Can J Physiol Pharmacol 86: 485–498, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Battistini B, Berthiaume N, Kelland N, Webb D, Kohan D. Profile of past and current clinical trials involving endothelin receptor antagonists: the novel “-sentan” class of drug. Exp Biol Med (Maywood) 231: 653–695, 2006 [PubMed] [Google Scholar]
- 8.Bremnes T, Paasche JD, Mehlum A, Sandberg C, Bremnes B, Attramadal H. Regulation and intracellular trafficking pathways of the endothelin receptors. J Biol Chem 275: 17596–17604, 2000 [DOI] [PubMed] [Google Scholar]
- 9.Bugaj V, Mironova E, Kohan DE, Stockand JD. Collecting duct-specific endothelin B receptor knockout increases ENaC activity. Am J Physiol Cell Physiol 302: C188–C194, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bugaj V, Pochynyuk O, Mironova E, Vandewalle A, Medina JL, Stockand JD. Regulation of the epithelial Na+ channel by endothelin-1 in rat collecting duct. Am J Physiol Renal Physiol 295: F1063–F1070, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cernacek P, Legault L, Stewart DJ, Levy M. Specific endothelin binding sites in renal medullary collecting duct cells: lack of interaction with ANP binding and cGMP signalling. Can J Physiol Pharmacol 70: 1167–1174, 1992 [DOI] [PubMed] [Google Scholar]
- 12.Chow LH, Subramanian S, Nuovo GJ, Miller F, Nord EP. Endothelin receptor mRNA expression in renal medulla identified by in situ PT-PCR. Am J Physiol Renal Fluid Electrolyte Physiol 269: F449–F457, 1995 [DOI] [PubMed] [Google Scholar]
- 13.Chun M, Lin HY, Henis YI, Lodish HF. Endothelin-induced endocytosis of cell surface ETA receptors. J Biol Chem 270: 10855–10860, 1995 [DOI] [PubMed] [Google Scholar]
- 14.Cuzzola F, Mallamaci F, Tripepi G, Parlongo S, Cutrupi S, Cataliotti A, Stancanelli B, Malatino LS, Bellanuova I, Ferri C, Galletti F, Filigheddu F, Glorioso N, Strazzullo P, Zoccali C. Urinary adrenomedullin is related to ET-1 and salt intake in patients with mild essential hypertension. Am J Hypertens 14: 224–230, 2001 [DOI] [PubMed] [Google Scholar]
- 15.D'Orleans-Juste P, Plante M, Honore JC, Carrier E, Labonte J. Synthesis and degradation of endothelin-1. Can J Physiol Pharmacol 81: 503–510, 2003 [DOI] [PubMed] [Google Scholar]
- 16.Davenport AP, Nunez DJ, Brown MJ. Binding sites for 125I-labelled endothelin-1 in the kidneys: differential distribution in rat,pig and man demonstrated by using quantitative autoradiography. Clin Sci 77: 129–131, 1989 [DOI] [PubMed] [Google Scholar]
- 17.Edwards RM, Stack EJ, Pullen M, Nambi P. Endothelin inhibits vasopressin action in rat inner medullary collecting duct via the ETB receptor. J Pharmacol Exp Ther 267: 1028–1033, 1993 [PubMed] [Google Scholar]
- 18.Edwards RM, Trizna W. Characterization of 125I-endothelin-1 binding to rat and rabbit renal microvasculature. J Pharmacol Exper Ther 274: 1064–1089, 1995 [PubMed] [Google Scholar]
- 19.Endemann D, Schweda F, Stubanus M, Ittner KP, Fischereder M, Kammerl MC, Kramer BK. Naftidrofuryl exerts antiserotonergic but no endothelin-receptor blocking effects in AS4.1 cells, juxtaglomerular cells and isolated perfused rat kidneys. J Cardiovasc Pharmacol 39: 1–8, 2002 [DOI] [PubMed] [Google Scholar]
- 20.Evans NJ, Walker JW. Sustained Ca2+ signaling and delayed internalization associated with endothelin receptor heterodimers linked through a PDZ finger. Can J Physiol Pharmacol 86: 526–535, 2008 [DOI] [PubMed] [Google Scholar]
- 21.Fattal I, Abassi Z, Ovcharenko E, Shimada K, Takahashi M, Hoffman A, Winaver J. Effect of dietary sodium intake on the expression of endothelin-converting enzyme in the renal medulla. Nephron Physiol 98: 89–96, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Francis B, Abassi Z, Heyman S, Winaver J, Hoffman A. Differential regulation of ETA and ETB in the renal tissue of rats with compensated and decompensated heart failure. J Cardiovasc Pharmacol 44: S362–S365, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Gallego MS, Ling BN. Regulation of amiloride-sensitive sodium channels by endothelin-1 in distal nephron cells. Am J Physiol Renal Fluid Electrolyte Physiol 271: F451–F460, 1996 [DOI] [PubMed] [Google Scholar]
- 24.Ge Y, Ahn D, Stricklett PK, Hughes AK, Yanagisawa M, Verbalis JG, Kohan DE. Collecting duct-specific knockout of endothelin-1 alters vasopressin regulation of urine osmolality. Am J Physiol Renal Physiol 288: F912–F920, 2005 [DOI] [PubMed] [Google Scholar]
- 25.Ge Y, Bagnall A, Stricklett PK, Strait K, Webb DJ, Kotelevtsev Y, Kohan DE. Collecting duct-specific knockout of the endothelin B receptor causes hypertension and sodium retention. Am J Physiol Renal Physiol 291: F1274–F1280, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Ge Y, Huang Y, Kohan DE. Role of the renin-angiotensin-aldosterone system in collecting duct-derived endothelin-1 regulation of blood pressure. Can J Physiol Pharmacol 86: 329–336, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Ge Y, Strait KA, Stricklett PK, Yang T, Kohan DE. Role of prostaglandins in collecting duct-derived endothelin-1 regulation of blood pressure and water excretion. Am J Physiol Renal Physiol 293: F1805–F1810, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Ge Y, Stricklett PK, Hughes AK, Yanagisawa M, Kohan DE. Collecting duct-specific knockout of the endothelin A receptor alters renal vasopressin responsiveness, but not sodium excretion or blood pressure. Am J Physiol Renal Physiol 289: F692–F698, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Gilmore ES, Stutts MJ, Milgram SL. SRC family kinases mediate epithelial Na+ channel inhibition by endothelin. J Biol Chem 276: 42610–42617, 2001 [DOI] [PubMed] [Google Scholar]
- 30.Girchev R, Backer A, Markova P, Kramer H. Impaired response of the denervated kidney to endothelin receptor blockade in normotensive and spontaneously hypertensive rats. Kidney Int 65: 982–989, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Helms MN, Yu L, Malik B, Kleinhenz DJ, Hart CM, Eaton DC. Role of SGK1 in nitric oxide inhibition of ENaC in Na+-transporting epithelia. Am J Physiol Cell Physiol 289: C717–C726, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Herrera M, Garvin J. A high-salt diet stimulates thick ascending limb eNOS expression by raising medullary osmolality and increasing release of endothelin-1. Am J Physiol Renal Physiol 288: F58–F64, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Hoffman A, Grossman E, Goldstein DS, Gill JRJ, Keiser HR. Urinary excretion rate of endothelin-1 in patients with essential hypertension and salt sensitivity. Kidney Int 45: 556–560, 1994 [DOI] [PubMed] [Google Scholar]
- 34.Hosoda K, Nakao K, Tamura N, Arai H, Ogawa Y, Suga S, Nakanishi S, Imura H. Organization, structure, chromosomal assignment, and expression of the gene encoding the human endothelin-A receptor. J Biol Chem 267: 18797–18804, 1992 [PubMed] [Google Scholar]
- 35.Hsieh TJ, Lin SR, Lee YJ, Shin SJ, Lai YH, Hsu CH, Tsai JH. Increased renal medullary endothelin-1 synthesis in prehypertensive DOCA- and salt-treated rats. Am J Physiol Renal Physiol 279: F112–F121, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Hughes AK, Cline RC, Kohan DE. Alterations in renal endothelin production in the spontaneously hypertensive rat. Hypertension 20: 666–673, 1992 [DOI] [PubMed] [Google Scholar]
- 37.Hwang YS, Hsieh TJ, Lee YJ, Tsai JH. Circadian rhythm of urinary endothelin-1 excretion in mild hypertensive patients. Am J Hypertens 11: 1344–1351, 1998 [DOI] [PubMed] [Google Scholar]
- 38.Hyndman KA, Boesen EI, Elmarakby AA, Brands MW, Huang P, Kohan DE, Pollock DM, Pollock JS. Renal collecting cuct NOS1 maintains fluid-electrolyte homeostasis and blood pressure. Hypertension 62: 91–98, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jackson RW, Treiber FA, Harshfield GA, Waller JL, Pollock JS, Pollock DM. Urinary excretion of vasoactive factors are correlated to sodium excretion. Am J Hypertens 14: 1003–1006, 2001 [DOI] [PubMed] [Google Scholar]
- 40.Karet FE, Kuc RE, Davenport AP. Novel ligands BQ123 and BQ3020 characterize endothelin receptor subtypes ETA and ETB in human kidney. Kidney Int 44: 36–42, 1993 [DOI] [PubMed] [Google Scholar]
- 41.Kitamura K, Tanaka T, Kato J, Eto T, Tanaka K. Regional distribution of immunoreactive endothelin in porcine tissue: abundance in inner medulla of kidney. Biochem Biophys Res Commun 161: 348–352, 1989 [DOI] [PubMed] [Google Scholar]
- 42.Kitamura K, Tanaka T, Kato J, Qgawa Y, Eto T, Tanaka K. Immunoreactive endothelin in rat kidney inner medulla: marked decrease in spontaneously hypertensive rats. Biochem Biophys Res Commun 162: 38–44, 1989 [DOI] [PubMed] [Google Scholar]
- 43.Kohan D. Endothelins in the normal and diseased kidney. Am J Kidney Dis 29: 2–26, 1997 [DOI] [PubMed] [Google Scholar]
- 44.Kohan DE, Fiedorek FTJ. Endothelin synthesis by rat inner medullary collecting duct cells. J Am Soc Nephrol 2: 150–155, 1991 [DOI] [PubMed] [Google Scholar]
- 45.Kohan DE, Hughes AK, Perkins SP. Characterization of endothelin receptors in the inner medullary collecting duct of the rat. J Biol Chem 267: 12336–12340, 1992 [PubMed] [Google Scholar]
- 46.Kohan DE, Padilla E. Endothelin-1 is an autocrine factor in rat inner medullary collecting ducts. Am J Physiol Renal Fluid Electrolyte Physiol 263: F607–F612, 1992 [DOI] [PubMed] [Google Scholar]
- 47.Kohan DE, Padilla E. Osmolar regulation of endothelin-1 production by rat inner medullary collecting duct. J Clin Invest 91: 1235–1240, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kohan DE, Padilla E, Hughes AK. Endothelin B receptor mediates ET-1 effects on cAMP and PGE2 accumulation in rat IMCD. Am J Physiol Renal Fluid Electrolyte Physiol 265: F670–F676, 1993 [DOI] [PubMed] [Google Scholar]
- 49.Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev 91: 1–77, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korbmacher C, Boulpaep EL, Giebisch G, Geibel J. Endothelin increases [Ca2+]i in M-1 mouse cortical collecting duct cells by a dual mechanism. Am J Physiol Cell Physiol 265: C349–C357, 1993 [DOI] [PubMed] [Google Scholar]
- 51.Kuc R, Davenport AP. Comparison of endothelin-A and endothelin-B receptor distribution visualized by radioligand binding versus immunocytochemical localization using subtype selective antisera. J Cardiovasc Pharmacol 44, Suppl 1: S224–S226, 2004 [DOI] [PubMed] [Google Scholar]
- 52.Kurokawa K, Yoshitomi K, Ikeda M, Uchida S, Naruse M, Imai M. Regulation of cortical collecting duct function: effect of endothelin. Am Heart J 125: 582–588, 1993 [DOI] [PubMed] [Google Scholar]
- 53.Kurtz A, Kaissling B, Busse R, Baier W. Endothelial cells modulate renin secretion from isolated mouse juxtaglomerular cells. J Clin Invest 88: 1147–1154, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lynch IJ, Welch AK, Kohan DE, Cain BD, Wingo CS. Endothelin-1 inhibits sodium reabsorption by ETA and ETB receptors in the mouse cortical collecting duct. Am J Physiol Renal Physiol 305: F568–F573, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lyon-Roberts B, Strait KA, van Peursem E, Kittikulsuth W, Pollock JS, Pollock DM, Kohan DE. Flow regulation of collecting duct endothelin-1 production. Am J Physiol Renal Physiol 300: F650–F656, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malatino LS, Bellanuova I, Cataliotti A, Cuzzola F, Mallamaci F, Tripepi G, Parlongo S, Cutrupi S, Mangiafico RA, Ferri C, Galletti F, Gloriso N, Strazzullo P, Zoccali C. Renal endothelin-1 is linked to changes in urinary salt and volume in essential hypertension. J Nephrol 13: 178–184, 2000 [PubMed] [Google Scholar]
- 57.Mann JF, Green D, Jamerson K, Ruilope LM, Kuranoff SJ, Littke T, Viberti G. Avosentan for overt diabetic nephropathy. J Am Soc Nephrol 21: 527–535, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57a.Martin CJ. Ernest Henry Starling, CMG, MD, FRS (Obituary). Br Med J 1: 900–906, 1927 [PMC free article] [PubMed] [Google Scholar]
- 58.Masaki T, Kimura S, Yanagisawa M, Goto K. Molecular and cellular mechanism of endothelin regulation. Implications for vascular function. Circulation 84: 1457–1468, 1991 [DOI] [PubMed] [Google Scholar]
- 59.Mattyus I, Zimmerhack LB, Schwarz A, Hentschel M, Brandis M, Milteny M, Tulassay T. Renal excretion of endothelin in children is influenced by age and diuresis. Acta Paediatr 83: 468–472, 1994 [DOI] [PubMed] [Google Scholar]
- 60.Migas I, Bäcker A, Meyer-Lehnert H, Kramer HJ. Endothelin synthesis by porcine inner medullary collecting duct cells. Am J Hypertens 8: 748–752, 1995 [DOI] [PubMed] [Google Scholar]
- 61.Moe O, Tejedor A, Campbell WB, Alpern R, Henrich WL. Effects of endothelin on in vitro renin secretion. Am J Physiol Endocrinol Metab 260: E521–E525, 1991 [DOI] [PubMed] [Google Scholar]
- 62.Nadler SP, Zimplemann JA, Hebert RL. Endothelin inhibits vasopressin-stimulated water permeability in rat terminal inner medullary collecting duct. J Clin Invest 90: 1458–1466, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Naruse M, Uchida S, Ogata E, Kurokawa K. Endothelin-1 increases cell calcium in mouse collecting tubule cells. Am J Physiol Renal Fluid Electrolyte Physiol 261: F720–F725, 1991 [DOI] [PubMed] [Google Scholar]
- 64.Oishi R, Nonoguchi H, Tomita K, Marumo F. Endothelin-1 inhibits AVP-stimulated osmotic water permeability in rat medullary collecting duct. Am J Physiol Renal Fluid Electrolyte Physiol 261: F951–F956, 1991 [DOI] [PubMed] [Google Scholar]
- 65.Omatsu S, Tomoyoshi T. Immunohistochemical study on endothelin in rat kidney. Hinyokika Kiyo 43: 109–114, 1997 [PubMed] [Google Scholar]
- 66.Ortiz PA, Garvin JL. Role of nitric oxide in the regulation of nephron transport. Am J Physiol Renal Physiol 282: F777–F784, 2002 [DOI] [PubMed] [Google Scholar]
- 67.Pandit MM, Strait KA, Matsuda T, Kohan DE. Na delivery and ENaC mediate flow regulation of collecting duct endothelin-1 production. Am J Physiol Renal Physiol 302: F1325–F1330, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pavlov TS, Chahdi A, Ilatovskaya DV, Levchenko V, Vandewalle A, Pochynyuk O, Sorokin A, Staruschenko A. Endothelin-1 inhibits the epithelial Na+ channel through betaPix/14-3-3/Nedd4-2. J Am Soc Nephrol 21: 833–843, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pochynyuk O, Rieg T, Bugaj V, Schroth J, Fridman A, Boss GR, Insel PA, Stockand JD, Vallon V. Dietary Na+ inhibits the open probability of the epithelial sodium channel in the kidney by enhancing apical P2Y2-receptor tone. FASEB J 24: 2056–2065, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ren Y, Garvin JL, Liu R, Carretero OA. Crosstalk between the connecting tubule and the afferent arteriole regulates renal microcirculation. Kidney Int 71: 1116–1121, 2007 [DOI] [PubMed] [Google Scholar]
- 71.Ritthaler T, Della Bruna R, Kramer BK, Kurtz A. Endothelins inhibit cyclic-AMP induced renin gene expression in cultured mouse juxtaglomerular cells. Kidney Int 50: 108–115, 1996 [DOI] [PubMed] [Google Scholar]
- 72.Ryan MJ, Black TA, Millard SL, Gross KW, Hajduczok G. Endothelin-1 increases calcium and attenuates renin gene expression in As4.1 cells. Am J Physiol Heart Circ Physiol 283: H2458–H2465, 2002 [DOI] [PubMed] [Google Scholar]
- 73.Saito I, Mizuno K, Niimura S, Fukuchi S. Urinary endothelin and sodium excretion in essential hypertension. Nephron 65: 152–153, 1993 [DOI] [PubMed] [Google Scholar]
- 74.Sakamoto A, Yanagisawa M, Sakurai T, Takuwa Y, Yanagisawa H, Masaki T. Cloning and functional expression of human cDNA for the ETB endothelin receptor. Biochem Biophys Res Commun 178: 656–663, 1991 [DOI] [PubMed] [Google Scholar]
- 75.Satlin L M, Sheng S, Woda CB, Kleyman TR. Epithelial Na+ channels are regulated by flow. Am J Physiol Renal Physiol 280: F1010–F1018, 2001 [DOI] [PubMed] [Google Scholar]
- 76.Schneider MP, Ge Y, Pollock DM, Pollock JS, Kohan DE. Collecting duct-derived endothelin regulates arterial pressure and Na excretion via nitric oxide. Hypertension 51: 1605–1610, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schramek H, Gstraunthaler G, Willinger CC, Pfaller W. Hyperosmolality regulates endothelin release by Madin-Darby canine kidney cells. J Am Soc Nephrol 4: 206–213, 1993 [DOI] [PubMed] [Google Scholar]
- 78.Speed JS, LaMarca B, Berry H, Cockrell K, George EM, Granger JP. Renal medullary endothelin-1 is decreased in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 301: R519–R523, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Strait KA, Stricklett PK, Kohan JL, Miller MB, Kohan DE. Calcium regulation of endothelin-1 synthesis in rat inner medullary collecting duct. Am J Physiol Renal Physiol 293: F601–F606, 2007 [DOI] [PubMed] [Google Scholar]
- 80.Strait KA, Stricklett PK, Kohan RM, Kohan DE. Identification of two nuclear factor of activated T-cells (NFAT)-response elements in the 5′-upstream regulatory region of the ET-1 promoter. J Biol Chem 285: 28520–28528, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stricklett PK, Hughes AK, Kohan DE. Endothelin-1 stimulates NO production and inhibits cAMP accumulation in rat inner medullary collecting duct through independent pathways. Am J Physiol Renal Physiol 290: F1315–F1319, 2006 [DOI] [PubMed] [Google Scholar]
- 82.Stricklett PK, Strait KA, Kohan DE. Novel regulation of endothelin-1 promoter activity by protein kinase C. Cell Biochem Biophys 61: 643–650, 2011 [DOI] [PubMed] [Google Scholar]
- 83.Stuart D, Chapman M, Rees S, Woodward SK, Kohan DE. Myocardial, smooth muscle, nephron and collecting duct gene targeting reveals the organ sites of endothelin A receptor antagonist fluid retention. J Pharmacol Exper Ther 346: 182–189, 2013 [DOI] [PubMed] [Google Scholar]
- 84.Stuart D, Rees S, Woodward SK, Koesters R, Strait KA, Kohan DE. Disruption of the endothelin A receptor in the nephron causes mild fluid volume expansion. BMC Nephrol 13: 166, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sullivan JC, Goodchild TT, Cai Z, Pollock DM, Pollock JS. Endothelin(A) (ET(A)) and ET(B) receptor-mediated regulation of nitric oxide synthase 1 (NOS1) and NOS3 isoforms in the renal inner medulla. Acta Physiol (Oxf) 191: 329–336, 2007 [DOI] [PubMed] [Google Scholar]
- 86.Takagi M, Tsukada H, Matsuoka H, Yagi S. Inhibitory effect of endothelin on renin release in vitro. Am J Physiol Endocrinol Metab 257: E833–E838, 1989 [DOI] [PubMed] [Google Scholar]
- 87.Takemoto F, Uchida S, Katagirl H, Oka Y, Nakao A, Kurokawa K. Desensitization of endothelin-1 binding by vasopressin via a cAMP-mediated pathway in rat CCD. Am J Physiol Renal Fluid Electrolyte Physiol 268: F385–F390, 1995 [DOI] [PubMed] [Google Scholar]
- 88.Takemoto F, Uchida S, Ogata E, Kurokawa K. Endothelin-1 and endothelin-3 binding to rat nephrons. Am J Physiol Renal Fluid Electrolyte Physiol 264: F827–F832, 1993 [DOI] [PubMed] [Google Scholar]
- 89.Teitelbaum I, Berl T. Increased cytosolic Ca2+ inhibits AVP-stimulated adenylyl cyclase activity in rat IMCT cells by activation of PKC. Am J Physiol Renal Fluid Electrolyte Physiol 266: F486–F490, 1994 [DOI] [PubMed] [Google Scholar]
- 90.Terada Y, Tomita K, Nonoguchi H, Yang T, Marumo F. Different localization and regulation of two types of vasopressin receptor messenger RNA in microdissected rat nephron segments using reverse transcription polymerase chain reaction. J Clin Invest 92: 2339–2345, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Terada Y, Yamada T, Takayama M, Nonoguchi H, Sasaki S, Tomita K, Marumo F. Presence and regulation of Raf-1-K (Kinase), MAPK-K, MAP-K, and S6-K in rat nephron segments. J Am Soc Nephrol 6: 1565–1577, 1995 [DOI] [PubMed] [Google Scholar]
- 92.Todd-Turla KM, Chen M, Kretzler M, Rose D, Smart A, Yu T, Zhu X, Fehes-Toth G, Briggs JP, Schnermann J. Synthesis and polarized secretion of endothelin in a cortical collecting duct cell line (M1). Hypertension 24: 408, 1994 [DOI] [PubMed] [Google Scholar]
- 93.Todd-Turla KM, Zhu XL, Shu X, Chen M, Yu T, Smart A, Killen PD, Fejes-Toth G, Briggs JP, Schnermann JB. Synthesis and secretion of endothelin in a cortical collecting duct cell line. Am J Physiol Renal Fluid Electrolyte Physiol 271: F330–F339, 1996 [DOI] [PubMed] [Google Scholar]
- 94.Tomita K, Nonguchi H, Terada Y, Marumo F. Effects of ET-1 on water and chloride transport in cortical collecting ducts of the rat. Am J Physiol Renal Fluid Electrolyte Physiol 264: F690–F696, 1993 [DOI] [PubMed] [Google Scholar]
- 95.Tomita K, Nonoguchi H, Marumo F. Effects of endothelin on peptide-dependent cyclic adenosine monophosphate accumulation along the nephron segments of the rat. J Clin Invest 85: 2014–2018, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tomita K, Nonoguchi H, Marumo F. Inhibition of fluid transport by endothelin through protein kinase C in collecting duct of rats. Basel: Karger, 1991, p. 207–215 [DOI] [PubMed] [Google Scholar]
- 97.Turner AJ, Murphy LJ. Molecular pharmacology of endothelin converting enzymes. Biochem Pharm 51: 91–102, 1996 [DOI] [PubMed] [Google Scholar]
- 98.Vijayaraghavan J, Scicli AG, Carretero OA, Slaughter C, Moomaw C, Hersh LB. The hydrolysis of endothelins by neutral endopeptidase 24.11 (enkephalinase). J Biol Chem 265: 14150–14155, 1990 [PubMed] [Google Scholar]
- 99.Vio CP, Figueroa CD, Caorsi I. Anatomical relationship between kallikrein-containing tubules and the juxtaglomerular apparatus in the human kidney. Am J Hypertens 1: 269–271, 1988 [DOI] [PubMed] [Google Scholar]
- 100.Vogel V, Backer A, Heller J, Kramer HJ. The renal endothelin system in the Prague hypertensive rat, a new model of spontaneous hypertension. Clin Sci 97: 91–98, 1999 [PubMed] [Google Scholar]
- 101.Waeber C, Hoyer D, Palacios JM. Similar distribution of [125I]sarafotoxin-6b and [125I]endothelin-1,-2,-3 binding sites in the human kidney. Eur J Pharmacol 176: 233–236, 1990 [DOI] [PubMed] [Google Scholar]
- 102.Wendel M, Knels L, Kummer W, Koch T. Distribution of endothelin receptor subtypes ETA and ETB in the rat kidney. J Histochem Cytochem 54: 1193–1203, 2006 [DOI] [PubMed] [Google Scholar]
- 103.Wilkes BM, Susin M, Mento PF, Macica CM, Giraldi EP, Boss E, Nord EP. Localization of endothelin-like immunoreactivity in rat kidneys. Am J Physiol Renal Fluid Electrolyte Physiol 260: F913–F920, 1991 [DOI] [PubMed] [Google Scholar]
- 104.Wong NL, Tsui JK. Angiotensin regulates endothelin-B receptor in rat inner medullary collecting duct. Metabolism 50: 661–666, 2001 [DOI] [PubMed] [Google Scholar]
- 105.Wong NL, Wong BP, Tsui JK. Vasopressin regulates endothelin-B receptor in rat inner medullary collecting duct. Am J Physiol Renal Physiol 278: F369–F374, 2000 [DOI] [PubMed] [Google Scholar]
- 106.Wong WH, Wong BP, Wong EF, Huang MH, Wong NL. Downregulation of endothelin B receptors in cardiomyopathic hamsters. Cardiology 89: 195–201, 1998 [DOI] [PubMed] [Google Scholar]
- 107.Woodcock EA, Land SL. Functional endothelin ETB receptors on renal papillary tubules. Eur J Pharmacol 247: 93–95, 1993 [DOI] [PubMed] [Google Scholar]
- 108.Worgall S, Manz F, Kleschin K, Feth F, Rascher W. Elevated urinary excretion of endothelin-like immunoreactivity in children with renal disease is related to urine flow rate. Clin Nephrol 41: 331–337, 1994 [PubMed] [Google Scholar]
- 109.Yamamoto T, Suzuki H, Kubo Y, Matsumoto A, Uemura H. Endothelin A receptor-like immunoreactivity on the basal infoldings of rat renal tubules and collecting ducts. Arch Histol Cytol 71: 77–87, 2008 [DOI] [PubMed] [Google Scholar]
- 110.Yamashita K, Discher D, Hu J, Bishopric N, Webster K. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem 276: 12645–12653, 2001 [DOI] [PubMed] [Google Scholar]
- 111.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332: 411–415, 1988 [DOI] [PubMed] [Google Scholar]
- 112.Yang T, Terada Y, Nonoguchi H, Ujiie K, Tomita K, Marumo F. Effect of hyperosmolality on production and mRNA expression of endothelin-1 in inner medullary collecting duct. Am J Physiol Renal Fluid Electrolyte Physiol 264: F684–F689, 1993 [DOI] [PubMed] [Google Scholar]
- 113.Yukimura T, Notoya M, Mizojiri K, Mizuhira V, Matsuura T, Ebara T, Miura K, Kim S, Iwao H, Song K. High resolution localization of endothelin receptors in rat renal medulla. Kidney Int 50: 135–147, 1996 [DOI] [PubMed] [Google Scholar]
- 114.Zeidel ML, Brady HR, Kone BC, Gullans SR, Brenner BM. Endothelin, a peptide inhibitor of Na+-K+-ATPase in intact tubular epithelial cells. Am J Physiol Cell Physiol 257: C1101–C1107, 1989 [DOI] [PubMed] [Google Scholar]
- 115.Zeiler M, Löffler BM, Bock HA, Thiel G, Basel K. Water diuresis increases endothelin-1 excretion in humans. J Am Soc Nephrol 6: 751, 1995 [Google Scholar]
- 116.Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel alpha. J Clin Invest 117: 773–783, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zoccali C, Leonardis D, Parlongo S, Mallamaci F, Postorino M. Urinary and plasma endothelin 1 in essential hypertension and in hypertension secondary to renoparenchymal disease. Nephrol Dial Transplant 10: 1320–1323, 1995 [PubMed] [Google Scholar]