

Abstract
The present study compared the progression of renal injury in Sprague-Dawley (SD) and Dahl salt-sensitive (SS) treated with streptozotocin (STZ). The rats received an injection of STZ (50 mg/kg ip) and an insulin pellet (2 U/day sc) to maintain the blood glucose levels between 400 and 600 mg/dl. Twelve weeks later, arterial pressure (143 ± 6 vs. 107 ± 8 mmHg) and proteinuria (557 ± 85 vs. 81 ± 6 mg/day) were significantly elevated in STZ-SS rats compared with the values observed in STZ-SD rats, respectively. The kidneys from STZ-SS rats exhibited thickening of glomerular basement membrane, mesangial expansion, severe glomerulosclerosis, renal interstitial fibrosis, and occasional glomerular nodule formation. In additional studies, treatment with a therapeutic dose of insulin (4 U/day sc) attenuated the development of proteinuria (212 ± 32 mg/day) and renal injury independent of changes in arterial pressure in STZ-SS rats. Since STZ-SS rats developed severe renal injury, we characterized the time course of changes in renal hemodynamics during the progression of renal injury. Nine weeks after diabetes onset, there was a 42% increase in glomerular filtration rate in STZ-SS rats vs. time-control SS rats with reduced renal blood flow. These results indicate that SS rats treated with STZ develop hyperfiltration and progressive proteinuria and display renal histological lesions characteristic of those seen in patients with diabetic nephropathy. Overall, this model may be useful to study signaling pathways and mechanisms that play a role in the progression of diabetes-induced renal disease and the development of new therapies to slow the progression of diabetic nephropathy.
Keywords: Type 1 diabetes, streptozotocin, hyperfiltration, glomerulosclerosis, renal fibrosis, Dahl S rats, renal hemodynamics, diabetes-associated renal disease
diabetes is the leading cause of end-stage renal disease (ESRD), yet the mechanisms involved in the development of diabetic nephropathy remain poorly understood. A confounding factor is that not all patients with diabetes develop renal disease, suggesting that hyperglycemia alone does not account for the renal injury. African-Americans are at a much greater risk for the development of diabetic nephropathy (35). This suggests that environmental factors, as well as genetic susceptibility, contribute to pathogenesis of diabetic nephropathy. Clearly, there is a critical need to better understand the genetic factors and mechanisms by which diabetes triggers development of glomerulosclerosis and renal interstitial fibrosis in patients that are genetically susceptible to renal injury.
The Dahl salt-sensitive (SS) rat is a salt-dependent model of hypertension that is highly susceptible to the development of renal disease when fed a high-salt (HS) diet (5, 8, 9, 23, 26–28, 31, 34). The development of glomerular disease is associated with impaired renal autoregulation, contributing to altered renal hemodynamics that causes elevations in glomerular hydrostatic pressure (Pgc) that precede the development of progressive renal injury in these animals (33). The glomerular lesions that develop resemble those seen in patients with hypertension-induced nephropathy. Thus, unlike most rodent models that are highly resistant to the development of diabetic nephropathy, such as the Type 1 diabetic streptozotocin (STZ)-treated Sprague-Dawley (STZ-SD) rat, we hypothesize that the SS rat would be more susceptible to the development of glomerular injury and renal fibrosis when treated with STZ (STZ-SS). In support of our hypothesis, Korner et al. (18) reported that SS rats treated with STZ develop hypertension, glomerular injury, and albuminuria. Therefore, the current study compared the development of renal injury in STZ-SS rats that are susceptible to renal injury vs. STZ-SD rats, which are resistant to renal disease. In addition, we also determined the temporal changes in renal hemodynamics during the progression of diabetes-induced renal injury in STZ-SS, since they develop similar renal histologic lesions to patients with diabetic nephropathy.
METHODS
General.
Experiments were performed on 96, 8–21-wk-old male SD and SS rats. SD rats were purchased from Taconic Farms (Germantown, NY). SS rats were obtained from in-house colonies of SS/Jr rats that had been maintained by brother-sister mating for 20 years from breeder pairs originally obtained from Dr. John Rapp at the University of Toledo. The rats were housed in the Laboratory Animal Facility at the University of Mississippi Medical Center, which is approved by the American Association for the Accreditation of Laboratory Animal Care. The rats had free access to food and water throughout the study. All protocols were approved by the Animal Care Committee of the University of Mississippi Medical Center.
Protocol 1. Comparison of the development of diabetes-induced renal disease in STZ-treated SD and Dahl SS rats.
Experiments were performed on 9-wk-old SD and SS rats that were treated with streptozotocin (STZ; 50 mg/kg ip) to induce diabetes and given one long-acting insulin implant (low dose, 2 U/day sc, recombinant human insulin; Linshin Canada, Ontario, Canada) to maintain blood glucose levels between 300 and 500 mg/dl. The rats were fed a 0.3% NaCl diet (Harlan Teklad 7034; Harlan Laboratories, Madison, WI) to minimize the development of hypertension. A catheter of a telemetry unit (model TA11PA-C40; Data Sciences International, St. Paul, MN) was implanted in the femoral artery, and the unit was placed under the skin on the back at 8 wk of age for the measurement of mean arterial pressure (MAP). After 1 wk of recovery, MAP was recorded for three consecutive days to obtain a baseline MAP at 9 wk of age, and then, the rats were injected with STZ. One week after STZ injection, diabetes was confirmed by the measurement of blood glucose levels, and the insulin pellets were implanted subcutaneously. MAP was measured in 3-wk intervals throughout the study for 12 wk. At each time point, urine was collected overnight to determine protein excretion, and blood was collected from the tail vein to measure blood glucose levels. At the end of the study, the kidneys were collected, weighed, and fixed in a 10% buffered formalin solution. Paraffin sections (3 μm) were prepared and stained with Periodic acid-Schiff and Masson's trichrome to assess the degree of glomerular injury and renal fibrosis, respectively, on ∼30 images per section. Thirty glomeruli per section were scored in a blinded fashion on a 0–4 scale with 0 representing a normal glomerulus, 1 representing a 25% loss, 2 representing a 50% loss, 3 representing a 75% loss, and 4 representing >75% loss of capillaries in the tuft. Five glomerular images at ×100 were taken from each section to determine the thickness of the glomerular basement membrane (GBM) by using the NIS-Elements D 3.0 software. Additional analysis was performed to determine the degree of renal fibrosis. Images were captured using a Nikon Eclipse 55i microscope equipped with a Nikon DS-Fi1 color camera (Nikon, Melville, NY) and analyzed for the percentage of the image stained blue (primarily collagen) in the Masson trichrome-stained sections using the software mentioned previously. Fifteen to twenty representative fields were analyzed per section.
Protocol 2. Effect of controlling blood glucose levels with insulin in STZ-treated Dahl SS rats on the development of proteinuria and renal injury.
We next determined whether controlling blood glucose levels with a therapeutic dose of insulin would prevent the development of renal disease in STZ-SS rats. Experiments were performed on two groups of 9 wk-old SS rats implanted with telemetry transmitters at 8 wk of age and treated with either vehicle or STZ. STZ-SS rats were separated into two groups and given either one insulin implant (low dose, 2 U/day sc, recombinant human insulin) to maintain hyperglycemia at 300–500 mg/dl or given two implants (therapeutic dose, 4 U/day sc, STZ-insulin-SS) that normalizes plasma glucose levels. Arterial pressure and protein excretion were measured every 3 wk until 12 wk after diabetes onset. At each time point, blood was collected for the measurement of blood glucose levels. At the end of the experiment, a final blood sample was collected to determine the plasma concentration of insulin (Mercodia Rat Insulin ELISA, Uppsala, Sweden). The kidneys also were weighed, collected, and fixed in a 10% buffered formalin solution for the assessment of renal injury as described above.
Protocol 3. Time course of changes in renal hemodynamics in control and STZ-treated Dahl SS rats.
Measurement of renal hemodynamics was performed on two groups of SS rats treated with either vehicle (Control-SS) or STZ (STZ-SS) with a low dose of insulin (2 U/day sc) to maintain blood glucose levels between 300 and 500 mg/dl during the control period and 3, 9, and 12 wk after induction of diabetes. On the day of the experiment, the rats were anesthetized with ketamine (30 mg/kg im; Phoenix Pharmaceuticals, St. Joseph, MO) and Inactin (50 mg/kg ip; Sigma, St. Louis, MO), and catheters were placed in the femoral artery and vein for the measurement of arterial pressure and the infusion of a 2% BSA solution containing FITC-labeled inulin (2 mg/ml; Sigma) in a 0.9% NaCl solution at a rate of 6 ml/h. Renal blood flow (RBF) was measured using a flowmeter (Transonic Systems, Ithaca, NY), and glomerular filtration rate (GFR) was measured from the clearance of FITC-labeled inulin. After a 30-min equilibration, urine and plasma samples were collected during a 30-min collection period. At the end of the experiment, the left kidney was removed and weighed, and the concentrations of inulin in the urine and plasma samples were determined with a microplate fluorometer (BioTek Instruments, Winooski, VT).
Statistical analysis.
Mean values ± SE are presented. The significance of differences in control and experimental values within the same animal was determined by a paired t-test. The significance of differences in the mean values between groups was determined by either a one-way or two-way repeated-measures ANOVA followed by Holm-Sidek test. P < 0.05 was considered to be significant.
RESULTS
Protocol 1. Comparison of the development of diabetes-induced renal injury in STZ-treated SD and SS rats.
A comparison of development of renal injury in STZ-treated SD and SS rats is presented in Fig. 1. Baseline blood glucose levels were similar in SD and SS rats (99 ± 4 and 96 ± 7 mg/dl, respectively) (Fig. 1A). After 12 wk of diabetes, blood glucose levels increased to 500 ± 7 and 469 ± 35 mg/dl, respectively, in STZ-treated SD and SS rats. After 12 wk of diabetes, MAP rose from 117 ± 1 to 143 ± 6 mmHg in STZ-SS rats, while it remained unaltered in STZ-SD rats (107 ± 8 mmHg) (Fig. 1B). Over the course of the study, proteinuria increased to 557 ± 85 mg/day in STZ-SS rats but only increased to 81 ± 10 mg/day in STZ-SD rats (Fig. 1C).
Fig. 1.

Time course measurements of blood glucose (A), mean arterial pressure (MAP; B), and proteinuria (C) in streptozotocin (STZ; 50 mg/kg ip)-treated Sprague Dawley (SD) and Dahl salt-sensitive (SS) rats with a low dose of insulin (2 U/day sc) during 12 wk of diabetes. Numbers in parentheses indicate the number of rats studied per group. Values are expressed as means ± SE. *Significant difference from the corresponding value within the same strain at week 0. †Significant difference from the corresponding value in STZ-SD rats at the same time period.
A comparison of the degree of renal injury in STZ-SD and STZ-SS rats is represented in Fig. 2. The kidneys from STZ-SS rats exhibited severe renal injury with thickening of the glomerular basement membrane, mesangial expansion, severe glomerulosclerosis, and renal interstitial fibrosis (Fig. 2, B and D). We also observed occasional formation of glomerular nodules (Fig. 2B, arrow) in the kidneys of STZ-SS rats. In contrast, kidneys from STZ-SD rats exhibited only a minor expansion of the mesangial matrix in the glomerulus (Fig. 2A) and little if any renal interstitial fibrosis (Fig. 2C). The glomerular injury score was significantly greater in STZ-SS rats compared with STZ-SD rats (Fig. 2E). The percentage of renal fibrosis (blue staining) (Fig. 2G) and GBM thickness (Fig. 2H) was significantly elevated in STZ-SS rats compared with values seen in STZ-SD rats after 12 wk of diabetes. The glomerular injury score in STZ-SS rats averaged 2.7, indicating that ∼66% of the glomerular capillary area available for filtration was lost. This could be due to focal glomerular sclerosis with very severe injury to some glomeruli and less involvement in others or global injury to most glomeruli. To address the issue, we measured the percentage of the glomeruli with severe injury (score >3) and found that 66% of the glomeruli were severely injured. This indicates that the STZ-SS rats exhibit global rather than focal glomerulosclerosis after 12 wk of diabetes in (Fig. 2F).
Fig. 2.
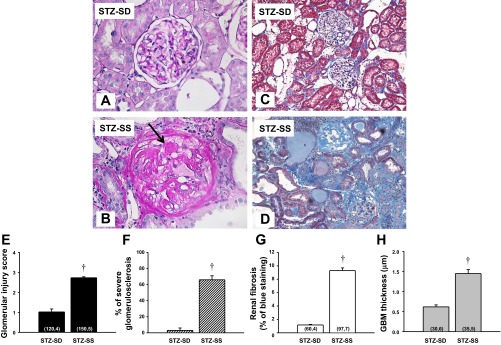
Comparison of the renal histopathology: Periodic acid-Schiff (PAS) staining (A and B), Masson's trichrome (C and D), glomerular injury score (E), percentage of severe glomerulosclerosis (F), renal fibrosis (G), and glomerular membrane thickness (GBM; H) in STZ (50 mg/kg ip)-treated SD and Dahl SS rats with a low dose of insulin (2 U/day sc) after 12 wk of diabetes. Arrow indicates areas of glomerular nodule formation. Numbers in parentheses indicate the number of glomeruli and rats studied per group. Values are expressed as means ± SE. †Significant difference from the corresponding value in STZ-SD rats at the same time period.
Protocol 2. Effect of effective control of blood glucose levels with chronic insulin therapy on the development of renal injury in STZ-SS rats.
Plasma insulin levels were significantly lower in STZ-SS rats treated with the low dose of insulin compared with control SS rats (0.59 ± 0.04 vs. 0.95 ± 0.05 ng/ml, respectively). Increasing the dose of insulin normalized plasma insulin levels in the STZ-insulin-SS rats to 1.04 ± 0.08 ng/ml. The effects of controlling blood glucose levels with a therapeutic dose of insulin on the development of hypertension and proteinuria in STZ-SS rats are presented in Fig. 3. Baseline blood glucose levels were similar in all groups (89 ± 4 mg/dl) (Fig. 3A). One week after STZ injection, blood glucose levels increased to the same extent in STZ-SS and STZ-insulin-SS groups (516 ± 34 mg/dl), while blood glucose levels remained normal for the duration of the study in control SS rats (86 ± 4 mg/dl). Administration of a therapeutic dose of insulin effectively controlled blood glucose levels in the STZ-insulin-SS rats to the same levels seen in control SS rats. The induction of diabetes in STZ-SS rats caused proteinuria to increase to 544 ± 60 mg/day, whereas proteinuria only increased to 248 ± 38 mg/day in control SS rats. Normalization of glucose levels with a therapeutic dose of insulin prevented the increase in proteinuria seen in STZ-SS rats (Fig. 3C).
Fig. 3.

Comparison of blood glucose levels (A), mean arterial pressure (MAP; B), and proteinuria (C) in control and STZ (50 mg/kg ip)-treated Dahl SS rats with either a low-dose (2 U/day sc, STZ-SS) or therapeutic dose (4 U/day sc, STZ-insulin-SS) of insulin during 12 wk of diabetes. Numbers in parenthesis indicate the number of rats studied per group. Values are expressed as means ± SE. *Significant difference from the corresponding value within the same strain at week 0; †Significant difference from the corresponding value in control SS rats at the same time period. #Significant difference from the corresponding value in STZ-SS rats at the same time period.
A comparison of the degree of renal injury in control, STZ, and STZ-insulin-SS rats treated for 12 wk is presented in Fig. 4. The kidneys from STZ-SS rats exhibited severe glomerulosclerosis, renal fibrosis, tubular necrosis, and inflammation (Fig. 4A). The glomerular injury score (Fig. 4B) and the percentage of renal fibrosis (Fig. 4C) were significantly elevated in STZ-SS rats compared with values observed in control SS rats. Administration of a therapeutic dose of insulin prevented the increase in the glomerular injury score but was only partially effective in reducing the degree of renal interstitial fibrosis in STZ-insulin-SS rats. We observed a significant increase in kidney weight (renal hypertrophy) in STZ-SS rats vs. the values seen in control SS rats (2.30 ± 0.12 vs. 1.41 ± 0.04 g, respectively) (data not shown). Treatment with a therapeutic dose of insulin reduced the degree of renal hypertrophy in STZ-insulin-SS rats by 26% (1.70 ± 0.11 g).
Fig. 4.

Comparison of the renal histopathology (A), glomerular injury scores (B), and renal fibrosis (C) in control and STZ (50 mg/kg ip)-treated Dahl SS rats with either a low-dose (2 U/day sc, STZ-SS) or therapeutic dose (4 U/day sc, STZ-insulin-SS) of insulin after 12 wk of diabetes. Numbers in parentheses indicate the number of glomeruli and rats studied per group. Values are expressed as means ± SE. *Significant difference from the corresponding value in control SS rats. †Significant difference from the corresponding value in STZ-SS rats.
Protocol 4. Time course of changes in renal hemodynamics in control SS and STZ-SS rats.
The results of these experiments are represented in Fig. 5. We did not observe any differences in MAP (measured under Inactin anesthesia) between control SS and STZ-SS rats, and both groups averaged 145 ± 5 mmHg by the end of the study (data not shown). Left kidney weight-to-body weight ratio (LKW/BW) was similar at baseline between the two groups (data not shown). However, 3 wk after diabetes onset, LKW/BW ratio rose by 55% in STZ-SS rats compared with values observed in control SS rats and remained at this elevated level for the duration of the study. After 3 wk of hyperglycemia, STZ-SS rats' RBF increased by 35% vs. the values observed in control SS rats. However, when normalized for the increase in kidney weight, there was no difference in RBF in STZ-SS rats and SS rats at 3 wk of age, indicating that the rise in total RBF was secondary to renal hypertrophy. Normalized RBF decreased significantly over the remainder of study in STZ-SS rats compared with time-control SS rats (Fig. 5A). Similarly, there was a 40% increase in total GFR in the first 3 wk after induction of diabetes in the STZ-SS rats that was secondary to the initial renal hypertrophy, but GFR factored per gram kidney weight was not altered. Normalized GFR increased by 42% in STZ-SS rats that were diabetic for 9 wk compared with values seen in control SS rats of the same age (Fig. 5B). Thereafter, GFR was reduced by 44% in STZ-SS rats vs. control SS rats after 12 wk of diabetes, which likely reflects the progression of glomerulosclerosis and loss of filtration area and the increase in renal vascular resistance (Fig. 5D). Filtration fraction (FF) was the same in both groups at baseline and after 3 wk of diabetes (Fig. 5C). FF was significantly elevated in STZ-SS rats vs. control SS rats when animals reached 9 wk of diabetes. However, 12 wk after diabetes induction, FF was similar in both groups.
Fig. 5.

Temporal changes in renal hemodynamics in control and STZ (50 mg/kg ip)-treated Dahl SS rats with a low dose (2 U/day sc) of insulin during 12 wk of diabetes: renal blood flow (RBF; ml·min−1·g kidney wt−1) (A), glomerular filtration rate (GFR; μl·min−1·g kidney wt−1) (B), filtration fraction (FF; %) (C), and renal vascular resistance (RVR; mmHg·ml−1·min·g kidney wt) (D). The number of rats studied was 5–8 animals per group. Values are expressed as means ± SE. *Significant difference from the corresponding value within the same group at week 0. †Significant difference from the corresponding value in Control SS rats within the same time period.
DISCUSSION
The SS rat has been commonly used to investigate mechanisms involved in the development of hypertension-induced renal disease. Williams et al. (33) observed that the development of renal injury in SS rats fed a high-salt diet was associated with a marked elevation in Pgc, suggesting they exhibit an impairment in long-term autoregulation of RBF and GFR. This was further supported by preliminary studies by Roman and colleagues (14) demonstrating that SS rats exhibit an impaired myogenic response in afferent arterioles of SS rats. Therefore, we hypothesized that they might be more susceptible to the development of proteinuria and glomerulosclerosis following induction of hyperglycemia, unlike other strains of rats and mice (i.e., SD rat). The present study compared the development of diabetes-induced renal injury in SD and SS rats treated with STZ. Administration of STZ to SD and SS rats raised blood glucose levels to a similar extent; however, hyperglycemia stimulated the development of hypertension, progressive proteinuria, and severe glomerular disease in STZ-SS rats following 12 wk of diabetes but had little effect in SD rats treated with STZ. Normalizing blood glucose levels by administration of a therapeutic dose of insulin prevented the development of proteinuria and the degree of glomerular injury in STZ-SS rats, indicating the changes seen were dependent on hyperglycemia and not due to a nephrotoxic effect of STZ. Because we observed that STZ-SS developed most of the characteristics of human diabetic nephropathy, including renal hypertrophy, thickening of the glomerular basement membrane, expansion of the mesangial matrix, severe glomerulosclerosis, and renal interstitial fibrosis, we next examined the time course changes in renal hemodynamics during the progression of renal injury. We observed hyperfiltration at 9 wk of diabetes that was associated with the development of progressive proteinuria and renal injury in STZ-SS rats. Later, after 12 wk of diabetes, SS-STZ rats experienced a decline in GFR, and this is consistent with the progression of severe glomerulosclerosis and loss of filtration area seen in a large percentage (>66%) of glomeruli during this time period.
Several factors have been reported to contribute to the progression of renal injury in diabetic nephropathy, including hyperglycemia, hypertension, proteinuria, and genetic background (17). The ethnic background plays a major role as well because African-American patients are susceptible to hypertension, diabetic nephropathy, and ESRD (13). In the present study, we compared the progression of diabetes-induced renal injury in SS rats that are more susceptible to renal injury than SD rats, which are resistant to renal disease when treated with STZ. We found that proteinuria was five times higher in STZ-SS rats compared with STZ-SD rats after 12 wk of diabetes. The kidneys from STZ-SS rats displayed histological lesions that are characteristic of those patients with diabetic nephropathy. We believe the increase in susceptibility in African-Americans and SS rats may be due to alterations in renal hemodynamics, in which the autoregulation of either RBF or GFR in response to increases in arterial pressures is impaired. In response to elevations in arterial pressure by either increased salt intake (19) or norepinephrine infusion (24), GFR increases substantially more in African-Americans compared with their Caucasian counterparts. Similarly, previous studies have shown that the kidneys of SS fed a high-salt diet for a week or so exhibit elevations in Pgc caused by reductions in afferent arteriolar resistance, which allows greater transmission of systemic pressure to the glomerulus to initiate the development of proteinuria and renal injury (2). Previous studies by Hayashi et al. (15) using the hydronephrotic kidney preparation and more recent preliminary studies by Ge et al. (14) have suggested that the myogenic response of the afferent arteriole is impaired in SS rats fed either a low-salt (LS) or HS diet. These data suggest that glomerular hyperfiltration in response to hyperglycemia may be a mechanism contributing to the increased susceptibility to develop diabetic nephropathy.
Previous investigators have reported that renal vasodilation and hyperfiltration precedes the development of proteinuria and diabetic nephropathy, whereas in later stages, diabetic patients develop chronic kidney disease and hypertension (36). However, most rodent models of diabetic nephropathy, especially mouse and rat models treated with STZ, do not develop progressive proteinuria or severe glomerular injury. Moreover, the characterization of the early changes in renal hemodynamics in most of these models has not been well described. In the current study, we found that the development of renal injury in STZ-SS rats was associated first with a rise in RBF and GFR during the first 3 wk of induction of diabetes. However, RBF and GFR were not elevated when normalized for kidney weight, and these early increases in RBF and GFR likely reflect the adaptive hypertrophy of the kidney to hyperglycemia. After the initial renal hypertrophy, RBF declined over the course of the experiment, but there was a twofold increase in GFR (normalized for kidney weight) after 9 wk of diabetes compared with the values seen in time control SS rats of the same age, indicating that the STZ-SS rats developed glomerular hyperfiltration between 3 and 9 wk of age. This may be due to the hyperglycemia-mediated stimulation of the renin-angiotensin system to constrict the efferent arteriole, since we found that FF was elevated after 9 wk of diabetes in STZ-SS rats compared with control SS rats. Thereafter, STZ-SS rats displayed a decline in renal function and GFR, which likely reflects the progressive loss of capillary filtration area in most of the glomeruli of the kidney that eventually overcomes the initial renal vasodilation and glomerular hypertrophy. Overall, the current work is one the first studies that shows early increases and sequential changes in renal hemodynamics in a rodent model of diabetes-induced renal injury. These data suggest that the initial changes in renal hemodynamics during the early phase of diabetes are due to adaptive renal hypertrophy, which is later followed by an increase in GFR (hyperfiltration), leading to the development of renal injury and chronic kidney disease.
STZ has been extensively used to induce diabetes in many strains of rats, including SD (1, 20, 21, 25), Wistar-Kyoto (WKY) (6, 32), and spontaneously hypertensive (SHR) strains (6, 7, 10–12, 16). Previous studies have demonstrated that the development of renal injury is greater in STZ-treated SHR rats compared with WKY rats. This likely is secondary to the hypertension in SHR rats that is transmitted to the glomerulus after the onset of diabetes (6). However, even in the SHR model, the development of the renal disease occurs very gradually over an 8-mo period, and it remains to be determined whether the renal injury in this model truly reflects diabetic nephropathy and whether the renal injury can be prevented by normalizing glucose levels or blood pressure. In an attempt to enhance the development of diabetes-induced renal injury, other investigators have induced diabetes with STZ in uninephrectomized strains of rats and mice (16, 32). However, the time frame for the development of renal histological lesions varies among the strains from 4 to 8 mo after STZ administration.
In the current study, proteinuria increased in the first 3 wk following administration of STZ to SS rats. Over the course of the study, hyperglycemia increased arterial pressure by 20 mmHg and augmented the development of renal injury in this model. By 12 wk, the kidneys from STZ-SS rats showed histological evidence of renal injury that was at least equal to and in most cases far greater than that reported in any other strain of rats or mice treated with STZ. Similar results have been observed in the two-kidney, one-clipped diabetic model in which the combination of diabetes and elevations in arterial pressure exacerbates renal disease (22). These data indicate that the SS rats are more susceptible to the development of renal injury than other strains, and they may be a useful model to study therapeutic interventions to slow the progression of diabetic nephropathy.
To determine whether the development of renal injury was due to elevations in blood glucose levels, STZ-SS rats were treated with a therapeutic dose of insulin. The tight control of blood glucose levels with insulin following the initial onset of diabetes prevented the development of renal injury in STZ-SS rats. There are two possible mechanisms by which treatment with insulin could have a renoprotective effect other than reducing blood glucose levels: 1) preventing the hyperglycemia-mediated increase in GFR and 2) reducing renal hypertrophy. Chang et al. (4) recently reported the prevention of hyperglycemia with insulin normalized GFR and inhibited the development of renal injury in diabetic mice. This may suggest that insulin, in restoring normoglycemia, prevents the increased glucose-stimulated hyperfiltration in diabetes by blunting tubuloglomerular feedback and causing dilation of the afferent arteriole (30). The inhibition of increased GFR decreases the reabsorption of filtered protein contributing to renal hypertrophy. While treatment with a therapeutic dose of insulin prevented most signs of renal injury, it had a minimal effect on reducing renal interstitial fibrosis in STZ-SS rats. This may be due to an inflammatory response by exogenous r-human insulin used in the current study that causes fibrosis for 12 wk. Overall, these data indicate that treating diabetes early with insulin prevents hyperglycemia, elevations in GFR (hyperfiltration), and inhibits renal hypertrophy, which inhibits the progression of diabetes-associated renal disease.
Patients with Type 1 diabetes are at higher risk to develop diabetic nephropathy than Type 2 diabetic patients. Currently, there are no rodent models of Type 1 diabetic complications that express all of the characteristics of diabetic nephropathy that are observed in patients. Recently, the Animal Models of Diabetic Complications Consortium defined characteristics for validating rodent models of diabetic nephropathy: 1) progressive proteinuria with a decline in renal function compared with control animals of the same age, 2) glomerulosclerosis with thickening of the glomerular basement membrane, 3) mesangial matrix deposition, 4) development of hypertension, and 5) formation of nodules (3). The current study demonstrated that the STZ-SS model meets all of the criteria for diabetic nephropathy (Table 1). STZ-SS rats developed progressive proteinuria, thickening of the glomerular basement membrane, expansion of the mesangial matrix, renal and glomerular hypertrophy, severe glomerulosclerosis and renal interstitial fibrosis, and occasional formation of glomerular nodules. Moreover, proteinuria develops faster and the degree of renal injury is more severe in STZ-SS rats than in most other strains of rats and mice treated with STZ (i.e., STZ-SD rats). The mechanism by which renal disease develops in this model remains to be determined, but it is associated with early renal vasodilation and hyperfiltration characterized by a marked increase in GFR. Over time, STZ-SS rats develop hypertension and severe renal injury, which eventually leads to a fall in RBF and GFR when taking into account renal hypertrophy. Overall, this model may be useful in studying signaling pathways and mechanisms that play a role in the progression of diabetes-induced renal disease and the development of new therapies that slow the progression of diabetic nephropathy.
Table 1.
Comparison of the criteria for diabetic nephropathy in human patients and streptozotocin-treated Dahl salt-sensitive rats
| Criteria | Human | STZ-SS |
|---|---|---|
| Early increase in GFR (hyperfiltration) | √ | √ |
| Progressive proteinuria | √ | √ |
| Hypertension | √ | √ |
| Mesangial matrix expansion | √ | √ |
| Thickening of glomerular basement membrane | √ | √ |
| Renal interstitial fibrosis | √ | √ |
| Nodular glomerular lesions | √ | √ |
| Decline in GFR | √ | √ |
GFR, glomerular filtration rate; STZ-SS, streptozotocin-treated Dahl salt-sensitive.
GRANTS
This work was supported in part by a National Institutes of Health Grant HL-094446 and the Robert M. Hearin Foundation awarded to M. Garrett, National Institutes of Health Grants HL-36279, HL-29587, and HL-2958727S1 awarded to R. Roman, as well as an American Heart Association Scientist Development Grant and a PhRMA Foundation Starter Grant awarded to J. Williams.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: T.N.S., A.P., D.S., N.K., P.B.K., and J.M.W. performed experiments; T.N.S., A.P., D.S., N.K., and J.M.W. analyzed data; T.N.S., N.K., M.R.G., R.J.R., and J.M.W. interpreted results of experiments; T.N.S. and J.M.W. prepared figures; T.N.S. and J.M.W. drafted manuscript; T.N.S., N.K., M.R.G., R.J.R., and J.M.W. edited and revised manuscript; T.N.S., A.P., D.S., N.K., M.R.G., R.J.R., and J.M.W. approved final version of manuscript; J.M.W. conception and design of research.
REFERENCES
- 1.Allen TJ, Cao Z, Youssef S, Hulthen UL, Cooper ME. Role of angiotensin II and bradykinin in experimental diabetic nephropathy. Functional and structural studies. Diabetes 46: 1612–1618, 1997 [DOI] [PubMed] [Google Scholar]
- 2.Azar SLC, Iwai J, Weller D. Single nephron dynamics during high Na+ intake and early hypertension in Dahl rats. Jpn Heart J 20: 138–140, 1979 [Google Scholar]
- 3.Brosius FC, 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T, and the Animal Models of Diabetic Complications Consortium Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang SY, Chen YW, Chenier I, Tran Sle M, Zhang SL. Angiotensin II type II receptor deficiency accelerates the development of nephropathy in type I diabetes via oxidative stress and ACE2. Exp Diabetes Res 2011: 521076, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen PY, St John PL, Kirk KA, Abrahamson DR, Sanders PW. Hypertensive nephrosclerosis in the Dahl/Rapp rat. Initial sites of injury and effect of dietary l-arginine supplementation. Lab Invest 68: 174–184, 1993 [PubMed] [Google Scholar]
- 6.Cooper ME, Allen TJ, Macmillan P, Bach L, Jerums G, Doyle AE. Genetic hypertension accelerates nephropathy in the streptozotocin diabetic rat. Am J Hypertens 1: 5–10, 1988 [DOI] [PubMed] [Google Scholar]
- 7.Cooper ME, Allen TJ, O'Brien RC, Macmillan PA, Clarke B, Jerums G, Doyle AE. Effects of genetic hypertension on diabetic nephropathy in the rat—functional and structural characteristics. J Hypertens 6: 1009–1016, 1988 [DOI] [PubMed] [Google Scholar]
- 8.Dahly-Vernon AJ, Sharma M, McCarthy ET, Savin VJ, Ledbetter SR, Roman RJ. Transforming growth factor-beta, 20-HETE interaction, and glomerular injury in Dahl salt-sensitive rats. Hypertension 45: 643–648, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Dahly AJ, Hoagland KM, Flasch AK, Jha S, Ledbetter SR, Roman RJ. Antihypertensive effects of chronic anti-TGF-β antibody therapy in Dahl S rats. Am J Physiol Regul Integr Comp Physiol 283: R757–R767, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Davis BJ, Forbes JM, Thomas MC, Jerums G, Burns WC, Kawachi H, Allen TJ, Cooper ME. Superior renoprotective effects of combination therapy with ACE and AGE inhibition in the diabetic spontaneously hypertensive rat. Diabetologia 47: 89–97, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Elmarakby AA, Faulkner J, Baban B, Saleh MA, Sullivan JC. Induction of hemeoxygenase-1 reduces glomerular injury and apoptosis in diabetic spontaneously hypertensive rats. Am J Physiol Renal Physiol 302: F791–F800, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elmarakby AA, Faulkner J, Baban B, Sullivan JC. Induction of hemeoxygenase-1 reduces renal oxidative stress and inflammation in diabetic spontaneously hypertensive rats. Int J Hypertens 2012: 957235, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freedman BI, Spray BJ, Tuttle AB, Buckalew VM., Jr The familial risk of end-stage renal disease in African-Americans. Am J Kidney Dis 21: 387–393, 1993 [DOI] [PubMed] [Google Scholar]
- 14.Ge Y, Fan F, Murphy SR, Williams JM, Liu R, Falck J, Roman RJ. Deficiency in the production of 20-HETE contributes to impaired myogenic and TGF responses in the afferent arteriole of Dahl S rats (Abstract 36). Hypertension 60: A36, 2012 [Google Scholar]
- 15.Hayashi K, Epstein M, Saruta T. Altered myogenic responsiveness of the renal microvasculature in experimental hypertension. J Hypertens 14: 1387–1401, 1996 [DOI] [PubMed] [Google Scholar]
- 16.Kang MJ, Ingram A, Ly H, Thai K, Scholey JW. Effects of diabetes and hypertension on glomerular transforming growth factor-beta receptor expression. Kidney Int 58: 1677–1685, 2000 [DOI] [PubMed] [Google Scholar]
- 17.Klein R, Klein BE, Moss SE, Davis MD, DeMets DL. The Wisconsin epidemiologic study of diabetic retinopathy. IV. Diabetic macular edema. Ophthalmology 91: 1464–1474, 1984 [DOI] [PubMed] [Google Scholar]
- 18.Korner A, Jaremko G, Eklof AC, Aperia A. Rapid development of glomerulosclerosis in diabetic Dahl salt-sensitive rats. Diabetologia 40: 367–373, 1997 [DOI] [PubMed] [Google Scholar]
- 19.Kotchen TA, Piering AW, Cowley AW, Grim CE, Gaudet D, Hamet P, Kaldunski ML, Kotchen JM, Roman RJ. Glomerular hyperfiltration in hypertensive African-Americans. Hypertension 35: 822–826, 2000 [DOI] [PubMed] [Google Scholar]
- 20.Manigrasso MB, Sawyer RT, Marbury DC, Flynn ER, Maric C. Inhibition of estradiol synthesis attenuates renal injury in male streptozotocin-induced diabetic rats. Am J Physiol Renal Physiol 301: F634–F640, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mankhey RW, Bhatti F, Maric C. 17beta-Estradiol replacement improves renal function and pathology associated with diabetic nephropathy. Am J Physiol Renal Physiol 288: F399–F405, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Mauer SM, Steffes MW, Azar S, Sandberg SK, Brown DM. The effects of Goldblatt hypertension on development of the glomerular lesions of diabetes mellitus in the rat. Diabetes 27: 738–744, 1978 [DOI] [PubMed] [Google Scholar]
- 23.O'Donnell MP, Kasiske BL, Katz SA, Schmitz PG, Keane WF. Lovastatin but not enalapril reduces glomerular injury in Dahl salt-sensitive rats. Hypertension 20: 651–658, 1992 [DOI] [PubMed] [Google Scholar]
- 24.Parmer RJ, Stone RA, Cervenka JH. Renal hemodynamics in essential hypertension. Racial differences in response to changes in dietary sodium. Hypertension 24: 752–757, 1994 [DOI] [PubMed] [Google Scholar]
- 25.Prabhu A, Xu Q, Manigrasso MB, Biswas M, Flynn E, Iliescu R, Lephart ED, Maric C. Expression of aromatase, androgen and estrogen receptors in peripheral target tissues in diabetes. Steroids 75: 779–787, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raij L, Azar S, Keane W. Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int 26: 137–143, 1984 [DOI] [PubMed] [Google Scholar]
- 27.Roman RJ, Alonso-Galicia M, Wilson TW. Renal P450 metabolites of arachidonic acid and the development of hypertension in Dahl salt-sensitive rats. Am J Hypertens 10: 63S–67S, 1997 [PubMed] [Google Scholar]
- 28.Roman RJ, Ma YH, Frohlich B, Markham B. Clofibrate prevents the development of hypertension in Dahl salt-sensitive rats. Hypertension 21: 985–988, 1993 [DOI] [PubMed] [Google Scholar]
- 29.Takenaka T, Forster H, De Micheli A, Epstein M. Impaired myogenic responsiveness of renal microvessels in Dahl salt-sensitive rats. Circ Res 71: 471–480, 1992 [DOI] [PubMed] [Google Scholar]
- 30.Thomson SC, Vallon V, Blantz RC. Kidney function in early diabetes: the tubular hypothesis of glomerular filtration. Am J Physiol Renal Physiol 286: F8–F15, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Tolins JP, Raij L. Comparison of converting enzyme inhibitor and calcium channel blocker in hypertensive glomerular injury. Hypertension 16: 452–461, 1990 [DOI] [PubMed] [Google Scholar]
- 32.Utimura R, Fujihara CK, Mattar AL, Malheiros DM, Noronha IL, Zatz R. Mycophenolate mofetil prevents the development of glomerular injury in experimental diabetes. Kidney Int 63: 209–216, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Williams JM, Sarkis A, Hoagland KM, Fredrich K, Ryan RP, Moreno C, Lopez B, Lazar J, Fenoy FJ, Sharma M, Garrett MR, Jacob HJ, Roman RJ. Transfer of the CYP4A region of chromosome 5 from Lewis to Dahl S rats attenuates renal injury. Am J Physiol Renal Physiol 295: F1764–F1777, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson TW, Alonso-Galicia M, Roman RJ. Effects of lipid-lowering agents in the Dahl salt-sensitive rat. Hypertension 31: 225–231, 1998 [DOI] [PubMed] [Google Scholar]
- 35.Young BA, Maynard C, Boyko EJ. Racial differences in diabetic nephropathy, cardiovascular disease, and mortality in a national population of veterans. Diabetes Care 26: 2392–2399, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Zelmanovitz T, Gerchman F, Balthazar AP, Thomazelli FC, Matos JD, Canani LH. Diabetic nephropathy. Diabetol Metab Syndrome 1: 10, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]