

Abstract
Wilson disease (WD) is an autosomal recessive disorder resulting in pathological progressive copper accumulation in liver and other tissues. The worldwide prevalence (P) is about 30/million, while in Sardinia it is in the order of 1/10 000. However, all of these estimates are likely to suffer from an underdiagnosis bias. Indeed, a recent molecular neonatal screening in Sardinia reported a WD prevalence of 1:2707. In this study, we used a new approach that makes it possible to estimate the allelic frequency (q) of an autosomal recessive disorder if one knows the proportion between homozygous and compound heterozygous patients (the homozygosity index or HI) and the inbreeding coefficient (F) in a sample of affected individuals. We applied the method to a set of 178 Sardinian individuals (3 of whom born to consanguineous parents), each with a clinical and molecular diagnosis of WD. Taking into account the geographical provenance of the parents of every patient within Sardinia (to make F computation more precise), we obtained a q=0.0191 (F=7.8 × 10−4, HI=0.476) and a corresponding prevalence P=1:2732. This result confirms that the prevalence of WD is largely underestimated in Sardinia. On the other hand, the general reliability and applicability of the HI approach to other autosomal recessive disorders is confirmed, especially if one is interested in the genetic epidemiology of populations with high frequency of consanguineous marriages.
Keywords: Wilson disease, genetic epidemiology, consanguinity, homozygosity index
Introduction
Wilson disease (WD, OMIM #277900) is an autosomal recessive disorder caused by mutations in the ATP7B gene (13q14.3, MIM 606882),1 which encode a copper transporting Cpx-type ATPase.2 Such mutations are associated with alterations of copper metabolism resulting in pathological progressive copper accumulation in liver and other tissues. The manifestations of liver disease vary from clinically asymptomatic, with only biochemical abnormalities, to acute liver failure. Similarly, the spectrum of neurologic manifestations ranges from normal or mild disturbances to a rapid and severe progression of neurological disability, including neuropsychiatric symptoms. Biochemical parameters typically include a low level of serum ceruloplasmin, and increased urinary and hepatic copper.3 Owing to the extreme clinical heterogeneity and to the late onset, molecular testing is always warranted to confirm a clinical diagnosis of WD.4
Approximately 520 mutations have so far been identified in the ATP7B gene.5 WD mutational spectra vary significantly between populations, with the most frequent mutation in Eastern Europe (H1069Q) showing a peculiar decreasing frequency gradient toward South-Western Europe while it is absent in Sardinia.2 By comparison, in Sardinia a characteristic founder mutation (−441/−427del) is highly prevalent (67%), with all other mutations showing a relative frequency below 10%.6, 7
The worldwide prevalence of WD has been reported as approximately 30/million, with a gene frequency of 0.56% and a carrier frequency of 1 in 90.8 In contrast to mainland Europe, the Sardinian population shows a WD prevalence that has been estimated to be 1/10 000–1/7000, one of the highest worldwide.6, 9 All these estimates have been inferred through classical approaches, such as the frequencies of clinically diagnosed cases, or from the findings of autopsy series,8 and as such they are likely to suffer from an underdiagnosis bias. In support of this hypothesis, a recent molecular neonatal screening in the Sardinian population reported a WD gene frequency of 1.92% and a resulting prevalence of 1/2707 live births.7 However, this screening program was performed for only 4 consecutive months on a total of 5290 births (representing approximately 1/3 of the annual number of births in Sardinia) and it needs replication.
In the present work, we used a new molecular-based approach, the homozygosity index (HI) method,10 which makes it possible to estimate the allelic frequency (q) of an autosomal recessive disorder if one knows the proportion of homozygous patients, the inbreeding coefficient (F) and the mutational spectrum of a sample of affected individuals. This method had already been successfully tested on Phenylketonuria (PKU) and Familial Mediterranean Fever (FMF),10 showing notable advantages over traditional descriptive epidemiology approaches.
Materials and methods
We collected molecular data from a set of 192 Sardinian patients with a diagnosis of WD through the Ospedale Regionale per le Microcitemie, Cagliari, which is the leading center for WD diagnosis and treatment in the island. The diagnosis of WD was based on low ceruloplasmin and copper-serum concentrations, increased urinary copper excretion, and a high liver copper concentration. In addition, a mutation analysis of the ATP7B gene was performed for each patient on DNA extracted from peripheral blood.11 Every sample was first investigated for the six most common reported mutations in Sardinia, and then, if no mutation was found, by single-strand conformation polymorphisms and Sanger sequencing of all exons and of the flanking intronic regions in the ATP7B gene.12
Data on the geographical provenance of the parents of every patient and their year of birth were also collected. The study was approved by the local ethical committee and all participants provided informed consent for the analysis.
To make the computation of F (and therefore of q) as precise as possible, we calculated F taking into account the birthplaces of the parents of patients (see online Supplementary Information). In Sardinia, the frequency of consanguineous marriages and hence the inbreeding coefficient of the population is highly correlated with altitude.13, 14 The most remote areas of the island (namely mountains and internal hills) where genetic isolation is stronger coincide with a higher local prevalence of recessive disorders like WD.9 Furthermore, the presence of a small number of individuals born to consanguineous parents was taken into account in the computation of F (for further details, see online Supplementary Information).
Of the 192 patients diagnosed with WD, 9 apparent heterozygotes (with only one mutation identified in the ATP7B locus) were removed from the study, since they did not fit the basic assumptions of the model (see below).
In order to compute F based on the geographical origins of the patients within Sardinia, a further five patients with non-detailed or ambiguous geographical provenance of their parents were excluded from the sample.
All of the computations were run in Excel 2010 and SPSS 16.0 (IBM Corp., Armonk, NY, USA). Assuming that: (1) patients can only be homozygous or compound heterozygous for the disease alleles of a single gene; (2) these alleles are identified only by a single disease-associated variant (ie, different haplotypes of the same variant are not considered as different alleles) and (3) they strictly act in a recessive manner (with no phenotypic effect in heterozygotes); the HI method computes the allelic frequency (q) of a given autosomal recessive disorder as:
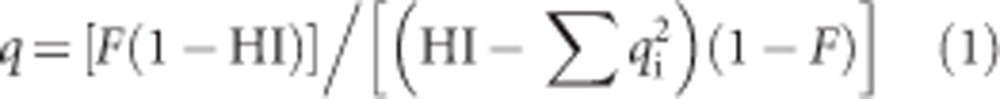 |
where HI is the HI of the subset (ie, the number of homozygotes over the total number of patients) and qi is the relative frequency of the ith disease allele (with i=0,1,2,..,n−1,n). This formula makes use of the inverse correlation between the prevalence of a recessive disease and the relative frequency of homozygous patients to estimate the allelic frequency of the disease gene in a population (the higher HI, the rarer the disease).
Results
The mutational spectrum of the Sardinian WD patients included in the study is summarized in Figure 1, which shows clearly that the deletion −441/−427 in the ATP7B gene is by far the most frequent WD mutation in Sardinia (representing 66.5% of all the WD alleles), followed by mutations V1146M and 2463delC (8.5% and 6.5%, respectively). All the other mutations give a smaller contribution to the mutational spectrum of WD in Sardinia, with six mutations with relative frequency around 1–2% and 16 below 1% (for further details, see Supplementary Table S4).
Figure 1.

Mutational spectrum of the ATP7B gene in the sample of WD Sardinian patients included in the analysis (ie, patients with unambiguous genotype and detailed geographical provenance of parents, N=178). Only the mutations reported with relative frequencies ≥1% were considered in the computation of q. *Other alleles with relative frequencies <1% (for further details, see Supplementary Table S4).
The inbreeding coefficient (F) and the HI (Figure 2) were calculated in order to get an estimate of the allelic frequency (q) of WD in Sardinia through the HI approach. The results of the analysis are summarized in Table 1.
Figure 2.

Proportions of homozygotes (HOM) versus compound heterozygotes (CH) within the subset of WD patients with detailed geographical provenance of parents (N=178).
Table 1. Allelic frequency (q) and resulting prevalence (P=q 2) computed for the WD sample through the HI method.
| Sample size | F | HI | q | P |
|---|---|---|---|---|
| 178a | 7.8 × 10−4 | 0.476 | 0.0191 | 1:2732 |
Abbreviations: HI, homozygosity index; WD, Wilson disease.
Excluding individuals with generic or ambiguous information on the geographical origin of parents within Sardinia.
The F for the subset of 178 individuals with detailed information on the geographical provenance of parents was basically computed relying on their distribution in the areas of Sardinia defined by altitude (for further details, see Table 2 and Supplementary Table S2). In Table 2, comparing the distribution of the affected individuals in the sample and of the general Sardinian population at four different levels of altitude, a clear bias is apparent in the relative frequency of affected individuals, who were mostly resident in the most isolated areas, that is, internal hills and mountains. Indeed, collectively they account for >35% of the WD sample, while representing <25% of the total Sardinian population.
Table 2. Breakdown of the WD patients by altitude of residence and comparison with the general Sardinian population (absolute counts are first reported, with percentages in parentheses).
| Areaa | Patients sampleb | Sardinian populationc |
|---|---|---|
| Plains | 87 (48.88) | 816 442 (48.82) |
| Coastal hills | 28 (15.73) | 442 291 (26.45) |
| Internal hills | 48 (26.97) | 351 942 (21.04) |
| Mountains | 15 (8.43) | 61 729 (3.69) |
| Total | 178 (100) | 1 672 404 (100) |
Abbreviation: WD, Wilson disease.
See online Supplementary Information (Supplementary Table S1) for details on the definition criteria of altitudinal areas.
Detailed and unambiguous information on the geographical origins was available only for 178 patients.
Original data from Italian National Institute of Statistics.15
Discussion
This study provides new insights in the genetic epidemiology of WD in Sardinia and confirms the accuracy and general applicability of the HI approach to study the genetic epidemiology of autosomal recessive disorders. The unique genetic history and population structure of the island, characterized by persistent isolation, strong endogamy and frequent consanguineous matings,13, 14 have traditionally made Sardinia very interesting for genetic studies. Furthermore, the availability of detailed frequencies of consanguineous marriages (for every degree of kinship) broken down into zones of altitude, has made it possible to use precise estimates of inbreeding in our sample.
Descriptive studies based on clinical diagnoses of WD have often been suspected to be biased by underdiagnosis. This hypothesis was confirmed by the results of a preliminary molecular screening in Sardinia conducted on 5290 newborns for 4 consecutive months, which reported a WD gene frequency of 1.92% and a resulting prevalence of 1/2707 live births7 that is about 3–4 times higher than previously estimated. In this study, by including only WD patients with both clinical and molecular diagnosis, and with detailed information on their geographical provenance (ie, birthplace of both parents), we were able to compute F for the sample in an acceptably accurate manner (for further details, see online Supplementary Information). This allowed us to optimize the computations through the HI method, which yields a q=1.91% and a P=1:2732 (Table 1), a result highly consistent with the data reported by Zappu et al.7
The high frequency, the expected prevention by preclinical diagnosis and early treatment of the devastating effect on the nervous system and liver tissue, make WD in Sardinia a model example for other autosomal recessive disorders, which can be prevented by newborn screening once their social impact is established by well established methods of genetic epidemiology.
In a previous work, we tested the HI method on different samples of patients affected with two other autosomal recessive disorders, namely FMF and PKU, born either to first cousins or to unrelated parents.10 This study represents an even stronger confirmation of the validity of the HI method for two reasons. First because the results could be compared with data from a preliminary neonatal molecular screening. Second because the method was tested on a larger group of patients with a very well-established F whose parents were known to be consanguineous in only three cases. In fact the real stumbling block in the application of the HI method is the calculation of F especially when a sample of children of apparently unrelated parents is analyzed. The calculation of F in the general control population was already the main problem when only the demographic approach based on consanguinity was possible to study the genetic epidemiology of autosomal recessive disorders16, 17, 18 and before the molecular analysis of mutations became a routine practice in diagnostics. This problem was bypassed in Italy by the availability of a reliable source of frequencies of consanguineous marriages in the general control population.14 Today the widespread use of the molecular genetic analysis of causative mutations to confirm clinical diagnoses has considerably improved the possibility of using the consanguinity approach through the HI method which, as shown in this paper, has become reliable, accurate and inexpensive. The argument used against this conclusion is that the calculation of F is problematic because of lack of data on the inbreeding necessary to calculate F for apparently unrelated parents of patients (α for the general population). This is certainly true if the number of well defined consanguineous marriages (eg, among first and second cousins) in the sample of patients' parents is scanty. However, we have shown in our previous study10 that only 25 of such consanguineous marriages are needed to apply the HI method in an effective manner. This is therefore no longer a stumbling block when studying the populations of the so-called ‘consanguinity belt' (1.2 billion people) where the frequency of consanguineous marriages can run up to 30–50%.19 Using even small patient samples from these populations, the HI method can then be applied effectively to investigate which autosomal recessive disorders are most frequent and to establish priorities for screening and intervention policies, as in the case of WD in Sardinia. This is not a trivial result for communities where autosomal recessive ‘rare' disorders can be not so rare and have a strong social impact.
Eventually, the question remains whether the HI method can be used efficiently in non-inbred populations where consanguineous marriages are becoming rarer and rarer. The first answer to this question is that it is always possible to select from data collected by large international Consortia studying specific autosomal recessive disorders a small sample of patients born to consanguineous parents who can provide a reliable estimate of F. This selection does not introduce any bias in the final calculation of q, which is not dependent on being calculated from patients born to consanguineous or unrelated parents. The second answer will come from the availability of high-density genome scans, which today make it possible to estimate individual autozygosity from data on runs of homozygosity (ROHs). Termed F(roh), this estimate is defined as the proportion of the autosomal genome in runs of homozygosity above a specified length.20 Approaches such as this will provide a more useful definition of F than has hitherto been possible and will certainly make the HI method more applicable in non-inbred populations.
At a time when the technology available for genetic analysis is rapidly evolving, consanguinity studies, once based essentially on demography and classical population genetics,14, 21 are making a strong come-back in human and medical genetics. This happens not only because the characterization of well-defined phenotypes in consanguineous families makes gene discovery for autosomal recessive disorders much easier,22 but also because of the increasing possibilities of preventing autosomal recessive disorders in populations living in the ‘consanguinity belt'.19 Immigrants from such populations represent nowadays an important component of many European societies and a rapid reduction in the preference for consanguineous unions by first- and second-generation immigrant families currently appears improbable.19 To this end, international collaborations between population-rich and resource-rich countries have to be developed to contribute to the understanding of human disease-associated genetic variation, as envisaged for the Mediterranean sea basin.23
In this context, the HI method offers the advantage over traditional descriptive epidemiology studies of generating community-specific epidemiological estimates free of underdiagnosis bias utilizing data already available, that is, mutational records and pedigree information, with no need to gather very large samples or additional mutation data from unaffected control individuals.
Acknowledgments
We would like to thank Dr Gianna Zei and Professor Antonio Moroni for providing detailed data on the historic series of consanguineous marriages in Sardinia, as well as clinicians who referred WD patients from all over Sardinia for molecular analysis. This study was funded by CHERISH project (http://www.cherishproject.eu/) to GR, Unità Operativa di Genetica Medica, Policlinico Sant'Orsola Malpighi, Bologna, Italy.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Tanzi RE, Petrukhin K, Chernov I, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- Ferenci P. Regional distribution of mutations in the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet. 2006;120:151–159. doi: 10.1007/s00439-006-0202-5. [DOI] [PubMed] [Google Scholar]
- Ala AP, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson's disease. Lancet. 2007;369:397–408. doi: 10.1016/S0140-6736(07)60196-2. [DOI] [PubMed] [Google Scholar]
- Ferenci P, Caca K, Loudianos G, et al. Diagnosis and phenotypic classification of Wilson disease. Final report of the proceedings of the working party at the 8th international meeting on Wilson disease and Menkes disease, Leipzig/Germany, 2001. Liver Int. 2003;23:139–142. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- Wilson Disease Mutation Database, University of Alberta http://www.wilsondisease.med.ualberta.ca/database.asp ).
- Loudianos G, Dessi V, Lovicu M, et al. Molecular characterization of Wilson disease in the Sardinian population—evidence of a founder effect. Hum Mutat. 1999;14:294–303. doi: 10.1002/(SICI)1098-1004(199910)14:4<294::AID-HUMU4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Zappu A, Magli O, Lepori MB, et al. High incidence and allelic homogeneity of Wilson Disease in 2 isolated populations: a prerequisite for efficient disease prevention programs. J Pediatr Gastroenterol Nutr. 2008;47:334–338. doi: 10.1097/MPG.0b013e31817094f6. [DOI] [PubMed] [Google Scholar]
- Scheinberg I, Sternlieb I.Wilson diseaseIn: Lloyd H, Smith J, (eds): Major Problems in Internal Medicine Vol. 23Philadelphia: WB Saunders; 1984 [Google Scholar]
- Ghiagheddu A, Demella L, Puggioni G, et al. Epidemiologic study of hepatolenticular degeneration (Wilson's disease) in Sardinia (1902-1983) Acta Neurol Scand. 1985;72:43–55. doi: 10.1111/j.1600-0404.1985.tb01546.x. [DOI] [PubMed] [Google Scholar]
- Gialluisi A, Pippucci T, Anikster Y, et al. Estimating the allele frequency of autosomal recessive disorders through mutational records and consanguinity: the homozygosity index (HI) Ann Human Genet. 2012;76:159–167. doi: 10.1111/j.1469-1809.2011.00693.x. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepori MB, Lovicu M, Dessí V, et al. Twenty-four novel mutations in Wilson disease patients of predominantly Italian origin. Genet Test. 2007;11:328–332. doi: 10.1089/gte.2007.0015. [DOI] [PubMed] [Google Scholar]
- Moroni A, Anelli A, Anghinetti W, Lucchetti E, Rossi O, Siri E. La consanguineità umana nell'isola di Sardegna dal secolo XVIII al secolo XX. Ateneo Parmese. 1972;8:69–82. [Google Scholar]
- Cavalli-Sforza LL, Moroni A, Zei G. Consanguinity, Inbreeding and Genetic Drift in Italy. Princeton, NJ, USA: Princeton University Press; 2004. [Google Scholar]
- Annuario Statistico Italiano http://www3.istat.it/dati/catalogo/20101119_00/ ) Italian National Institute of Statistics,2010
- Romeo G, Menozzi P, Ferlini A, et al. Incidence of Friedreich ataxia in Italy estimated from consanguineous marriages. Am J Hum Gen. 1983;35:523–529. [PMC free article] [PubMed] [Google Scholar]
- Romeo G, Menozzi P, Ferlini A, et al. Incidence of classic PKU in Italy estimated from consanguineous marriages and from neonatal screening. Clin Genet. 1983;24:339–345. [PubMed] [Google Scholar]
- Romeo G, Bianco M, Devoto M, et al. Incidence in Italy, genetic heterogeneity and segregation analysis of cystic fibrosis. Am J Hum Gen. 1985;37:338–349. [PMC free article] [PubMed] [Google Scholar]
- Bittles AH. Consanguinity in Context. Cambridge University Press; 2012. [Google Scholar]
- McQuillan R, Leutenegger AL, Abdel-Rahman R, et al. Runs of homozygosity in European populations. Am J Hum Gen. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg G. Mathematical Methods for Population Genetics. Basel: Karger Pbl; 1947. pp. 61–65. [Google Scholar]
- Pippucci T, Benelli M, Magi A, et al. EX-HOM (EXome-HOMozygosity): a proof of principle. Human Heredity. 2011;72:45–53. doi: 10.1159/000330164. [DOI] [PubMed] [Google Scholar]
- Ozcelik T, Kanaan M, Avraham KB, et al. Collaborative genomics for human health and cooperation in the Mediterranean region. Nat Genet. 2010;42:641–645. doi: 10.1038/ng0810-641. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.