

Abstract
In the hippocampus, activity-dependent changes of synaptic transmission and spike-timing coordination are thought to mediate information processing for the purpose of memory formation. Here, we investigated the self-tuning of intrinsic excitability and spiking reliability by CA1 hippocampal pyramidal cells via changes of their GABAergic inhibitory inputs and endocannabinoid (eCB) signaling. Firing patterns of CA1 place cells, when replayed in vitro, induced an eCB-dependent transient reduction of spontaneous GABAergic activity, sharing the main features of depolarization-induced suppression of inhibition (DSI), and conditioned a transient improvement of spike-time precision during consecutive burst discharges. When evaluating the consequences of DSI on excitatory postsynaptic potential (EPSP)-spike coupling, we found that transient reductions of uncorrelated (spontaneous) or correlated (feedforward) inhibition improved EPSP-spike coupling probability. The relationship between EPSP-spike-timing reliability and inhibition was, however, more complex: transient reduction of correlated (feedforward) inhibition disrupted or improved spike-timing reliability according to the initial spike-coupling probability. Thus eCB-mediated tuning of pyramidal cell spike-time precision is governed not only by the initial level of global inhibition, but also by the ratio between spontaneous and feedforward GABAergic activities. These results reveal that eCB-mediated self-tuning of spike timing by the discharge of pyramidal cells can constitute an important contribution to place-cell assemblies and memory formation in the hippocampus.
Keywords: short-term plasticity, DSI, feedforward inhibition, spontaneous inhibition, endocannabinoids, spike timing, EPSP-spike coupling, CA1 place cell
learning-related brain computations rely on complex interactions between excitatory and inhibitory circuits and possibly involve activity-dependent synaptic plasticity to refine the spike timing underlying firing synchronization and temporal coordination of cell assemblies. In the hippocampus, a key circuit for memory, pyramidal cells called “place cells” fire in specific regions of the environment (i.e., place fields), and their joint activity is thought to provide representations of space used to guide behavior (O'Keefe and Conway 1978; O'Keefe and Dostrovsky 1971; Wilson and McNaughton 1993). Cell assemblies forming distinct representations are thought to arise as a result of changes in synaptic weights between active place cells, and precise spike timing is expected to be instrumental for their temporal coordination (Kentros et al. 1998; O'Neill et al. 2008; Robbe et al. 2006; Wilson and McNaughton 1994). However, the mechanisms by which place cells could refine the spike-time precision of their firing patterns remain unknown.
In the hippocampus, GABAergic interneurons orchestrate the activity of principal cells, notably through feedforward inhibition (FFI) and feedback inhibition (for review, see Kullmann 2011). The timing of inhibition, related to specific GABAergic innervation of distinct subcellular domains of pyramidal cells, may be tightly linked to excitation and may sharpen excitatory postsynaptic potential (EPSP)-spike coupling (Glickfeld and Scanziani 2006; Pouille and Scanziani 2001). However, variable firing of interneurons (Klausberger et al. 2005) and action-potential-independent (for review, see Ramirez and Kavalali 2011) and asynchronous release (for review, see Jonas and Hefft 2010) provide fluctuating inhibitory bombardment of principal cells that participate in action-potential jitter in response to excitatory responses (Caillard 2011).
Depolarization-induced suppression of inhibition (DSI) has been suggested to represent an important mechanism for learning and memory (for review, see Freund et al. 2003; Kano et al. 2009). This form of plasticity is mediated by endocannabinoid (eCB) signaling involving the cannabinoid type 1 receptor (CB1R), the activation of which is required for hippocampus-dependent long-term memory (Han et al. 2012; Puighermanal et al. 2009; Varvel et al. 2005). CB1Rs are predominantly expressed on axon terminals of cholecystokinin-expressing (CCK) interneurons (Katona et al. 1999; Tsou et al. 1999) known to provide local inhibition to pyramidal cells by means of asynchronous release of GABA (Daw et al. 2009; Foldy et al. 2006; Karson et al. 2009; Losonczy et al. 2004). During DSI, hippocampal CA1 pyramidal neurons release eCBs that transiently inhibit afferent GABAergic activity, and the CB1R agonist 2-arachidonoylglycerol (2-AG) is known to mediate such a retrograde control (Gao et al. 2010; Tanimura et al. 2010). In knockout mice deficient for the 2-AG-degrading enzyme monoacylglycerol lipase, sustained levels of 2-AG correlate with enhanced spatial learning and hippocampal long-term potentiation (Pan et al. 2011). As these knockout mice exhibit prolonged DSI of hippocampal CA1 pyramidal neurons (Pan et al. 2011), it was suggested that DSI may be an important mechanism for learning and memory. Yet, whether in vivo hippocampal place-cell firing patterns induce eCB-mediated DSI is controversial (Hampson et al. 2003). Moreover, the effects of DSI on spike-time precision remain to be determined.
Here, we established the firing requirement for DSI using in vivo place-cell patterns monitored during a spatial learning task (Dupret et al. 2010) and evaluated the effect of DSI on CA1 pyramidal cell firing and spike-timing properties. We found that the in vitro replay of place-cell discharge elicited an eCB-mediated transient reduction in GABA release depending on the number of action potentials fired and improved their spike-train precision as measured by a decrease in the coefficient of variation during burst discharge. Moreover, we employed dynamic current injections to mimic synaptic input events with computational modeling to identify the parameters and decipher the mechanisms underlying self-tuning of spike-time precision by coexisting correlated and uncorrelated GABAergic activities. We revealed that EPSP-spike coupling was governed by FFI imposing a biphasic tuning of spike-time precision, ultimately prevented by high-level spontaneous inhibition.
MATERIALS AND METHODS
Ethics statement.
All rats were maintained on a 12:12-h light-dark cycle with food and water provided ad libitum. Animal research was approved by the local direction of veterinary services [Direction Départementale des Services Vétérinaires, Préfecture des Bouches du Rhône, France (O. Caillard, license no. A13-493)]. All experiments were carried out according to the European and institutional guidelines for the care and use of laboratory animals (Council Directive 86/609/EEC and French National Research Council).
Hippocampal slice preparation and electrophysiological recordings.
Transverse hippocampal slices (350 μm thick) were obtained from 14- to 20-day old Wistar rats as previously described (Debanne et al. 2008). Experiments were performed at 32°C in artificial cerebrospinal fluid containing the following (in mM): 125 NaCl, 26 NaHCO3, 3 CaCl2, 2.5 KCl, 2 MgCl2, 0.8 NaH2PO4, and 10 d-glucose; and saturated with 95% O2 and 5% CO2. CA1 pyramidal cells were visualized using an Olympus BX51WI microscope equipped with differential interference contrast optics under infrared illumination and a water immersion lens (×60, 0.9 NA; Olympus). Electrophysiological recordings were performed in whole cell configuration with a MultiClamp 700B amplifier (Axon Instruments) filtered at 5 kHz and digitized at 20 kHz via a PCI-6220 National Instruments interface controlled by IGOR Pro (WaveMetrics, Lake Oswego, OR) and/or Digidata 1322A interface controlled by pCLAMP software (Axon Instruments). Patch pipettes had a resistance of 4–10 MΩ when filled with a KCl solution containing (in mM): 140 KCl, 5 HEPES, 2 EGTA, 2 Na2ATP, 0.3 NaGTP, and 2 MgCl2, pH 7.4 or with a low-chloride solution containing (in mM): 140 K-gluconate, 10 HEPES, 2 EGTA, 2 Na2ATP, 0.3 NaGTP, and 2 MgCl2, pH 7.4. With the KCl-based solution, the reversal potential for GABAA receptors, determined by local pressure ejection of isoguvacine (0.5 mM) at different holding membrane potentials (Vm), was 3.7 ± 2.9 mV (n = 4). With the low-chloride-based solution, the reversal potential for GABAA receptors determined by the same method was −73.7 ± 1.7 mV (n = 9).
Cells were recorded if the series resistance, measured throughout experiments, remained stable and <40 MΩ. Stimulating pipettes were filled with extracellular saline. Picrotoxin (PTX; 100 mM) was prepared in ethanol and stored at −80°C. Kynurenate (200 mM) and carbachol (5 mM) were prepared in distilled water and stored at −20°C. N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251; 10 mM) and tetrahydrolipstatin (THL; 1 mM) were prepared in DMSO and stored at −20°C. Stock solutions were thawed and diluted into the extracellular solution before use. Final DMSO concentrations were 0.05 and 0.1% for extracellular and intracellular solutions, respectively. At these concentrations, DSI was not affected by DMSO. DSI induced by depolarization of the cell in voltage-clamp at 0 mV for 1 s was 27 ± 4% of control GABAergic charge (n = 16) in the absence of DMSO. DSI was of comparable magnitude when cells were recorded with the intracellular or extracellular DMSO concentrations used in this study, respectively [28 ± 5%, U(22) = 41, z = 0.52, P = 0.303; 32 ± 3%, U(21) = 22, z = 1.49, P = 0.069].
Stimuli.
Voltage or direct-current (DC) steps and GABAA synaptic conductances were generated using IGOR Pro software (WaveMetrics) and converted to analog signals via a PCI-6723 National Instruments interface.
DSI was induced in two different modes. When DSI was induced in voltage-clamp mode, the cell was depolarized from a holding Vm of −80 to 0 mV for 1 s. Otherwise, DSI was induced by a series of action potentials elicited in current-clamp mode. The transient switch to current-clamp mode was under the control of an analog signal, constructed with IGOR Pro software and injected to the external mode command of the MultiClamp 700B amplifier contemporaneously with the voltage-clamp or current-clamp external input commands. The holding current was preliminarily set on the amplifier such that the resting Vm was kept at around −80 mV for 125 ms when switching to current-clamp mode before the depolarizing DC steps.
The firing patterns presented in Figs. 1, 2, and 4 were extracted from the data set used in Dupret et al. (2010). These patterns corresponded to three successive learning trials (trials 21, 22, and 23) during each of which the rat had to retrieve three food rewards hidden in a cheeseboard maze before returning to the start box and collecting an additional reward. All procedures were carried out in accordance with the Animals (Scientific Procedures) Act 1986 (United Kingdom) and associated procedures under an approved project license. In vitro replay of these place-cell firing patterns was obtained by delivering sequences of brief depolarization (1.25 ms/2 nA) followed by short hyperpolarization (1.25 ms/2 nA) at specific times (Fig. 1 and Fig. 2, C–F) to drive cell discharges at very short interspike intervals (ISI; see Fig. 2C for instance). The whole cell amplifier switched to current-clamp mode 125 ms before each action potential and back to voltage-clamp mode unless the interval to the next spike to be triggered was <250 ms. Following the elicited action-potential discharge (34 ± 7 mV, mean peak ± SE; n = 5), the Vm was transiently further hyperpolarized (−78.0 ± 3.3 mV) compared with the preceding holding potential (−76.2 ± 1.4 mV). In behaving rats, CA1 pyramidal cells classically exhibit bursts of spikes (Ranck Jr 1973) with ISI ranging from 2 to 6 ms (Harris et al. 2001). In line with this, the CA1 pyramidal cell activity from the study of Dupret et al. (2010) also revealed short ISI, as low as 2.5 ms. Hence, the protocols we used were the most reliable for driving action potentials at precise times. The DC injected to hyperpolarize the cell was also set to avoid uncontrolled action-potential discharge, which may be facilitated by the depolarizing envelope that follows the fast phase of action-potential repolarization (Schwartzkroin 1975; Storm 1987) and depolarizing GABAergic postsynaptic potentials that occurred frequently in our recording conditions [5 μM carbachol and GABAA reversal potential = 3.7 ± 2.9 mV (n = 4)].
Fig. 1.
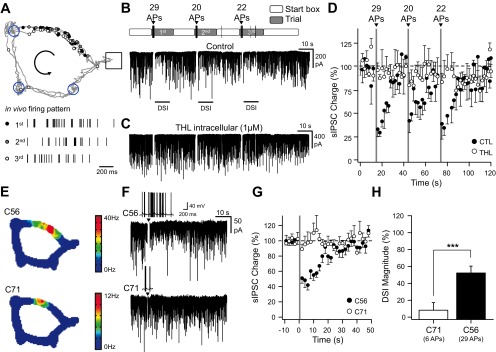
In vivo firing patterns induce depolarization-induced suppression of inhibition (DSI) in vitro. A, top: scheme representing the position at which action potentials (APs) were emitted by a CA1 place cell during 3 consecutive trials when evaluating spatial memory on a cheeseboard maze (Dupret et al. 2010). During learning, the animal is given consecutive trials to locate 3 hidden food rewards (blue circles). The start box is opened for each trial, and the animal harvests the 3 rewards before returning to the start box and collects a 4th pellet. Each dot corresponds to the location at which place cell emitted an individual spike and is superimposed on the animal's path. A, bottom: the bars represent the time at which APs were emitted by a CA1 place cell during the 1st, 2nd, and 3rd trial. B: voltage-clamp recordings [holding membrane potential (Vhold) = −80 mV] of spontaneous GABAergic activity interrupted by transient current-clamp switches during which brief direct-current (DC) injections were performed to replay the whole sequence of APs that were recorded in vivo (including the time spent in the start box). Above and with a similar time scale are depicted the time spent in the start box and the cheeseboard maze during the 3 consecutive trials together with the numbers of APs emitted during the crossing of the place field at a time depicted by an arrowhead. C: same experiment as in B except that the cell was dialyzed with tetrahydrolipstatin (THL; 1 μM). D: time course of spontaneous GABAergic charge during the replay of the whole AP sequence depicted in B and C in control (CTL; black, n = 3) and THL (1 μM, white, n = 4). The time spent in the place field is denoted in gray. sIPSC, spontaneous inhibitory postsynaptic current. E: color-coded rate map of simultaneously recorded place cells for the last 20 trials starting from those depicted in B for clusters C56 (top) and C71 (bottom). C56 is the same place cell as the 1 depicted in A. F: voltage-clamp recordings (Vhold = −80 mV) of spontaneous GABAergic activity interrupted by transient current-clamp switches during which brief DC injections were performed to replay a sequence of APs that were recorded in vivo during a single trial in 2 different place cells (C56 and C71). The insets represent the sequence of APs that was evoked in the in vitro recorded cells. G: mean time course of spontaneous GABAergic charge before and after the replay of the different AP sequences depicted in F (C56, n = 6; C71, n = 4). H: DSI magnitude measured in the next 4 s following AP discharge for the cells depicted in G. ***P < 0.001.
Fig. 2.

Pyramidal cell firing induces endocannabinoid-mediated suppression of GABAergic activity. A: voltage-clamp recordings of spontaneous GABAergic activity (Vhold = −80 mV). After a control period, the cell is transiently switched to current-clamp mode to allow the firing of APs either by a series of brief DC injections to replay a sequence of APs that were recorded in vivo during a single trial in 2 different place cells (top and bottom left, same patterns as in Fig. 1) or a 1-s DC step injection (top and bottom right). B: DSI magnitude as a function of the number of spikes evoked during a 1-s DC step injection in control (black, n = 20) or in the presence of AM251 (5 μM, gray, n = 8) or THL (1 μM, white, n = 3). The average change was measured between 1 and 5 s after the end of neuronal discharge. C: voltage-clamp recordings (Vhold = −80 mV) of spontaneous GABAergic activity interrupted by transient current-clamp switches during which brief DC injections were performed to evoke a series of APs at various rates and durations. Insets represent voltage fluctuations during each paradigm. D: DSI magnitude measured at various spike frequency rates and number of APs. The number of spikes was 1 (white), 5 (gray), or 30 (black). **P < 0.01; n.s., not significant. E1: voltage-clamp recordings of spontaneous GABAergic activity (Vhold = −50 mV) interrupted by episodic current-clamp switches, during which 1-s duration DC injections were performed to evoke a series of APs at various rates (insets). The last but 1 series of APs was evoked at a precise time to replay a place-cell discharge. E2: spike rate vs. DC injection for the cell depicted in E1 and measured for DC injections evoked every 30 s (black) or 5 s after the last DC injection of the series (white). E3: coefficient of variation of the interspike interval (CVISI) vs. spike rate for the cell depicted in A. F: pooled normalized changes in excitability observed during DSI induced by an in vivo spike pattern (n = 4 black circles) or a large DC step (n = 6 gray circles). G: pooled normalized changes in CVISI observed during DSI. The control CVISI was interpolated from the spike rate measured during DSI (n = 4 and 6 as for F).
Fig. 4.

Pyramidal cell firing tunes spike timing in CA1 pyramidal neurons. A: current-clamp recording of a cell receiving fluctuating GABAergic dynamic inputs mimicking DSI after the replay of an in vivo place-cell firing pattern and a series of excitatory postsynaptic conductances (EPSG) before (control) and during dynamic DSI (dDSI) and during recovery from dDSI. Stim, stimulus. B: 5 superimposed Vm fluctuations in response to an EPSG recorded in the presence of spontaneous dynamic-clamp IPSGs, in control conditions, and during DSI and recovery (same cell as and taken from times marked by the arrowheads in A). C: excitatory postsynaptic potential (EPSP)-spike latencies for the cell depicted in F in control and during DSI and recovery. D: summary for EPSP-spike jitter measured in different cells for the same protocol as the 1 depicted in A (n = 4). The filled circles correspond to the mean spike jitter. *P < 0.05.
Neuronal and synaptic modeling.
GABAergic and glutamatergic synaptic events were modeled as a conductance with a dual-exponential time course of the form [1−exp(−t/τrise)·exp(−t/τdecay)] where τrise/decay is the rise/decay time constant. Spontaneous activity was created by convolution of the synaptic event template with the Poisson train occurrence of spontaneous events. τrise And τdecay were 1 and 5.5 ms for excitatory postsynaptic conductances (EPSGs) and 1.5 and 14.7 ms for inhibitory postsynaptic conductances (IPSGs), respectively (Caillard 2011). IPSG amplitude varied according to a binomial distribution with n = 5 and P = 0.2. Feedforward IPSGs (FF-IPSGs) were sent with a delay of 2 ms after EPSGs (Pouille and Scanziani 2001). The conductance instruction was then either converted to an analog signal to instruct the dynamic-clamp amplifier or injected into the computational model. The latter was based on a leaky integrate-and-fire model with random threshold (RT-LIF; Caillard 2011; Holden 1976; Manwani et al. 2002): Cm·dVi/dt = σi·gi·(Vo − Vi) + σi·I with Cm = 400 pF; Vo = −65 mV. A spike is generated when Vi reaches the action-potential threshold (−45 ± 1 mV, mean ± SD), and the Vm resets to Vo. Resting membrane resistance was set at 200 MΩ. GABAergic and glutamatergic synaptic events, affecting both gi and I when present, were included according to the equations IGABA = gGABA·(Vm−EGABA) and IGLU = gGLU·(Vm−EGlu). EGABA and EGlu were respectively set at −70 and 0 mV. All modeling was performed with IGOR Pro software.
Dynamic-clamp.
To add artificial inhibitory postsynaptic potentials (IPSPs) and EPSPs, we used a fast dynamic-clamp system with National Instruments data acquisition cards (PCI-6052E or PCI-6251) and NI-DAQmx 9.21, controlled by the QuB software (http://www.qub.buffalo.edu/; Milescu et al. 2008). The dynamic-clamp system was running on an independent personal computer mounted with a dual eight-core Xeon processor at 2.67 GHz. The feedback loop (>90 kHz) continuously read the Vm from the MultiClamp 700B, EPSG, and/or IPSG and the DC command from the PCI-6723 NIDAQ card controlled by IGOR Pro software on a separate computer and generated an output according to the equation Isum = EPSG·(Vm−EGlu) + IPSG·(Vm−EGABA) + DC. EGlu and EGABA were set respectively at 0 and −75 mV.
Dynamic-clamp experiments were performed in the presence of kynurenate (2 mM) and PTX (100 μM). DSI was modeled by an initial reduction of presynaptic activity to 10% of control, followed by a 30-s exponential recovery of GABAergic charge. A minimum of six dynamic DSI (dDSI) trials, with place-cell in vivo firing patterns associated with every other trial, were performed per recorded cell to evaluate the contribution of both dDSI and in vivo place-cell firing to EPSP-spike coupling. EPSP-spike coupling was evaluated by measuring both EPSP-spike latency and temporal jitter in control conditions with dynamic GABAergic activity reduced to approximately 20 and 80% for DSI and recovery, respectively.
For dynamic FFI experiments, the following protocol was performed: EPSG peak amplitude was set so that spikes were successfully triggered with an average latency of ∼5 ms after each EPSG; the largest FF-IPSG amplitude was then defined to prevent action-potential firing when recording 20 successive EPSG/IPSG sequences. A preliminary EPSP-spike probability curve was drawn online after testing a few IPSG amplitudes several times. It served to define, for a given FF-IPSG amplitude, the number of EPSG/IPSG sequences that should theoretically be injected into the cell to get one successful coupling, with the limit probability of 0.05. Thereafter, EPSG/IPSG sequences with variable FF-IPSG amplitudes were injected in random order, such that EPSP-spike latencies at low EPSP-spike-coupling probabilities could successfully be collected.
Analysis.
Spontaneous synaptic activity and spike analysis were performed with IGOR Pro (WaveMetrics). A collection of IGOR Pro functions (NeuroMatic, J. Rothman, http://www.neuromatic.thinkrandom.com) were used in addition to homemade functions.
To quantify spontaneous GABAergic activity, we integrated the current area under baseline for each 500-ms bin of recordings to determine the total synaptic charge received by the cell over time. This parameter captured changes in both the frequency and the amplitude of synaptic events. DSI was measured as the change in the average charge in the 4 s that followed action-potential discharge or depolarization to 0 mV. DSI magnitude depicted in Figs. 1 and 2 are defined as the percentage of reduction of GABAergic charge during this period of time.
Spike threshold was detected when dV/dt exceeded 10 mV/ms. Changes in excitability were evaluated for equivalent DC steps. Measurements of coefficient of variation of ISI (CVISI) were performed by averaging CV for each sequential interval along the discharge starting from the second spike. With this method, the reproducibility of the full discharge from one trial to another could be evaluated independently of spike-frequency adaptation that was observed experimentally (Caillard 2011). During the recordings dedicated to the measurement of CVISI, small and large DC steps alternated to allow DSI recovery and prevent its buildup over time.
Measurement of spike-time jitter at 20 spikes per second was performed by interpolating the CVISI vs. average spike discharge with either the best linear or exponential fit. For EPSP-spike-coupling experiments, action-potential coupling was evaluated, and latencies between the beginning of EPSG and spike threshold were collected. Jitter corresponded to the SD of the mean latency.
Statistical comparisons were made using the Mann-Whitney U test. The degree of freedom (df), the value of the U test (U), as well as z and P are provided in results using the format of “U(df) = U, z = z value, P = P value.” Differences were considered significant when P < 0.05. Latencies are reported as means ± jitter where jitter corresponds to SD of the mean. Other data are reported as means ± SE.
RESULTS
Place-cell firing induces eCB-mediated decrease of GABAergic transmission.
To determine whether waking firing patterns of place cells can induce DSI, spiking activity recorded in vivo was replayed in vitro while monitoring GABAergic activity in CA1 pyramidal cells. Neurons were recorded in the presence of kynurenate (2 mM) to block ionotropic glutamatergic receptors and monitor the temporal dynamics of GABAergic activity together with carbachol (5 μM) to promote spontaneous inhibitory postsynaptic currents (IPSCs; Pitler and Alger 1992) and an elevated intracellular chloride concentration to detect GABAA events reliably. In these conditions, GABAA synaptic activity was detected as large inward currents when neurons were recorded at Vm = −80 mV. The CA1 place-cell firing patterns were obtained from rats performing a spatial learning task on a cheeseboard maze (Dupret et al. 2010) and converted into short current steps to elicit action potentials in vitro (see materials and methods). Infield place-cell discharge (i.e., occurring inside the place field) observed across successive learning trials was followed in vitro by a transient reduction of the GABAergic input activity (Fig. 1, A and B). The time integral of the spontaneous occurring currents, referred to as GABAergic charge (see materials and methods), was reduced within the 4 s following each place field traversal [Fig. 1B; 31 ± 4, 45 ± 5, and 36 ± 3% of baseline GABAergic charge after each replayed trial; P < 0.05 for each place field traversal; U(8) = 0, z = 2.31, P = 0.014]. This induction of DSI by infield place-cell firing patterns was prevented by adding to the intracellular solution the THL (1 μM), an inhibitor of sn-1-diacylglycerol lipase responsible for 2-AG synthesis [Fig. 1, C and D; P > 0.05 for each place field traversal; U(8) = 5, z = 0.87, P = 0.24; U(8) = 2, z = 1.73, P = 0.06; U(8) = 6, z = 0.58, P = 0.34]. Thus the DSI induced by the replay of CA1 place-cell discharge involved the endogenous CB1R agonist 2-AG. Hippocampal place cells can exhibit substantial differences in their discharge (McNaughton et al. 1983). We then replayed the patterns recorded from another, less active cell (Fig. 1, E–H). In this case, however, the discharge of fewer spikes failed to induce DSI [6 spikes discharged; DSI: 8 ± 6%; U(18) = 36, z = 0.40, P = 0.35], suggesting that only the discharge of the most active place cells can induce DSI.
We next investigated how spiking activity governs the induction of the DSI and its magnitude. Pyramidal cells were driven with action-potential bursts defined by the number and frequency of action potentials (Fig. 2). The DSI induced by the replay of an in vivo discharge of 29 action potentials [Fig. 2A, top left; 53 ± 7%, U(18) = 0, z = 3.58, P < 0.001] and by a depolarizing current eliciting a comparable number of spikes [Fig. 2A, top right; 55 ± 8%, U(12) = 0, z = 2.88, P = 0.001] were similar [U(15) = 27, z = 0.0, P = 0.500]. In both cases, a lower number of elicited spikes failed to induce a DSI [Fig. 2A, bottom; in vivo replay: U(13) = 11, z = 1.08, P = 0.14; constant depolarizing current: U(8) = 8, z = 0.0, P = 0.56]. The DSI was indeed governed by the number of spikes discharged (Fig. 2, B–D), and its induction was prevented both by the CB1R antagonist AM251 [Fig. 2B; 5 μM; 0 ± 3%; U(10) = 10, z = 0.52, P = 0.34] and the intracellular dialysis of THL [Fig. 2B; 1 μM; 1 ± 3%; U(6) = 3, z = 0.65, P = 0.35]. Within the 10- to 250-Hz frequency range, however, a given number of action potentials induced a DSI of similar magnitude (Fig. 2D).
Taken together, these results showed that place-cell firing patterns replayed in vitro can induce an eCB-mediated DSI provided that the number of action potentials emitted is sufficiently high.
eCBs mediate self-tuning of spike timing in CA1 pyramidal neurons.
Next, we determined the consequence of DSI on action-potential firing and timing of CA1 pyramidal cells. We evoked sequences of bursts of action potentials at various intraburst rates using DC injections every 30 s and monitored spontaneous GABAergic activity in between (Fig. 2E1). We measured spike rate as a function of DC injection (Fig. 2E2) and calculated the mean CVISI along the discharge (see materials and methods; Fig. 2E3) to evaluate spike-train precision as a function of discharge rate before DSI. To evaluate excitability and CVISI during DSI, the last burst of the sequence was elicited 5 s after the next to last burst, when DSI was maximal. When cells were recorded with a physiological, hyperpolarizing GABAA reversal potential, the DSI induced either by a constant depolarizing current or place-cell patterns did not significantly change cell excitability [Fig. 2F; 98 ± 3%, U(20) = 40, z = 0.76, P = 0.22] but improved intraburst spike-time precision as shown by a smaller CVISI [Fig. 2G; 81 ± 5%, U(20) = 10, z = 3.02, P = 0.001]. To confirm that CB1-mediated reduction in GABAergic activity was required for the improvement of spike-train precision during DSI, we performed similar protocols while recording cells with an elevated intracellular chloride concentration to detect GABAA events reliably. In control conditions, the mean CVISI decreased following sustained burst discharge and related maximal DSI [Fig. 3A; 78 ± 5% of control, U(20) = 10, z = 3.02, P = 0.001]. In the presence of AM251 (Fig. 3B; 5 μM), sustained discharge did not induce DSI nor improved spike-train precision [97 ± 5% of the CVISI measured in AM251, U(16) = 24, z = 0.84, P = 0.200]. If the reduced GABAergic activity observed during DSI was responsible for the improvement in spike-train precision, DSI should not improve spike-train precision in the presence of the GABAA receptor blockers. This was indeed the case when cells were recorded in the presence of PTX [100 μM; Fig. 3C; 98 ± 2% of the CVISI measured in PTX; U(16) = 32, z = 0.0, P = 0.50].
Fig. 3.

Endocannabinoid-mediated self-tuning of spike timing in pyramidal neurons. A1: voltage-clamp recordings (Vhold = −80 mV) of spontaneous GABAergic activity interrupted by episodic current-clamp switches, during which 1-s duration DC injections were performed to evoke a series of APs at various rates (top insets). The last series of APs was performed 4 s after the previous 1 to evaluate spike timing during DSI. A2: CVISI vs. spike rate for the cell depicted in A1 in control (black) and during DSI (white). A3: summary of changes in CVISI for the discharge rate during DSI vs. an equivalent rate of discharge during the control period (n = 10). B: same experiments as in A but in the presence of AM251 (5 μM, n = 8). C: same experiments as in A but in the presence of picrotoxin (PTX; 100 μM, n = 8). D1: waterfall view of membrane-potential (Vm) fluctuations from the 4th to the 5th spike of the discharge in response to a DC step (1 s, 150 pA) in the absence (blue) and when spontaneous inhibitory postsynaptic conductances (SP-IPSGs) were dynamically injected at a rate of 33 events per second (orange). The 5th spike of each discharge was set as the time reference (arrowheads). D: CVISI vs. spike firing rate (D2) and firing rate vs. DC step (D3) in control (blue) and in the presence of SP-IPSGs randomly injected at a rate of 10 (green), 33 (orange), or 100 (red) events per second (evts/s). E: changes in excitability (black) and CVISI (white) when neurons received different rates of SP-IPSGs. F: summary of changes in CVISI for a discharge rate of 20 spikes per second in control conditions (n = 10), after DSI, in the presence of AM251 (5 μM, n = 8) and in the presence of PTX and SP-IPSGs injected dynamically (Dyn) at a rate of 0 (n = 8), 10, 33, and 100 events per second (n = 5). *P < 0.05, **P < 0.01.
These results suggest that the improvement of spike-train precision following sustained burst discharges required a reduction in spontaneous GABAergic activity under the control of eCBs. We addressed this directly by dynamically injecting random IPSG waveforms (spontaneous IPSGs, SP-IPSGs) into CA1 pyramidal cells recorded in the presence of kynurenate (2 mM) and PTX (100 μM; Fig. 3D) to evaluate the consequence of spontaneous GABAergic activity on excitability and spike precision. For the purpose of these dynamic-clamp experiments, the GABAA reversal potential was set at −75 mV. Changes in excitability were normalized for each individual cell by the average change in spike number at each DC step when SP-IPSGs were present. We found that the rate of spontaneous GABAergic activity predicted both changes in excitability and CVISI, although GABAergic activity affected spike-train precision (CVISI) more than excitability (Fig. 3E). The level of spontaneous GABAergic activity correlated with the change in CVISI (Fig. 3F): in the presence of AM251 (5 μM), pyramidal cells received a higher level of spontaneous GABAergic activity [148 ± 23% of the mean drug-free GABAergic charge; U(18) = 21, z = 1.69, P = 0.046]; in the presence of the GABAA receptor blocker PTX (100 μM), CVISI was reduced relative to the drug-free condition [57 ± 5% of control, U(18) = 14.5, z = 2.27, P = 0.012]. When comparing CVISI measured in different pharmacological or dynamic-clamp conditions, we found that CVISI in drug-free conditions before and during DSI corresponded to spontaneous dynamic-clamp GABAergic activity of 33 events per second [U(15) = 15, z = 1.22, P = 0.11] and 10 events per second [U(15) = 18, z = 0.86, P = 0.19], respectively. Moreover, the CVISI measured in AM251 (5 μM) was comparable with the CVISI measured with a spontaneous dynamic-clamp GABAergic activity of 100 events per second [U(13) = 18, z = 0.29, P = 0.38]. These results demonstrate that spontaneous GABAergic activity set the initial level of spike-train precision and conditioned, under the control of eCB signaling, the improvement of spike-train precision following sustained neuronal discharge.
Tuning of EPSP-spike coupling by spontaneous inhibition and FFI.
Next, we addressed how DSI affected the tuning of neuronal discharge in response to EPSPs, a phenomenon referred to as EPSP-spike coupling. To avoid the trial-to-trial fluctuation of EPSP amplitude, we dynamically elicited EPSPs by injecting EPSGs to CA1 neurons together with randomly injected IPSGs (Fig. 4, A and B; see materials and methods). dDSI was simulated by an initial 10% reduction of IPSG rate followed by a 30-s exponential recovery. Following dDSI, EPSP-spike latency remained stable [92 ± 1% of control latency, U(8) = 4, z = 1.15, P = 0.171; Fig. 4C], but EPSP-spike jitter decreased [61 ± 5% of control jitter, U(8) = 0, z = 2.31, P = 0.014; Fig. 4D], showing that spontaneous GABAergic activity received by CA1 neurons correlated with their spike-time precision.
In the hippocampus, eCB CB1Rs are predominantly expressed on the axon terminals of CCK/CB1-expressing interneurons that partially mediate the FFI of CA1 neurons (Glickfeld and Scanziani 2006; Katona et al. 1999; Tsou et al. 1999). We next aimed to evaluate how a transient suppression of FFI might tune EPSP-spike coupling during DSI. We first placed an extracellular stimulation electrode in the stratum radiatum 200–400 μm from the recorded CA1 pyramidal cell body (Fig. 5A) to stimulate Schaffer collaterals, evoke both a monosynaptic excitatory postsynaptic current (EPSC) and a disynaptic IPSC (Pouille and Scanziani 2001), and evaluate the consequence of a depolarization on both amplitude responses (Fig. 5B). We observed that a 1-s depolarization to 0 mV reliably elicited within 5 s a transient reduction of the GABAergic response (Fig. 5C) explained by a reduced disynaptic IPSC [Fig. 5D; evoked IPSC; 76 ± 2%; U(8) = 0, z = 2.31, P = 0.014] while the monosynaptic EPSC remained unaffected [Fig. 5D; evoked EPSC; 105 ± 6%; U(8) = 6, z = 0.58, P = 0.34]. Thus the depolarization of CA1 pyramidal cells transiently decreased FFI while the excitatory drive from the Schaffer collaterals remained constant. To evaluate how this change in excitatory/inhibitory balance affected spike timing of CA1 pyramidal cells, neurons were next driven by dynamic EPSGs followed with a 2-ms delay by FF-IPSGs. In this case, both EPSP-spike latency and jitter could be evaluated for the full range of EPSP-spike-coupling probabilities (see materials and methods). We found that increased FF-IPSG amplitude reduced EPSP-spike-coupling probability (Fig. 5, E and F). In contrast, we observed a bell-shaped response curve of EPSP-spike latency as a function of IPSG amplitude (Fig. 5G). A similar bell-shaped relationship was observed between the EPSP-spike precision (as measured by the spike jitter) and the spike-coupling probability in all recorded cells, with the maximum jitter for a mean coupling probability of 0.50 ± 0.06 (Fig. 5H; n = 4).
Fig. 5.

Biphasic tuning of EPSP-spike timing by feedforward inhibition (FFI). A: schema of the stimulating and recording (REC) configuration for evoking FFI. SC, Schaffer collaterals. B: superimposed current responses recorded in a CA1 pyramidal cell following the extracellular stimulation of the Schaffer collateral pathway in control and 1 and 30 s after DSI. Stimulation intensity was set to record a sequence of inward and outward currents mediated by glutamatergic and GABAergic synapses, respectively, while recording in a voltage-clamp configuration at −50 mV with a low intracellular chloride concentration. C: fluctuations (gray circles) and average (black circles) evoked IPSC (eIPSC) amplitudes over time before and after a 1-s depolarization to 0 mV for 5 successive DSI protocols. Shown is the same cell as the 1 depicted in B. IPSC amplitude was normalized to 1 during the control period. D: average fluctuations of normalized evoked excitatory postsynaptic current (eEPSC; red) and eIPSC amplitudes (blue) over time before and after a 1-s depolarization to 0 mV (n = 4). E, from top to bottom: superimposed Vm fluctuations in response to an EPSG (peak amplitude 6.25 nS) followed after 2 ms by feedforward IPSGs (FF-IPSGs) of various amplitudes (black, 1 nS; light gray, 2 nS; dark gray, 2.5 nS). Vm fluctuations associated with a spike emission failure are also displayed. The lower traces correspond to the EPSG and IPSG fluctuations over time for the 3 different conditions. F: EPSP-spike-coupling probability vs. the amplitude of feedforward IPSG. The black and gray circles correspond to conditions depicted in E. G: individual EPSP-spike latencies (red) superimposed with the mean EPSP-spike latency ± jitter vs. the amplitude of feedforward IPSG for the cell depicted in E. The black and gray circles correspond to conditions depicted in E. The black and red traces correspond to 3rd-order polynomial fits of mean latency and mean latency ± jitter, respectively. H: 3rd-order polynomial fits of EPSP-spike jitter vs. the probability of EPSP-spike coupling measured in different cells.
These results imply that the integration time window for Schaffer collateral EPSPs depended on the recruitment of GABAergic interneurons mediating disynaptic inhibition and followed a biphasic tuning conditioned to the initial EPSP-spike-coupling probability. Since a DSI of FFI was observed in CA1 pyramidal neurons, spike-time precision changed according to the initial contribution of FFI to EPSP-spike coupling.
Theoretical contribution of spontaneous inhibition and FFI to EPSP-spike coupling.
The coexistence of correlated (evoked) and uncorrelated (spontaneous) GABAergic inhibitions implies that the integration time window of excitatory inputs by CA1 pyramidal neurons may fluctuate depending on the relative contribution of both types of inhibition before and after place-cell discharge activity. To evaluate how spontaneous inhibition and FFI tune EPSP-spike coupling, we used a RT-LIF model that focuses on subthreshold membrane properties while excluding the mechanisms underlying action-potential generation (Holden 1976; Manwani et al. 2002). Such a model also fairly reproduces the spike-time jitter attributed to the stochastic properties of voltage-dependent ion channels (Caillard 2011).
In a 1st series of simulations, the model received excitatory inputs represented by EPSGs of different amplitudes (0–100 nS) combined or not with spontaneous GABAergic activity, referred to as SP-IPSG (0–250 events per second; Fig. 6, A–D). In the absence of inhibition, the ESPG amplitude determined both EPSP-spike-coupling probability and spike-time precision. Once spontaneous GABAergic activity was added to the model, both EPSP-spike latency and jitter (as expressed by SD of latency; Fig. 6B) increased with SP-IPSG rate. Thus spontaneous GABAergic activity reduces EPSP-spike-coupling probability (Fig. 6C) and increases spike latency and jitter at any given EPSG amplitude (Fig. 6D).
Fig. 6.

Modeling tuning of EPSP-spike coupling by spontaneous and feedforward GABAergic activity. A, from left to right: superimposed (25) Vm fluctuations in response to a constant EPSG (80 nS) in the absence of spontaneous GABAergic activity in the presence of SP-IPSGs occurring at a rate of 50 and 200 events per second. On the top of each group of traces is represented the mean latency (Lat) ± jitter (n = 250). The EPSP-spike-coupling probability and latency ± jitter (in milliseconds) are written under Vm. Only Vm traces where a spike was successfully emitted are displayed. SP-IPSG fluctuations associated with a spike emission success or failure are shown in blue and gray, respectively (25 traces each). The EPSG is colored in black. B: EPSP-spike-coupling probability (red) and latency ± jitter (black) vs. the rate of randomly occurring GABAA events per second. EPSG amplitude was set at 80 nS (n = 250). C: EPSP-spike-coupling probability displayed on a pseudocolor scale vs. SP-IPSG rate and EPSG amplitude. The dashed line corresponds to the conditions displayed in A and B. D: EPSP-spike-coupling jitter displayed on a pseudocolor scale vs. SP-IPSG rate and EPSG amplitude. The dashed line corresponds to the conditions displayed in A and B. E, from left to right: superimposed (25) Vm fluctuations in response to a constant EPSG (80 nS) in the absence of FF-IPSG with FF-IPSG amplitude of 38.5 and 60 nS. On the top of each group of traces is represented the mean latency ± jitter (n = 250). The EPSP-spike-coupling probability and latency ± jitter (in milliseconds) are written under Vm. The EPSG (black) and FF-IPSG (red) are displayed and aligned with Vm. F: EPSP-spike-coupling probability (red) and latency ± jitter (black) vs. the amplitude of the FF-IPSG. EPSG amplitude was set at 80 nS (n = 250). G: EPSP-spike-coupling probability displayed on a pseudocolor scale vs. FF-IPSG and EPSG amplitudes. The dashed line corresponds to the conditions displayed on E and F. H: EPSP-spike-coupling jitter displayed on a pseudocolor scale vs. FF-IPSG and EPSG amplitudes. The dashed line corresponds to the conditions displayed in E and F. I, left: Vm fluctuations for a constant EPSG (80 nS) in the absence of FF-IPSG (red) with FF-IPSG amplitude of 39 (blue) and 60 nS (green). In gray is represented the fluctuating threshold. The dashed lines represent the upper and lower limits for spike threshold when considering its Gaussian distribution and its SD value (1 mV). The firing was cancelled in this simulation to see the integration of conductances over time in the absence of firing. Right traces show Vm fluctuations around spike threshold on an expanded scale. The superimposed bold traces represent the distribution of the time and Vm at which AP generation were generated in each condition. J: cumulative histograms for AP latencies for the 3 conditions depicted in I. Same color code as in I.
In the second series of simulations, the same range of EPSGs was injected into the model in association with FF-IPSGs, referred to as FF-IPSG of various amplitudes (0–100 nS; Fig. 6, E–H). Both spike-time latency and jitter (Fig. 6F) increased with the amplitude of FF-IPSG until EPSP-spike probability dropped to 0.5 (Fig. 6, F and G); however, a further increase of FF-IPSG then decreased these two key features of EPSP-spike coupling.
To identify the mechanism underlying this biphasic tuning of EPSP-spike coupling, we next determined how FFI affected EPSP amplitude and kinetics and correlated these parameters with spike coupling. In the absence of FFI (Fig. 6E, left), EPSPs crossed the spike threshold rapidly (Fig. 6I; 0 nS), and as a consequence both the mean spike latency and jitter were small (Fig. 6J; 2.6 ± 0.1 ms). Increasing FFI reduced EPSP amplitude and shortened its decay time constant (Fig. 6I). With a FFI of 39 nS, EPSP-spike-coupling probability fell to 0.5 (Fig. 6E, middle). Because EPSP peak remained in the lower part of the Gaussian noise distribution of spike threshold, spike latency was distributed in the widest range (Fig. 6J; 3.1 ± 0.4 ms). For stronger FFI (60 nS), EPSP peak amplitude was low with respect to spike threshold, and the fast repolarization shortened the time window during which EPSP could elicit an action potential (Fig. 6I; 60 nS). As a consequence, spikes were rarely fired (Fig. 6E, right) and occurred within a short and narrow latency range (Fig. 6J; 2.6 ± 0.3 ms). Therefore, both the amplitude and the repolarization rate of the EPSP/IPSP sequence determined the broadness of the integration time window with respect to the fluctuating spike threshold for action-potential generation. With these mechanisms, the RT-LIF model matched the experimental data showing that FFI can tune EPSP-spike-time precision and that it correlated with EPSP-spike-coupling probability.
Finally, we determined how the simultaneous presence of spontaneous and FFI affected EPSP-spike coupling (Fig. 7). We 1st found that increasing spontaneous activity (range 0–100 SP-IPSG events per second) while maintaining FFI constant (39-nS FF-IPSG) reduced EPSP-spike-coupling probability below its 0.5 initial value but had no effect on spike-time latency and precision (Fig. 7, A and B). Thus a DSI of spontaneous GABAergic activity would not be associated with any change in EPSP-spike-time precision if FFI is present and insensitive to DSI. However, increasing FFI (range 0- to 70-nS FF-IPSGs) while maintaining constant the spontaneous inhibition rate (50 SP-IPSG events per second) reduced the EPSP-spike-coupling probability below its 0.5 initial value as previously but decreased EPSP-spike jitter (Fig. 7, C and D). In contrast to the simulation performed without spontaneous inhibition (Fig. 6F), FFI was only associated with an improvement of spike-time precision as the biphasic tuning of EPSP-spike timing was not observed (Fig. 7, C and D). Thus a DSI of FFI would be associated with a disruption in EPSP-spike-time precision if spontaneous GABAergic activity is present and insensitive to DSI. Last, when both FFI and spontaneous inhibition were simultaneously present and varied with a constant SP-IPSG-to-FF-IPSG ratio, they biphasically tuned EPSP-spike latencies and jitter responses (Fig. 7, E and F). The largest spike-time jitter was observed at a spike-coupling probability of 0.5. Thus tuning of spike-time precision was governed by the relative contribution of the two combined forms of inhibition during DSI: DSI of FFI imposed a biphasic response of spike-time jitter, unless spontaneous inhibition was sufficiently high to prevent this biphasic time code (Fig. 7, G and H).
Fig. 7.

Modeling tuning of EPSP-spike coupling by combined spontaneous inhibition and FFI. A, from left to right: superimposed (25) Vm fluctuations in the presence of constant FF-IPSG of 39 nS and various rates of SP-IPSGs (0–30-65 events per second). The EPSG amplitude was set at 80 nS. Only Vm traces where a spike was successfully emitted are displayed. On the top of each group of traces is represented the mean latency ± jitter (n = 250). The EPSP-spike-coupling probability and latency ± jitter are written under Vm. The EPSG is colored in black, FF-IPSG in red, and SP-IPSG fluctuations in blue and gray for spike success and failures, respectively. B: EPSP-spike-coupling probability (red) and latency ± jitter (black) vs. rate of spontaneous GABAergic activity. The EPSG amplitude was set at 80 nS. FF-IPSG was set at 39 nS (n = 250). C: same conditions as in A except that the model received a SP-IPSG rate of 50 events per second and FF-IPSGs of various amplitudes (0-34-44 nS). D: same graph as in B except that the model received an SP-IPSG rate of 50 events per second and FF-IPSGs of various amplitudes (n = 250). E: same conditions as in A except that the model received SP-IPSG rates and FF-IPSGs at constant ratio (7.7 events per second and 6 nS; 28.2 events per second and 22 nS; and 50 events per second and 39 nS for SP-IPSG rate and FF-IPSGs, respectively). F: EPSP-spike-coupling probability (red) and latency ± jitter (black) vs. the percentage of inhibition. One hundred percent inhibition was set when SP-IPSG rate and FF-IPSGs were 50 events per second and 39 nS, respectively. SP-IPSG rates and FF-IPSGs were changed in similar proportions from 0 to 120% of control. G: EPSP-spike-coupling probability displayed on a pseudocolor scale vs. FF-IPSG amplitude and SP-IPSG rates. The horizontal, vertical and diagonal lines correspond to the conditions displayed on A and B, C and D, and E and F, respectively. H: EPSP-spike-coupling jitter displayed on a pseudocolor scale vs. FF-IPSG amplitude and SP-IPSG rates. The horizontal, vertical, and diagonal lines correspond to the conditions displayed on A and B, C and D, and E and F, respectively.
DISCUSSION
In this study, we established that firing patterns of place cells recorded in vivo induce DSI in CA1 pyramidal cells in vitro. This DSI is mediated by the activation of CB1Rs. A major consequence of the eCB-mediated transient suppression of GABAergic inputs is the fine tuning of spike timing.
GABAergic contribution to spike-time precision.
DSI is the most widely studied form of eCB-mediated short-term plasticity, but whether it can be induced by physiological firing patterns remained to be determined. Here, we successfully induced DSI injecting waking patterns of CA1 place cells discharge recorded during a spatial task (Dupret et al. 2010). CB1Rs are expressed in axons and terminals of CCK-expressing GABAergic interneurons (Katona et al. 1999, 2000; Nyiri et al. 2005; Pan et al. 2011; Tsou et al. 1999). CCK interneurons are a heterogeneous family of interneurons that innervate pyramidal cells on their somatic and dendritic domains (for reviews, see Klausberger 2009; Klausberger and Somogyi 2008). Spontaneous GABAergic activity recorded in pyramidal cells may arise from CCK-inhibitory inputs targeting both subcellular domains (Cope et al. 2002). Nevertheless, because Schaffer collateral-associated dendritic GABAergic synapses are less sensitive to DSI than basket cell synapses (Lee et al. 2010) and as demonstrated with extracellular stimulations of dendritic GABAergic inputs (Morishita and Alger 2001), it is likely that the CB1-sensitive component of spontaneous GABAergic inhibition originates primarily from perisomatic synapses. Our experiments revealed that a transient reduction in CB1R-dependent spontaneous GABAergic activity improved spike-time precision of discharging pyramidal cells. This self-tuned improvement of spike-time precision was achieved: 1) during DSI; 2) after pharmacological blockade of GABAA receptors; and 3) when the rate of dynamic GABAA events was reduced. However, DSI was not associated with an increase in pyramidal cell firing. Although carbachol (5 μM, applied in the bath) reduced Ca2+-dependent K+ conductances activated during DSI (Pitler and Alger 1992), we observed transient shifts in the holding current. Such shifts likely reflected the incomplete blockade of these conductances known to control excitability after repetitive discharges in a feedback manner (Madison and Nicoll 1984). It is likely that DSI counterbalanced the reduction in intrinsic excitability. In fact, dynamic injection of IPSPs at various rates demonstrated that spontaneous GABAergic activity was associated with a reduction of excitability.
To evaluate the impact of DSI on EPSP-spike coupling, we have considered time, frequency, and amplitude variations in GABAergic conductance for constant excitatory conductance. A consensual correlation exists between the transience of an excitatory signal and its ability to time a spike precisely when the excitatory input signal amplitude is scaled up or down to maintain a constant spike-coupling probability (Axmacher and Miles 2004; Cudmore et al. 2010; Gastrein et al. 2011; Jaeger and Bower 1999; Mainen and Sejnowski 1995; Mittmann et al. 2005; Pouille and Scanziani 2001). Measuring simultaneously Schaffer collateral EPSCs and disynaptic IPSCs showed no significant changes in EPSC amplitude during DSI. The transient changes in EPSC amplitude observed in a subset of cells were likely due to the temporal overlap of the reduced IPSC (Wagner and Alger 1996). In hippocampal cell cultures, both excitatory and inhibitory synapses can be depressed by exogenous cannabinoid agonists or postsynaptic depolarization, but the cannabinoid sensitivity of excitatory synapses appeared lower than GABAergic synapses (Ohno-Shosaku et al. 2002; Xu et al. 2010) such that the physiological conditions for eCB-mediated transient reduction in excitatory synaptic transmission may be reached but only under conditions allowing a large increase in postsynaptic calcium concentration (Nakamura et al. 1999).
When GABAergic and glutamatergic activities were uncorrelated, i.e., in the presence of spontaneous activity, the disruption of spike-time jitter was proportional to the rate of spontaneous GABAergic activity, as represented by our modeling study (Fig. 6). Therefore, during DSI, a transient improvement in spike-time precision was observed either when the cell integrated phasic glutamatergic inputs or when it integrated summed uncorrelated inputs, giving rise to a repetitive discharge of action potentials. In contrast, when excitatory and inhibitory inputs correlated in time through the disynaptic recruitment of presynaptic interneurons, the relationship between the amplitude of the IPSG and EPSP-spike parameters were more complex and depended on the initial EPSP-spike-coupling probability (Figs. 5 and 6). In our model, voltage fluctuations before spike initiation explained how modulation of EPSP amplitude and decay kinetics by disynaptic inhibition affected EPSP-spike timing in a biphasic manner. During DSI, the reduction in disynaptic inhibition together with the initial EPSP-spike-coupling probability were critical for defining qualitatively and quantitatively the impact on EPSP-spike timing. When FFI was present and reduced coupling probability to ≥50%, spike timing was exclusively controlled by disynaptic inhibition (Fig. 7B). As FFI is only partly mediated by CB1R-expressing interneurons (Glickfeld et al. 2008; Glickfeld and Scanziani 2006), this earlier component of disynaptic inhibition should be relatively insensitive to DSI compared with feedback inhibition. Enhanced firing probability during DSI would therefore be mainly supported by uncorrelated inhibition. Nevertheless, as observed in our recordings, coincident reduction in both uncorrelated and correlated inhibition during DSI should affect spike-time precision in a biphasic manner (Fig. 7F).
Place-cell activity, DSI firing requirement, and eCBs.
We confirm here that spike-train duration determines the magnitude of DSI (Pitler and Alger 1992). However, the broad time window observed for the buildup of DSI contrasted with the firing requirements, especially at low (<20-Hz) firing frequencies, that were previously reported in the hippocampus (Hampson et al. 2003; Zhuang et al. 2005) but not in the neocortex (Fortin et al. 2004). The DSI observed at low (10-Hz) firing frequencies together with the slow time course for DSI recovery suggest that underlying postsynaptic calcium transients are large in amplitude and display slow kinetics (Lenz and Alger 1999; Myoga et al. 2009; Wang and Zucker 2001).
Carbachol enhances both the frequency of spontaneous IPSCs (Pitler and Alger 1992) and the magnitude of DSI in CA1 pyramidal neurons (Kim et al. 2002; Martin et al. 2001; Martin and Alger 1999; Pitler and Alger 1994) through the activation of postsynaptic M1 and M3 receptors (Ohno-Shosaku et al. 2003). The number of action potentials required for DSI induction may thus be linked to endogenous release of acetylcholine. Indeed, endogenous release of acetylcholine affected GABA release and DSI magnitude (Martin et al. 2001; Nagode et al. 2011; Pitler and Alger 1994). Moreover, activation of the cholinergic system in the hippocampus appears relevant when studying synaptic transmission and plasticity in vitro (Isaac et al. 2009). In fact, microdialysis measurements of acetylcholine in the hippocampus have revealed marked increases in the basal levels during locomotion, exploration, and spatial learning (for review, see Pepeu and Giovannini 2004).
Different reasons can account for the low activity threshold for DSI induction reported here compared with a previous report (Hampson et al. 2003). Not only magnitude, but also recovery time course increases with the duration of postsynaptic depolarization or action-potential number during discharge (Fortin et al. 2004; Pitler and Alger 1992; Zhuang et al. 2005). In the present study, we have measured DSI in the 4 s that followed action-potential discharge. Therefore, the firing discharge required for successfully inducing a significant DSI will appear to be lower than expected from previous studies where DSI magnitude was evaluated later following DSI induction (4–7 s after the end of induction protocol; Hampson et al. 2003). Next, the age of the animals is critical for DSI induction. In immature animals, DSI has been reported to be weak (Zhu and Lovinger 2010; Zhuang et al. 2005). The place-cell activity may not be sufficient to induce DSI in a 1-wk-old animal (Hampson et al. 2003) but adequate later in development (Langston et al. 2010; Scott et al. 2011; Wills et al. 2010).
CCK-expressing interneurons preferentially fire on the ascending phase of theta-oscillations at times when CA1 place cells with place fields in front of the animal also discharge (Dragoi and Buzsaki 2006; Huxter et al. 2008; Klausberger et al. 2005; Klausberger and Somogyi 2008; O'Keefe and Recce 1993). Moreover, place cells also exhibit a gradual phase shift relative to theta-rhythm as the animal passes through the place field (phase precession; O'Keefe and Recce 1993). Therefore, the activity-dependent transient suppression of CCK-expressing cell outputs by eCB released following place-cell infield discharge can improve spike-time precision on consecutive theta-cycles, support the segregation of spatially selective pyramidal cells forming distinct assemblies, and contribute to theta-paced dynamic flickering of hippocampal place-cell maps (Dupret et al. 2013; Jezek et al. 2011). Many place cells fire at goal locations during goal-oriented tasks, indicating that salient locations are represented in the hippocampal code (Hok et al. 2007; Hollup et al. 2001; Markus et al. 1995; McKenzie et al. 2013). Place cells encoding such locations showed increased firing synchronization during waking sharp-wave/ripple (SWR; 150–250 Hz) events (Dupret et al. 2010; O'Neill et al. 2006). Therefore, coincident firing of place cells related to goal locations may provide additional eCB levels and DSI. During SWRs, individual CCK-expressing interneurons exhibited highly variable firing responses in contrast to the reliable increased firing of PV-expressing basket cells, for instance (Klausberger et al. 2005). Moreover, during repetitive discharges, CCK interneurons display an asynchronous mode of GABA release (Ali and Todorova 2010; Foldy et al. 2006; Hefft and Jonas 2005; Karson et al. 2008; Lee et al. 2010; Losonczy et al. 2004). Because asynchronous GABA release from CCK-expressing outputs outlasts ripple episodes (Karson et al. 2009), DSI may provide an efficient retrograde presynaptic mechanism to shut down asynchronous GABA release from CCK-expressing cells. As suggested by Ali and Todorova (2010), decreasing asynchronous release would certainly reduce spike jitter. Nevertheless, spike jitter cannot be directly correlated to asynchronous release because, as shown here with FFI in dynamic-clamp and modeling experiments, a delayed inhibition, even if perfectly synchronized, can tune up and down spike jitter according to EPSP-spike-coupling probability.
In summary, our data show that place-related firing activity observed during spatial learning can drive an eCB-mediated transient reduction of synaptic inhibition received by pyramidal cells in vitro. This DSI of inhibition not only affected pyramidal cell excitability, but also spike-time precision in such a way that self-tuning of CB1R-dependent GABAergic inputs following place-cell activity may be physiologically relevant for both spike-timing coordination and network oscillations in the hippocampus.
GRANTS
This work was supported by the Institut National de la Santé et de la Recherche Médicale (Inserm), the Centre National de la Recherche Scientifique (CNRS), and a grant from the Fondation Française pour la Recherche sur l'Épilepsie (FFRE). Part of the equipment used in this study was funded by European Community (LSHM-CT-2004-511995), Agence Nationale de la Recherche (ANR 06-Neuro-014-01), and Région PACA (APO Plexin) grants attributed to D. Debanne. Salary support of F. Dubruc was provided by Région PACA (APO Plexin) Doctoral Studentship and Neuroservice (B. Buisson). D. Dupret was supported by a Medical Research Council (MRC) Intramural Programme Grant (U138197111).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
F.D. and O.C. conception and design of research; F.D. and O.C. performed experiments; F.D., D.D., and O.C. analyzed data; F.D., D.D., and O.C. interpreted results of experiments; F.D., D.D., and O.C. prepared figures; F.D., D.D., and O.C. drafted manuscript; F.D., D.D., and O.C. edited and revised manuscript; F.D., D.D., and O.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank J. Csicsvari for sharing spike-time occurrence of place cells during in vivo recordings of behavioral tasks, M. Manko for constructive comments on a previous version of the manuscript, L. S. Milescu for help in installing and tuning QuB dynamic-clamp system, L. Fronzaroli-Molinieres and C. Gomez for technical assistance, the members of Inserm UMR_S 1072 for helpful comments and suggestions during completion of this work, and D. Debanne, K. Lamsa, and M. Seagar for critical reading of the manuscript.
REFERENCES
- Ali AB, Todorova M. Asynchronous release of GABA via tonic cannabinoid receptor activation at identified interneuron synapses in rat CA1. Eur J Neurosci 31: 1196–1207, 2010 [DOI] [PubMed] [Google Scholar]
- Axmacher N, Miles R. Intrinsic cellular currents and the temporal precision of EPSP-action potential coupling in CA1 pyramidal cells. J Physiol 555: 713–725, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillard O. Pre & postsynaptic tuning of action potential timing by spontaneous GABAergic activity. PLoS One 6: e22322, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope DW, Maccaferri G, Marton LF, Roberts JD, Cobden PM, Somogyi P. Cholecystokinin-immunopositive basket and Schaffer collateral-associated interneurones target different domains of pyramidal cells in the CA1 area of the rat hippocampus. Neuroscience 109: 63–80, 2002 [DOI] [PubMed] [Google Scholar]
- Cudmore RH, Fronzaroli-Molinieres L, Giraud P, Debanne D. Spike-time precision and network synchrony are controlled by the homeostatic regulation of the d-type potassium current. J Neurosci 30: 12885–12895, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daw MI, Tricoire L, Erdelyi F, Szabo G, Mcbain CJ. Asynchronous transmitter release from cholecystokinin-containing inhibitory interneurons is widespread and target-cell independent. J Neurosci 29: 11112–11122, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Boudkkazi S, Campanac E, Cudmore RH, Giraud P, Fronzaroli-Molinieres L, Carlier E, Caillard O. Paired-recordings from synaptically coupled cortical and hippocampal neurons in acute and cultured brain slices. Nat Protoc 3: 1559–1568, 2008 [DOI] [PubMed] [Google Scholar]
- Dragoi G, Buzsaki G. Temporal encoding of place sequences by hippocampal cell assemblies. Neuron 50: 145–157, 2006 [DOI] [PubMed] [Google Scholar]
- Dupret D, O'Neill J, Csicsvari J. Dynamic reconfiguration of hippocampal interneuron circuits during spatial learning. Neuron 78: 166–180, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupret D, O'Neill J, Pleydell-Bouverie B, Csicsvari J. The reorganization and reactivation of hippocampal maps predict spatial memory performance. Nat Neurosci 13: 995–1002, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foldy C, Neu A, Jones MV, Soltesz I. Presynaptic, activity-dependent modulation of cannabinoid type 1 receptor-mediated inhibition of GABA release. J Neurosci 26: 1465–1469, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DA, Trettel J, Levine ES. Brief trains of action potentials enhance pyramidal neuron excitability via endocannabinoid-mediated suppression of inhibition. J Neurophysiol 92: 2105–2112, 2004 [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017–1066, 2003 [DOI] [PubMed] [Google Scholar]
- Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, Samad TA, Doherty P. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J Neurosci 30: 2017–2024, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastrein P, Campanac E, Gasselin C, Cudmore RH, Bialowas A, Carlier E, Fronzaroli-Molinieres L, Ankri N, Debanne D. The role of hyperpolarization-activated cationic current in spike-time precision and intrinsic resonance in cortical neurons in vitro. J Physiol 589: 3753–3773, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Atallah BV, Scanziani M. Complementary modulation of somatic inhibition by opioids and cannabinoids. J Neurosci 28: 1824–1832, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickfeld LL, Scanziani M. Distinct timing in the activity of cannabinoid-sensitive and cannabinoid-insensitive basket cells. Nat Neurosci 9: 807–815, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampson RE, Zhuang SY, Weiner JL, Deadwyler SA. Functional significance of cannabinoid-mediated, depolarization-induced suppression of inhibition (DSI) in the hippocampus. J Neurophysiol 90: 55–64, 2003 [DOI] [PubMed] [Google Scholar]
- Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F, Koehl M, Abrous DN, Mendizabal-Zubiaga J, Grandes P, Liu Q, Bai G, Wang W, Xiong L, Ren W, Marsicano G, Zhang X. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell 148: 1039–1050, 2012 [DOI] [PubMed] [Google Scholar]
- Harris KD, Hirase H, Leinekugel X, Henze DA, Buzsaki G. Temporal interaction between single spikes and complex spike bursts in hippocampal pyramidal cells. Neuron 32: 141–149, 2001 [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8: 1319–1328, 2005 [DOI] [PubMed] [Google Scholar]
- Hok V, Lenck-Santini PP, Roux S, Save E, Muller RU, Poucet B. Goal-related activity in hippocampal place cells. J Neurosci 27: 472–482, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden AV. Models of Stochastic Activity of Neurones. New York: Springer-Verlag, 1976 [Google Scholar]
- Hollup SA, Molden S, Donnett JG, Moser MB, Moser EI. Accumulation of hippocampal place fields at the goal location in an annular watermaze task. J Neurosci 21: 1635–1644, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huxter JR, Senior TJ, Allen K, Csicsvari J. Theta phase-specific codes for two-dimensional position, trajectory and heading in the hippocampus. Nat Neurosci 11: 587–594, 2008 [DOI] [PubMed] [Google Scholar]
- Isaac JT, Buchanan KA, Muller RU, Mellor JR. Hippocampal place cell firing patterns can induce long-term synaptic plasticity in vitro. J Neurosci 29: 6840–6850, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger D, Bower JM. Synaptic control of spiking in cerebellar Purkinje cells: dynamic current clamp based on model conductances. J Neurosci 19: 6090–6101, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezek K, Henriksen EJ, Treves A, Moser EI, Moser MB. Theta-paced flickering between place-cell maps in the hippocampus. Nature 478: 246–249, 2011 [DOI] [PubMed] [Google Scholar]
- Jonas P, Hefft S. GABA release at terminals of CCK-interneurons: synchrony, asynchrony and modulation by cannabinoid receptors (commentary on Ali & Todorova). Eur J Neurosci 31: 1194–1195, 2010 [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev 89: 309–380, 2009 [DOI] [PubMed] [Google Scholar]
- Karson MA, Tang AH, Milner TA, Alger BE. Synaptic cross talk between perisomatic-targeting interneuron classes expressing cholecystokinin and parvalbumin in hippocampus. J Neurosci 29: 4140–4154, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karson MA, Whittington KC, Alger BE. Cholecystokinin inhibits endocannabinoid-sensitive hippocampal IPSPs and stimulates others. Neuropharmacology 54: 117–128, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Magloczky Z, Santha E, Kofalvi A, Czirjak S, Mackie K, Vizi ES, Freund TF. GABAergic interneurons are the targets of cannabinoid actions in the human hippocampus. Neuroscience 100: 797–804, 2000 [DOI] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19: 4544–4558, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kentros C, Hargreaves E, Hawkins RD, Kandel ER, Shapiro M, Muller RV. Abolition of long-term stability of new hippocampal place cell maps by NMDA receptor blockade. Science 280: 2121–2126, 1998 [DOI] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci 22: 10182–10191, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T. GABAergic interneurons targeting dendrites of pyramidal cells in the CA1 area of the hippocampus. Eur J Neurosci 30: 947–957, 2009 [DOI] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, O'Neill J, Huck JH, Dalezios Y, Fuentealba P, Suen WY, Papp E, Kaneko T, Watanabe M, Csicsvari J, Somogyi P. Complementary roles of cholecystokinin- and parvalbumin-expressing GABAergic neurons in hippocampal network oscillations. J Neurosci 25: 9782–9793, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science 321: 53–57, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM. Interneuron networks in the hippocampus. Curr Opin Neurobiol 21: 709–716, 2011 [DOI] [PubMed] [Google Scholar]
- Langston RF, Ainge JA, Couey JJ, Canto CB, Bjerknes TL, Witter MP, Moser EI, Moser MB. Development of the spatial representation system in the rat. Science 328: 1576–1580, 2010 [DOI] [PubMed] [Google Scholar]
- Lee SH, Foldy C, Soltesz I. Distinct endocannabinoid control of GABA release at perisomatic and dendritic synapses in the hippocampus. J Neurosci 30: 7993–8000, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz RA, Alger BE. Calcium dependence of depolarization-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol 521: 147–157, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A, Biro AA, Nusser Z. Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc Natl Acad Sci USA 101: 1362–1367, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J Physiol 354: 319–331, 1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mainen ZF, Sejnowski TJ. Reliability of spike timing in neocortical neurons. Science 268: 1503–1506, 1995 [DOI] [PubMed] [Google Scholar]
- Manwani A, Steinmetz PN, Koch C. The impact of spike timing variability on the signal-encoding performance of neural spiking models. Neural Comput 14: 347–367, 2002 [DOI] [PubMed] [Google Scholar]
- Markus EJ, Qin YL, Leonard B, Skaggs WE, McNaughton BL, Barnes CA. Interactions between location and task affect the spatial and directional firing of hippocampal neurons. J Neurosci 15: 7079–7094, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LA, Alger BE. Muscarinic facilitation of the occurrence of depolarization-induced suppression of inhibition in rat hippocampus. Neuroscience 92: 61–71, 1999 [DOI] [PubMed] [Google Scholar]
- Martin LA, Wei DS, Alger BE. Heterogeneous susceptibility of GABA(A) receptor-mediated IPSCs to depolarization-induced suppression of inhibition in rat hippocampus. J Physiol 532: 685–700, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie S, Robinson NT, Herrera L, Churchill JC, Eichenbaum H. Learning causes reorganization of neuronal firing patterns to represent related experiences within a hippocampal schema. J Neurosci 33: 10243–10256, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton BL, Barnes CA, O'Keefe J. The contributions of position, direction, and velocity to single unit activity in the hippocampus of freely-moving rats. Exp Brain Res 52: 41–49, 1983 [DOI] [PubMed] [Google Scholar]
- Milescu LS, Yamanishi T, Ptak K, Mogri MZ, Smith JC. Real-time kinetic modeling of voltage-gated ion channels using dynamic clamp. Biophys J 95: 66–87, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittmann W, Koch U, Hausser M. Feed-forward inhibition shapes the spike output of cerebellar Purkinje cells. J Physiol 563: 369–378, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita W, Alger BE. Direct depolarization and antidromic action potentials transiently suppress dendritic IPSPs in hippocampal CA1 pyramidal cells. J Neurophysiol 85: 480–484, 2001 [DOI] [PubMed] [Google Scholar]
- Myoga MH, Beierlein M, Regehr WG. Somatic spikes regulate dendritic signaling in small neurons in the absence of backpropagating action potentials. J Neurosci 29: 7803–7814, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagode DA, Tang AH, Karson MA, Klugmann M, Alger BE. Optogenetic release of ACh induces rhythmic bursts of perisomatic IPSCs in hippocampus. PLoS One 6: e27691, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials [see comments]. Neuron 24: 727–737, 1999 [DOI] [PubMed] [Google Scholar]
- Nyiri G, Cserep C, Szabadits E, Mackie K, Freund TF. CB1 cannabinoid receptors are enriched in the perisynaptic annulus and on preterminal segments of hippocampal GABAergic axons. Neuroscience 136: 811–822, 2005 [DOI] [PubMed] [Google Scholar]
- O'Keefe J, Conway DH. Hippocampal place units in the freely moving rat: why they fire where they fire. Exp Brain Res 31: 573–590, 1978 [DOI] [PubMed] [Google Scholar]
- O'Keefe J, Dostrovsky J. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res 34: 171–175, 1971 [DOI] [PubMed] [Google Scholar]
- O'Keefe J, Recce ML. Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus 3: 317–330, 1993 [DOI] [PubMed] [Google Scholar]
- O'Neill J, Senior T, Csicsvari J. Place-selective firing of CA1 pyramidal cells during sharp wave/ripple network patterns in exploratory behavior. Neuron 49: 143–155, 2006 [DOI] [PubMed] [Google Scholar]
- O'Neill J, Senior TJ, Allen K, Huxter JR, Csicsvari J. Reactivation of experience-dependent cell assembly patterns in the hippocampus. Nat Neurosci 11: 209–215, 2008 [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Matsui M, Fukudome Y, Shosaku J, Tsubokawa H, Taketo MM, Manabe T, Kano M. Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde endocannabinoid signalling in the hippocampus. Eur J Neurosci 18: 109–116, 2003 [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M. Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci 22: 3864–3872, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Wang W, Zhong P, Blankman JL, Cravatt BF, Liu QS. Alterations of endocannabinoid signaling, synaptic plasticity, learning, and memory in monoacylglycerol lipase knock-out mice. J Neurosci 31: 13420–13430, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepeu G, Giovannini MG. Changes in acetylcholine extracellular levels during cognitive processes. Learn Mem 11: 21–27, 2004 [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G protein involvement in a presynaptic mechanism. Neuron 13: 1447–1455, 1994 [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci 12: 4122–4132, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293: 1159–1163, 2001 [DOI] [PubMed] [Google Scholar]
- Puighermanal E, Marsicano G, Busquets-Garcia A, Lutz B, Maldonado R, Ozaita A. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat Neurosci 12: 1152–1158, 2009 [DOI] [PubMed] [Google Scholar]
- Ramirez DM, Kavalali ET. Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr Opin Neurobiol 21: 275–282, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranck JB., Jr Studies on single neurons in dorsal hippocampal formation and septum in unrestrained rats. I. Behavioral correlates and firing repertoires. Exp Neurol 41: 461–531, 1973 [DOI] [PubMed] [Google Scholar]
- Robbe D, Montgomery SM, Thome A, Rueda-Orozco PE, McNaughton BL, Buzsaki G. Cannabinoids reveal importance of spike timing coordination in hippocampal function. Nat Neurosci 9: 1526–1533, 2006 [DOI] [PubMed] [Google Scholar]
- Schwartzkroin PA. Characteristics of CA1 neurons recorded intracellularly in the hippocampal in vitro slice preparation. Brain Res 85: 423–436, 1975 [DOI] [PubMed] [Google Scholar]
- Scott RC, Richard GR, Holmes GL, Lenck-Santini PP. Maturational dynamics of hippocampal place cells in immature rats. Hippocampus 21: 347–353, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J Physiol 385: 733–759, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 65: 320–327, 2010 [DOI] [PubMed] [Google Scholar]
- Tsou K, Mackie K, Sanudo-Pena MC, Walker JM. Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing GABAergic interneurons in the rat hippocampal formation. Neuroscience 93: 969–975, 1999 [DOI] [PubMed] [Google Scholar]
- Varvel SA, Anum EA, Lichtman AH. Disruption of CB(1) receptor signaling impairs extinction of spatial memory in mice. Psychopharmacology (Berl) 179: 863–872, 2005 [DOI] [PubMed] [Google Scholar]
- Wagner JJ, Alger BE. Increased neuronal excitability during depolarization-induced suppression of inhibition in rat hippocampus. J Physiol 495: 107–112, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zucker RS. Photolysis-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol 533: 757–763, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills TJ, Cacucci F, Burgess N, O'Keefe J. Development of the hippocampal cognitive map in preweanling rats. Science 328: 1573–1576, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL. Dynamics of the hippocampal ensemble code for space. Science 261: 1055–1058, 1993 [DOI] [PubMed] [Google Scholar]
- Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science 265: 676–679, 1994 [DOI] [PubMed] [Google Scholar]
- Xu JY, Chen R, Zhang J, Chen C. Endocannabinoids differentially modulate synaptic plasticity in rat hippocampal CA1 pyramidal neurons. PLoS One 5: e10306, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM. Developmental alteration of endocannabinoid retrograde signaling in the hippocampus. J Neurophysiol 103: 1123–1129, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang S, Hampson RE, Deadwyler SA. Behaviorally relevant endocannabinoid action in hippocampus: dependence on temporal summation of multiple inputs. Behav Pharmacol 16: 463–471, 2005 [DOI] [PubMed] [Google Scholar]