

Abstract

In this Article, we have demonstrated the in vivo efficacy of D-512 and D-440 in a 6-OHDA-induced unilaterally lesioned rat model experiment, a Parkinson’s disease animal model. D-512 is a novel highly potent D2/D3 agonist, and D-440 is a novel highly selective D3 agonist. We evaluated the neuroprotective properties of D-512 and D-440 in the dopaminergic MN9D cells. Cotreatment of these two drugs with 6-OHDA and MPP+ significantly attenuated and reversed 6-OHDA- and MPP+-induced toxicity in a dose-dependent manner in the dopaminergic MN9D cells. The inhibition of caspase 3/7 and lipid peroxidation activities along with the restoration of tyrosine hydroxylase levels by D-512 in 6-OHDA-treated cells may partially explain the mechanism of its neuroprotective property. Furthermore, studies were carried out to elucidate the time-dependent changes in the pERK1/2 and pAkt, two kinases implicated in cell survival and apoptosis, levels upon treatment with 6-OHDA in presence of D-512. The neuroprotective property exhibited by these drugs was independent of their dopamine-agonist activity, which is consistent with our multifunctional drug-development approach that is focused on the generation of disease-modifying symptomatic-treatment agents for Parkinson’s disease.
Keywords: Dopamine, Parkinson’s disease, multifunctional drugs, dopamine agonist, MN9D cells, MPTP, 6-OHDA
Introduction
The impairment of movement in Parkinson’s disease (PD) results from the progressive loss of dopamine (DA) neurons in the substantia nigra (SN) and the neurotransmitter DA in the nigrostriatal dopaminergic pathway.1,2 PD is one of the major neurodegenerative diseases and is the second most prevalent neurodegenerative disease after Alzheimer’s disease. The loss of dopaminergic cells in the substantia nigra pars compacta gives rise to the motor symptoms of PD, which include bradykinesia, rigidity, and tremor.2−4
The etiology of the loss of nigral dopaminergic neurons in PD is not well understood. The complex pathogenesis of PD includes the presence of oxidative stress and environmental toxins that lead to mitochondrial dysfunction and cell death, have been implicated in sporadic PD cases.5−7 The production of reactive oxygen species (ROS) can be accelerated in the brain for different reasons.8,9 Some of these reasons include an imbalance in the level of dopamine, which results in the formation of oxidized species that can cause damage to the mitochondrial system, resulting in the production of ROS.10,11 The presence of a compromised natural antioxidant defense in PD results in the generation of ROS, which is considered to be an important component in the pathogenesis of the disease.
The dopamine precursor l-DOPA (l-3,4-dihydroxyphenylalanine) has been used as the gold-standard treatment agent for PD from the time of its discovery almost 40 years ago.12l-DOPA is α-carboxy dopamine, which, in the presence of dopamine decarboxylase, gets converted into dopamine in the surviving dopamine neurons.12,13 This therapy works well in the early stages of the disease process by restoring motor function in virtually all patients. However, long-term use of l-DOPA gives rise to motor fluctuations with dyskinesias as well as a decrease in the duration of the response to a given l-DOPA dose.14 Prolonged use of l-DOPA also gives rise to “on” and “off” episodes, resulting in additional complications. Long-term exposure to l-DOPA has been implicated in the acceleration of the dopamine neurodegeneration process.15,16
Dopamine agonists that provide an alternative to l-DOPA therapy have been used in therapy for PD.17 DA agonists have been shown to delay the complications associated with l-DOPA use. DA agonists such as pramipexole have been shown to possess neuroprotective properties in various in vitro cell-culture toxin models. The D3 receptor has been implicated in neuroprotection because the D3 receptor-preferring agonists protected DA neurons against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA) neurotoxicity more robustly than the less selective D3 receptor-preferring agonists.18−21
PD is a disease with a complex pathogenesis. Because of the complex nature of the pathogenesis of PD and neurodegeneration in general, drugs that target a single functional component have been shown to be inadequate, and a growing body of literature suggests that suitable multifunctional drugs might be more effective to act as disease-modifying agents.22−24 Thus, such multifunctional drugs with antioxidant property could slow the progression of PD while being symptomatically beneficial in alleviating motor dysfunction.25
Our goal of developing drugs with multifunctional properties prompted us to work with our hybrid template with D2/D3 agonist activity to explore whether the properties that are important for neuroprotection could be incorporated into these dopamine-agonist molecules.26−28 In this regard, we have recently developed multifunctional D2/D3 agonists with antioxidant and iron-chelation properties that have exhibited neuroprotection properties in different animal models.21,29 Continuing along this approach, we recently developed a lead indole molecule, D-512, as a potent agonist for both the D2 and D3 receptors as well as highly D3-receptor-selective agonist D-440 (Figure 1 and Table 1).30 In vitro antioxidant studies indicated the potent antioxidant activity of these two drugs. In this Article, we report the evaluation of the effect of the treatment with different doses of D-512 and D-440 on reversing 6-OHDA- or MPP+-induced toxicity in the dopaminergic MN9D cells. It is important to point out that the neuroprotection effect of these molecules is independent of their agonist activity because MN9D cells, although dopaminergic, do not express dopamine receptors. Furthermore, because these drugs are being developed as potential therapeutics for PD, we wanted to evaluate the in vivo efficacy of D-512 and D-440 in a Parkinson’s disease animal-model experiment.
Figure 1.

Molecular structures of D-512 and D-440.
Table 1. Stimulation of [35S]GTPγS Binding to the Cloned Human D2 and D3 Receptors Expressed in CHO Cells.
| hCHO-D2 |
hCHO-D3 |
||||
|---|---|---|---|---|---|
| compound | EC50 (nM) [35S]GTPγS | Percent of Emax | EC50 (nM) [35S]GTPγS | Percent of Emax | D2/D3 |
| DA | 227 ± 11 | 100 | 8.57 | 100 | 26.5 |
| (−)-D-440a | 114 ± 12 | 101 ± 5 | 0.26 ± 0.07 | 103 ± 10 | 438 |
| (−)-D-512a | 2.96 ± 0.3 | 107 ± 3 | 1.26 ± 0.2 | 93.1 ± 4.4 | 2.35 |
See ref (30).
Results and Discussion
D-512 and D-440 Evaluated in Rat Parkinson’s Disease Animal Models
We evaluated the in vivo efficacy of D-512 and D-440 in rats carrying a unilateral lesion in the medial forebrain bundle induced by the application of the neurotoxin 6-OHDA. The neurotoxin 6-OHDA induces the destruction of dopamine neurons as well as the development of the supersensitivity of dopamine receptors on the lesioned side. Such surgically modified rats, when challenged with a direct-acting dopamine agonist, produce contralateral rotations away from the lesioned side. This rat model is considered to be one of the standard models for the preclinical screening of drugs for possible antiparkinsonian property.31
It is evident from Figure 2 that both of the drugs were highly potent in improving the rotational activity in a dose-dependent manner compared to both the control groups and ropinirole.
Figure 2.
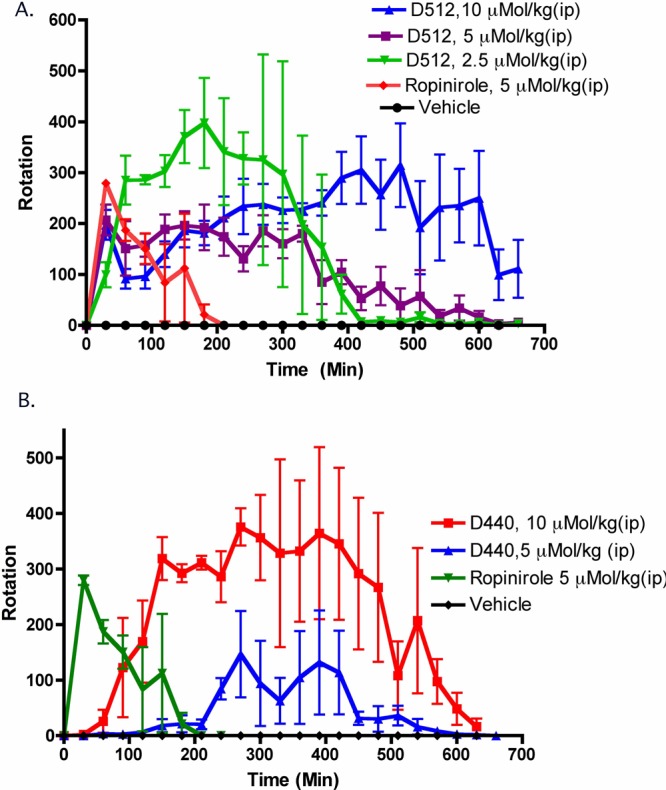
Effect on turning behavior of D-512, D-440, and ropinirole in 6-OHDA-induced unilaterally lesioned rats studied over 7.5 h. Each point is the mean ± SEM of four rats. All drugs were administered i.p. immediately before counting the rotational activity. (A) For three doses of D-512, one-way ANOVA analysis demonstrated a significant effect among the treatments, F(5, 95) = 13.74 (P < 0.0001). Dunnett’s analysis shows that the effect of three doses (2.5, 5, and 10 μMol/kg) of D-512 are significantly different compared to that of the vehicle (P < 0.01). (B) For two doses of D-440, one-way ANOVA analysis demonstrated a significant effect among the treatments, F(4, 95) = 27.49 (P < 0.0001). Dunnett’s analysis shows that the effect of two doses (5 and 10 μMol/kg) of D-440 are significantly different compared to that of the vehicle (P < 0.01).
D-512 and D-440 Protect MN9D Cells from 6-OHDA Toxicity
6-OHDA is known to cause cell death in a dose-dependent manner via the production of reactive oxygen species. We have employed various concentrations of the neurotoxin 6-OHDA against MN9D cells and found that 75 μM of 6-OHDA can cause 50–60% cell death in MN9D cells (Figure 2C). The dose-dependent effect of D-512 and D-440 on cell viability was carried out with MN9D cells, which indicated the induction of proliferation of the cells by both compounds at higher concentrations (Figure 3A,B). To examine whether D-512 and D-440 can protect MN9D cells from the exposure to 75 μM 6-OHDA, the cells were incubated with various concentrations (30, 20, 10, 5, 1, 0.1 μM) of D-512 and D-440 for 1 h followed by cotreatment with 75 μM 6-OHDA for an additional 24 h. As shown in Figure 3D,E, the data from the MTT assay clearly indicated that both D-512 and D-440 were able to protect MN9D cells significantly from 6-OHDA toxicity in a dose-dependent manner. The significant protection conferred by D-512 was exhibited in the concentration range of 5–30 μM of the drug. Similarly, D-440 was able to protect MN9D cells significantly compared to the 6-OHDA treatment alone, and the highest protection was exhibited at the three highest concentrations of the drug. It seems that the extent of the protection conferred by D-440 was slightly less than that of D-512. The reference compound Ropinirole did not provide any significant protection from the neurotoxicity of 6-OHDA under our experimental conditions (Figure 3F). N-acetyl cysteine (NAC), a potent antioxidant, failed to produce neuroprotection at 75 μM but produced significant neuroprotection at 2 mM, which was comparable to the neuroprotection produced by 5 μM D-512 (Figure 3G).
Figure 3.

Dose-dependent effect of the combination of a pretreatment followed by the cotreatment of D-512, D-440, ropinirole, and N-acetyl cystine (NAC) with 75 μM 6-OHDA on the cell viability of MN9D cells from toxicity caused by 75 μM 6-OHDA. (A, B) Dose-dependent effect of D-440 and D-512 on cell viability. (C) MN9D cells were treated with different concentrations of 6-OHDA (25–125 μM). (D–G) MN9D cells were pretreated with different doses of D-440, D-512, ropinirole, and NAC for 1 h followed by cotreatment with 75 μM 6-OHDA for 24 h. The values shown are the mean ± SD of three independent experiments performed with 4–6 replicates. One-way ANOVA analysis followed by Tukey’s multiple comparison posthoc test was performed. (**p < 0.001 compared to the 6-OHDA group and ##p < 0.001 compared to the control group).
D-512 and D-440 Protect Cells from MPP+ Toxicity
The dose-dependent effect of the treatment with D-512 and D-440 in reversing the toxicity of MPP+ to MN9D cells is demonstrated in Figure 4, and the morphological changes in these cells in response to the treatments are shown in Figure 5. From the dose-dependent experiment of MPP+, we chose 100 μM MPP+, which can induce 50–60% cell death, for our study (Figure 4A). To test whether D-512 and D-440 can protect MN9D cells from MPP+-induced toxicity, the cells were pretreated with various concentrations of D-512 and D-440 (30, 20, 10, 5, 1, and 0.1 μM) for 1 h followed by cotreatment with 100 μM MPP+ for an additional 24 h. The data from the MTT assay further indicated that both D-512 and D-440 can protect the MN9D cells in a dose-dependent manner. It is evident from the result in Figure 4B,C that at doses of 5–30 μM, D-512 conferred a significant protection from the toxicity caused by MPP+. At 20 μM, D-512 not only reversed the toxicity of MPP+ but also induced cell proliferation compared to the control treatment (Figure 4B). Similar to D-512, compound D-440 also exhibited a significant neuroprotection in the dose range of 5–30 μM. At a dose of 30 μM, D-440 was able to reverse the toxicity completely. However, unlike D-512, D-440 did not result in any additional cell proliferation (Figure 4C). The reference compound, Ropinirole, did not produce any neuroprotection effect under our experimental protocol against MPP+ toxicity (Figure 4D).
Figure 4.

Dose-dependent effect of the combination of a pretreatment followed by the cotreatment of D-512, D-440, and ropinirole with 100 μM MPP+ on the viability of MN9D cells from toxicity caused by 100 μM MPP+. (A) MN9D cells were treated with different concentration of MPP+ (25–125 μM). (B–D) MN9D cells were pretreated with different doses of D-440, D-512, and ropinirole for 1 h followed by cotreatment with 100 μM MPP+ for 24 h. The values shown are the mean ± SD of three independent experiments performed with 4–6 replicates. One-way ANOVA analysis followed by Tukey’s multiple comparison posthoc test was performed. (**p < 0.001 compared to the MPP+ group and ##p < 0.001 compared to the control group).
Figure 5.

Morphological changes of MN9D cells treated with control, 75 μM 6-OHDA (24 h), 20 μM D-512 + 75 μM 6-OHDA (1 h pretreatment/24 cotreatment), 100 μM MPP+ (24 h), 20 μM D-512 + 100 μM MPP+ (1 h pretreatment/24 cotreatment), and 20 μM D-512 (24 h) detected by light microscopy at 20× magnification.
D-512 Inhibits Lipid Peroxidation
Lipid peroxidation was measured to assess the oxidative stress generated by the treatment with sodium nitroprusside (SNP). The amount of thiobarbituric acid reactive species (TBARS) produced was evaluated fluorometrically, which was the measure of the level of oxidative stress in the dopaminergic MN9D cells. Treatment with 300 μM SNP resulted in a 100% increase in TBARS in the assay. D-512 dose-dependently reduced the SNP-induced increase of TBARS, with the highest dose (20 μM) producing an almost 100% reduction (Figure 6).
Figure 6.

Effects of pretreatment of different concentrations of D-512 on the production of thiobarbituric acid reactive substances induced by sodium nitroprusside (SNP, 300 μM). The data from three different experiments are presented as the mean ± SEM. The control data represents the group that was not treated with SNP and is represented as 100% with respect to the other groups. One-way ANOVA analysis followed by Bonferroni’s multiple comparison posthoc test was performed. (##p < 0.001 compared to the control group and **p < 0.001 compared to the SNP + drug group).
D-512 Inhibits Caspase 3/7 Activity
We have examined whether D-512 can inhibit caspase 3/7 activity induced by 6-OHDA. MN9D cells were pretreated with 20 and 1 μM of D-512 followed by cotreatment with 75 μM of 6-OHDA for 24 h. As shown in Figure 7, treatment with 6-OHDA significantly increased caspase 3 activity compared to control untreated cells. A nearly 2-fold decrease in caspase activity for the cotreated cells took place compared to the cells that were treated with 6-OHDA alone. Thus, the cotreatment with 20 μM D-512 resulted in a level of caspase 3 activity that was similar to the control cells. The inhibition of caspase activity was dose dependent, as the lower dose of 1 μM D-512 produced a smaller inhibition. This result confirms that D-512 has the ability to inhibit caspase 3/7 activity, protecting the cells from apoptosis in the presence of 6-OHDA.
Figure 7.

Dose-dependent inhibition of caspase 3 activity in 6-OHDA-treated MN9D cells by D-512. The values shown are the mean ± SD of three independent experiments performed with 4–6 replicates. One-way ANOVA analysis followed by Tukey’s multiple comparison posthoc test was performed. (##p < 0.01 control compared to the 6-OHDA alone group and ***p < 0.01 6-OHDA alone group compared to the 6-OHDA + D-512 groups).
D-512 Rescues 6-OHDA-Induced Changes in Nuclear Morphology
The treatment of MN9D cells with 75 μM 6-OHDA for 16 h produced apoptotic nuclear condensation. This nuclear fragmentation was reduced significantly by the treatment with 10 μM D-512 (Figure 8).
Figure 8.
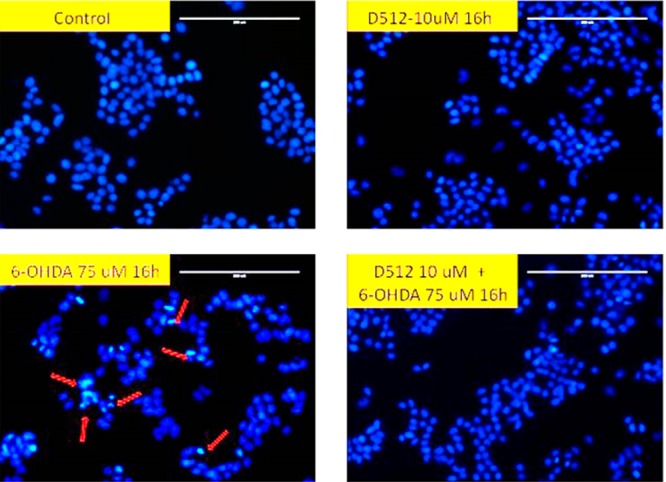
Effect of D-512 on 6-OHDA-induced nuclear morphology changes. Apoptotic nuclei were visualized Hoechst 33342 staining. Apoptotic cells are indicated by arrows. The scale bar is 200 μm.
Western Blot Analysis
Tyrosine hydroxylase (TH) is a key enzyme involved in dopamine biosynthesis and is considered to be a marker of DA neurons. In our goal of evaluating the ability of D-512 to restore the level of TH following the treatment with 6-OHDA, we carried out a Western blot experiment. The level of expression of TH was significantly reduced compared to the control upon treatment with 75 μM of 6-OHDA. However, a 1 h pretreatment with D-512 followed by cotreatment with 6-OHDA reversed and restored the level of TH significantly (Figure 9). Treatment with D-512 alone upregulated the level of TH compared to that of the control.
Figure 9.

TH protein level was determined by Western blot analysis. MN9D cells were pretreated with 20 μM D-512 for 1 h and co-treated with 75 μM 6-OHDA for 24 h. MN9D cells were also treated with control vehicle, D-512 and 6-OHDA alone. For quantification purpose protein level was normalized with respect to GAPDH protein. The values shown are means ± SDs of three independent experiments performed in triplicate. One way ANOVA analysis followed by Tukey’s Multiple Comparison post hoc test were performed. (**p < 0.05 control compared to the 6-OHDA group. **p < 0.05 6-OHDA compared to the D512 + 6OHDA group).
To elucidate further any role of cellular-signaling pathways in this neuroprotection, the activation of extracellular-signal-regulated kinase (ERK) and protein kinase Akt upon treatment with the drug and 6-OHDA was evaluated. The activation of the ERK signaling pathway in the survival of neuronal cells is not clear.32,33 However, the activation of protein kinase Akt has been shown to be linked to cell survival.34,35 Three treatment time points were chosen for our studies. The levels of total and phosphorylated ERK and Akt proteins were analyzed by Western blot studies using specific antibodies. Figure 10 indicates that activation of ERK took place at 2 and 4 h upon treatment with 6-OHDA alone, whereas at an early time point of 0.5 h, activation by 6-OHDA alone was reduced compared to both the control and D-512 treated alone groups. Cotreatment with D-512 lowered the level of p-ERK induced by 6-OHDA at 4 h. On the contrary, Akt was distinctly activated at 0.5 and 2 h time points with 6-OHDA and D512/6-OHDA-treated groups. At a later 4 h time point, Akt activation from all treatment groups was slightly higher compared to that of the 6-OHDA alone group.
Figure 10.

Determination of the activities of ERK and Akt by Western blot analysis. MN9D cells were pretreated with 10 μM D512 for 1 h followed by cotreatment with 75 μM 6-OHDA separately for 0.25, 2, and 4 h. GAPDH was used as an internal control protein. For quantification purposes, the protein level was normalized with respect to either the total Akt or ERK proteins. The values shown are the mean ± SD of three independent experiments performed in triplicate. One-way ANOVA analysis followed by Tukey’s multiple comparison posthoc test was performed.
Discussion
To follow up on our previous study and to further evaluate the efficacy of D-512 and D-440 in a Parkinson’s disease animal model, the compounds were tested in rats carrying a unilateral lesion in the medial forebrain bundle induced by application of the neurotoxin 6-OHDA.31 These rats produce contralateral rotations away from the lesioned side when they are challenged with direct-acting dopamine agonists. This model is considered to be one of the standard preclinical models for the early screening of PD drugs. D-512 produced the highest number of rotations (total rotations = 4555) at the highest dose of 10 μMol/kg, which lasted for more than 10 h (Figure 2). The rotational activity produced by the 2.5 μMol/kg dose of D-512 was slightly more efficacious than the 5 μMol/kg does, but it exhibited a shorter duration of action compared to both the 5 and 10 μMol/kg doses. It might be possible that because of the high potency of D-512 on the D2 receptor, the saturation of dopamine D2 receptor sites at higher doses may led to some inhibition of activity that did not occur with the lowest 2.5 μMol/kg dose. For D-440, the highest number of rotations (total rotations = 4666) was produced at a dose of 10 μMol/kg. Thus, both of the compounds were efficacious in this animal model experiment.
After evaluating the efficacy in the PD animal model, we next wanted to determine the neuroprotective property of these two compounds in a dopaminergic cell line. Thus, we describe the effect of novel dopamine agonists D-440 and D-512 on the protection of MN9D cells from toxicity induced by 6-OHDA and MPP+. Compound D-440 is a highly selective dopamine D3 agonist, and D-512 is a highly potent nonselective D2/D3 agonist.30 The MN9D cells are hybridoma cells derived via the somatic fusion of rostral mesencephalic neurons from embryonic C57BL/6J (E14) mice and N18TG2 neuroblastoma cells.36 Because the MN9D cells express a high level of tyrosine hydroxylase, have a high dopamine content, and exhibit other similarities with DA neurons, they represent one of the most suitable models for the in vitro study of PD.
Previously, we have shown in the DPPH (2,2-diphenyl-1-picrylhydrazyl) radical quenching assay that D-512 is a potent antioxidant with a 2-fold higher potency compared to that of ascorbic acid; however, D-440 showed a nearly 1.5-fold antioxidant potency compared to that of ascorbic acid.30 In our present study, we carried out experiments to evaluate the effect of treatment with D-512 and D-440 on the protection of the dopaminergic MN9D cells from toxicity induced by 6-OHDA and MPP+. Both 6-OHDA and MPP+ are known to cause dopamine cell death, possibly via different mechanisms. The mechanism of toxicity of 6-OHDA centers around the production of ROS after it gets transported into the cytosol via the dopamine transporter. The oxidation of 6-OHDA is known to produce p-quinones as well as free radicals such as hydrogen peroxides, superoxides, and hydroxyl radicals that induce apoptosis in cells.37,38 MPP+, which is a metabolite of MPTP, is a dopaminergic neurotoxin that destroys the nigrostriatal dopaminergic pathway and produces Parkinsonian syndrome with a massive loss of nigral DA neurons.39−41 The inhibition of mitochondrial complex I by MPP+, which increases the oxidative stress, is the central mechanism for its toxicity.40 In the animal model, the MPP+ level is positively correlated to MPTP toxicity.41
The initial profile of the dose-dependent toxicity of 6-OHDA on MN9D cells led us to choose 75 μM 6-OHDA for our experiments, which produced an approximately 50% cell death (Figure 3C). The dose-dependent treatment with D-512 and D-440 reversed the toxicity induced by 75 μM 6-OHDA. Cell viability data from MTT experiments indicated that at doses between 5 and 30 μM, both D-512 and D-440 increased cell viability significantly. D-512 showed a significant protection at the lower dose of 5 μM and fully reversed the toxicity at the two highest doses of 20 and 30 μM (Figure 3D). D-440 exhibited a similar profile as D-512, although it exhibited little less potency in reversing the toxicity compared to that of D-512 (Figure 3E).
Similarly, both D-512 and D-440 could reverse the toxicity of MPP+ dose dependently (Figure 4). The initial dose-response profile of MPP+ led us to choose a concentration of 100 μM for our study. In the case of MPP+, D-512 not only reversed the toxicity but also induced cell proliferation at doses of 10 and 20 μM. As shown for the 6-OHDA assay, D-512 was more efficient in reversing MPP+ toxicity compared to D-440. It is interesting to note that the both compounds, when treated alone, induced cell proliferation at higher doses. It has been previously reported that a compound with a trophic property can induce the proliferation of MN9D cells and is neuroprotective against 6-OHDA-induced cell death.42 The morphology of the cells under different treatment regimens with 6-OHDA and MPP+ is shown in Figure 5. Figure 5 demonstrates significant changes in the morphology of the cells under treatment with either 6-OHDA or MPP+ compared to control. Cotreatment with D-512 restored the cell morphology significantly, which correlates with the results from the cell viability experiments. To compare the observed neuroprotection of D-512 and D-440 to Ropinirole, which is an approved PD drug used in the clinic, we carried out neuroprotection studies under identical conditions. Interestingly, the results indicate that Ropinirole showed very weak to no neuroprotection in reversing the toxicity of both 6-OHDA and MPP+ (Figures 3F and 4D). This clearly underscores the inability of Ropinirole, unlike D-440 and D-512, to counteract the pathways involved in the mechanisms of toxicity of 6-OHDA and MPP+. Interestingly, it is evident from the treatment with antioxidant NAC at two higher doses, 75 and 2 mM, that a 400-fold increase in the dose (2 mM) of NAC is required to produce a neuroprotection effect that is comparable to the effect of 5 μM D-512 (Figure 3G). The lower 75 μM dose was not effective in producing neuroprotection. It may be possible that the antioxidant property alone may not explain the neuroprotection property of D512 and D-440, unless these molecules exhibit a more efficacious antioxidant activity. Interestingly, NAC treatment alone did not induce much cell proliferation (Figure 3G). As mentioned above, any additional property (e.g., trophic factors, etc.) of the test drugs that can induce such cell proliferation may also produce, besides a neuroprotective effect, a beneficial neurogenesis property. Further future studies will need to be carried out to understand the scope of any such properties.
One of the possible mechanisms behind the neuroprotection of D-512 and D-440 may involve the reduction of oxidative stress induced by both 6-OHDA and MPP+. To evaluate any contribution of the antioxidant activity of these compounds, we carried out a cell-based lipid peroxidation experiment with sodium nitroprusside (SNP). SNP is a strong oxidizing agent that is known to produce lipid peroxidation in vitro and in vivo. (43) As expected, treatment with SNP (300 μM) alone increased the level of lipid peroxidation by 100%. Pretreatment with different doses of D-512 for 1 h followed by cotreatment with different doses of D-512 and SNP (300 μM) could reduce the lipid peroxidation induced by SNP dose dependently (Figure 6). The induction of lipid peroxidation by SNP is due to the production of reactive oxygen species via the release of NO from SNP. The data clearly indicates that D-512 was able to reduce the formation of reactive oxygen species by SNP. This data together with the data on the DPPH radical quenching study that was carried out by us previously indicates the potent antioxidant property of D-512.
The caspases are a family of cysteine proteases. They are known to play crucial roles in programmed cell death or apoptosis.44,45 Among the caspases, the effector caspases (e.g., caspase-3 and caspase-7) can trigger the mitochondrial apoptosis cascade. It has been previously reported that 6-OHDA causes cell death via an intrinsic apoptotic pathway in MN9D cells.46,47 On the contrary, MPP+ causes cell death via necrosis in MN9D cells.46,47 To evaluate whether D-512 promotes cell survival in 6-OHDA treated cells via the inhibition of caspase 3/7 activity, we carried out a caspase 3/7 assay. The result indicates that D-512 dose dependently inhibited caspase 3/7 activity produced by treatment with 6-OHDA (Figure 7). The inhibitory effect was dose dependent, as the highest 20 μM dose produced the maximum inhibition. The results from this assay correlates well with Hoechst staining data that demonstrated the protection of nuclear morphology from toxicity caused by 6-OHDA (Figure 8). The exposure of MN9D cells to 6-OHDA led to the formation of apoptotic condensed nuclei, as marked in Figure 8, which was decreased significantly upon cotreatment with D-512 (Figure 8).
Next, we wanted to evaluate the effect of the treatment of 6-OHDA and D-512 on the level of TH. TH is a rate-limiting enzyme for dopamine synthesis. TH immunohistochemistry has been widely used as an important marker of dopaminergic neurons.48−50 Our results demonstrated that pre- and cotreatment with D-512 was able to restore the level of TH that was reduced significantly upon treatment with 6-OHDA alone (Figure 9). This indicates that D-512 could rescue the dopaminergic MN9D cells from the toxicity of 6-OHDA. Interestingly, the treatment with D-512 alone could upregulate the level of TH.
To understand the possible cellular-signaling pathways that lead to neuroprotection, we carried out a study to evaluate the activation of ERK and Akt. Although the role of ERK in neuroprotection is not clear, we wanted to study the effect of the treatment of the drug in presence of 6-OHDA on ERK protein levels. As reported earlier, we found that the treatment with 6-OHDA alone at 2 and 4 h increased the activated ERK (p-ERK) level compared to that of control, and this activation was somewhat attenuated by cotreatment with D-512 at 4 h. (51) However, at 0.5 h, the activation of ERK was reduced by treatment with 6-OHDA alone compared to both the control and D-512 groups, whereas the level of ERK activity was restored to a higher level in the D512/6-OHDA treated group. The activation of ERK from the treatment with 6-OHDA has been attributed to the production of oxidative stress.51 It is possible that the reduction of such activation of ERK by D-512 at 4 h might indicate a possible mechanism of neuroprotection.
To understand further any role of an intracellular-signaling mechanism in cell survival, we investigated the level of the activated phosphorylated form of the serine threonine kinase Akt upon treatment with the drug and 6-OHDA. The activated form of Akt has been implicated in cell survival. (52) Our results showed a clear enhancement of the p-Akt signal at 0.5 and 2 h in the 6-OHDA and D-512/6-OHDA cotreatment groups. At 4 h, the activation of Akt in the 6-OHDA group was less compared to the other groups, including the D512/6-OHDA group, indicating a possible neuroprotection mechanism. Interestingly, as observed earlier at a shorter time point (0.5 h), 6-OHDA treatment alone also enhanced the p-Akt signal. (42)
Conclusions
D-512, which is a novel highly potent D2/D3 agonist, and D-440, which is a novel highly selective D3 agonist, exhibited in vivo efficacy in the 6-OHDA-induced unilaterally lesioned rat model, a Parkinson’s disease animal model. To evaluate their neuroprotective property, we demonstrated that both D-512 and D-440 can attenuate 6-OHDA-induced toxicity in a dose-dependent manner in the dopaminergic MN9D cells. Similarly, dose-dependent neuroprotection was exhibited by D-512 and D-440 when the MN9D cells were treated with the dopaminergic neurotoxin MPP+. The potent inhibition of lipid peroxidation, the inhibition of caspase 3/7 activity, and the restoration of TH activity by D-512 in 6-OHDA-treated cells may partialy explain its neuroprotective property. The protection of nuclear morphology in drug-treated groups also indicates the survival of the cells. It is important to note that the neuroprotective property exhibited by these drugs was independent of their dopamine-agonist property, which is consistent with our multifunctional drug-development approach, indicating that these compounds could be disease-modifying symptomatic-treatment agents for Parkinson’s disease.
Methods
Materials
1-Methyl-4-phenylpyridinium (MPP+), 6-hydroxydopamine hydrochloride, thiazolyl blue tetrazolium bromide (MTT), poly-l-lysine (PLL), Dulbecco’s modified Eagle’s medium, sodium bicarbonate, hydrochloric acid, dimethyl sulfoxide (DMSO), methanol, Dulbecco’s phosphate buffer saline, and tissue culture grade water were obtained from Sigma-Aldrich (St. Louis, MO). Pennicilin/streptomycin and trypsin were purchased from Invitrogen (Grand Island, NY). Fetal clone III bovine serum (Hyclone, Logan, UT) was purchased from Fisher Scientific. 96-well plates were purchased from Greiner Bio-One (Monroe, NC). Apo-One Homogeneous Caspase 3/7 reagent was purchased from Promega Inc. (Madison, WI). RIPA lysis buffer and the anti-GAPDH antibody were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). BCA protein assay reagents as well as the horseradish peroxidase (HRP)-conjugated rabbit secondary antibody and antimouse secondary antibody were purchased from Thermo Fisher Scientific Inc. (Rockford, IL). The tyrosine hydroxylase (TH) primary antibody, phospho-ERK, total ERK, phospho-Akt, and total-Akt antibodies were purchased from the Cell Signaling Technology Inc. (Danvers, MA). Compounds D-512 ((6S)-N6-(2-(4-(4-chloro-1H-indol-5-yl)piperazin-1-yl)ethyl)-N6-propyl-3a,4,5,6,7,7a-hexahydrobenzo[d]thiazole-2,6-diamine) and D-440 ((4-(2-(((6S)-2-amino-3a,4,5,6,7,7a-hexahydrobenzo[d]thiazol-6-yl)(propyl)amino)ethyl) piperazin-1-yl)(1H-indol-2-yl)methanone) were synthesized in our laboratory as described in our recent publication.30 The corresponding trifluoroacetate or mesylate salt of D-512 and D-440 were used for our studies. Ropinirole hydrochloride was purchased from Sigma-Aldrich (St. Louis, MO).
Animal Experiments
Drugs and Chemicals
The following drugs were used in the experiment: the trifluoroacetate salts of compounds D-512, D-440, and ropinirole were dissolved in de-ionized (DI) water for both the locomotor and 6-OHDA rotational experiments. All compounds for this study were administered i.p. in a volume of 0.7–0.9 mL into each rat.
Animals
The lesioned rats (250–260 g) were purchased from Charles River (Wilmington, MA), and their unilateral lesion was checked twice by apomorphine challenge following the surgery. Animals were maintained in sawdust-lined cages in a temperature and humidity controlled environment at 22 ± 1 °C and 60 ± 5%, respectively, with a 12 h light/dark cycle (with the lights on from 6:00 a.m. to 6:00 p.m. The rats were group housed with unrestricted access to food and water. All experiments were performed during the light component. All animal use procedures were in compliance with the Wayne State University Animal Investigation Committee and were consistent with AALAC guidelines.
In Vivo Rotational Experiment with 6-OHDA Lesioned Rats
The first challenge with apomorphine was done 7 days postlesion with lesioned animals to observe a complete rotation session postadministration. In the second challenge with apomorphine (0.05 mg/kg) that was done 21 days postlesion, contralateral rotations were recorded for 30 min; apomorphine produced rotations in all four rats (average rotation >250), indicating a successful unilateral lesion. In these rats, the lesion was performed on the left side of the medial forebrain bundle in the brain, and the coordinates used from Bregma are: IB +2.5 AP −1.5 ML ± 1.8 DV −7.5 from the Dura on the basis of the Paxinos-Watson Second Addition Atlas. The rotations produced upon agonist challenge occurred in a clockwise direction. In this study, apomorphine was also used as a reference compound. The test drugs, including ropinirole, were dissolved in saline and were administered i.p. The rats were brought to the test room 1 h before the administration of the test drug, standard drug, or vehicle for acclimatization purposes. The rotations were measured over 7–10 h. For the control, the vehicle was administered alone. Rotations were measured with the Rotomax Rotometry System (AccuScan Instruments, Inc., Columbus, OH) equipped with a Rotomax Analyzer, high-resolution sensor, and animal chambers with harnesses. Data were analyzed with the Rotomax Window software program. Test drugs D-512 (5 and 10 μMol/kg), D-440 (5 and 10 μMol/kg), and ropinirole (5 μMol/kg) dissolved in saline were administered i.p. The rotations were measured in a rotational chamber immediately after the administration of the drugs. The data were collected at every 30 min. The data were analyzed by the GraphPad version 4 program. All drugs produced contralateral rotations in all lesioned rats which lasted for over 3–10 h.
Cell Culture and Treatments
The dopaminergic MN9D hybridoma cells are derived from the somatic fusion of rostral mesencephalic neurons from embryonic C57BL/6J (E14) mice with N18TG2 mouse cells. They were cultured in T-75 flasks (Greiner Bio-One, Frickenhausen) coated with 1 mg/mL of poly-l-lysine and maintained in DMEM (high glucose with phenol red) supplemented with 10% FetalClone III serum, penicillin (50 units/ml), and streptomycin (50 μg/mL) at 37 °C under a 5% CO2 atmosphere. Stock solutions of D-512, D-440, NAC, and Ropinirole were prepared in DMSO and stored at −20 °C for the period of the experiments. MN9D cells were pretreated with various concentrations of D-512, D-440, or Ropinirole for 1 h followed by cotreatment with 75 μM 6-OHDA (freshly prepared from a stock solution in DMSO stored at −20 °C immediately before it was added) or 100 μM MPP+ (freshly prepared from a stock solution in DMSO stored at −20 °C immediately before it was added) for 24 h. The control cells were treated with the above medium containing 0.01% DMSO (vehicle) only.
Assessment of Cell Viability
To evaluate the neuroprotection ability of the test compounds in the presence of the neurotoxins 6-OHDA and MPP+, the quantitative and colorimetric MTT (3,4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazoliumbromide) tetrazolium salt assay was used to assess cell viability. MN9D cells were seeded onto poly-l-lysine coated 96-well plates at 1 × 104 cells/well in 100 μL of medium. After the plate was equilibrated for 40 h, the old medium was taken out from each well and 160 μL of fresh medium (containing 0.01% DMSO) was added to the control wells as well as the wells to be treated with 6-OHDA or MPP+. A solution of 160 μL of D-512, D-440, or Ropinirole in the above medium without DMSO in 30, 20, 10, 5, 1, and 0.1 μM were added to the wells that would be cotreated with 6-OHDA or MPP+. In the case of NAC, 75 μM and 2 mM doses were used for the cotreatment with 6-OHDA. The plate was incubated for 1 h at 37 °C under a 5% CO2 atmosphere. At the end of the incubation, the required amount of 6-OHDA or MPP+ was added to each well (except the control wells) to maintain a final concentration of 75 or 100 μM, respectively. The plate was then incubated for 24 h at 37 °C under a 5% CO2 atmosphere. Next, 20 μL of the MTT stock solution (prepared in Dulbecco’s phosphate-buffered saline) was added to each well to maintain a final concentration of 0.5 mg/mL, and the plate was incubated for another 3 h at 37 °C under a 5% CO2 atmosphere. Next, the plate was centrifuged at 1500 rpm for 10 min, and the supernatants were removed carefully. The formazan crystals were dissolved in 100 μL of a 1:1 mixture of a DMSO/methanol solution by shaking gently at 400 rpm for 30 min at room temperature on a Thermomix R shaker (Eppendorf, Hamburg). The absorbance was measured at 570 and 690 nM using an Epoch microplate reader (BioTek, Winooski, VT). The background-corrected values (570–690 nM) were used to plot the graph. Data from at least three experiments were analyzed using GraphPad software (version 4).
Lipid Peroxidation Assay
MN9D cells were plated at 2 × 105 cells/well in 12-well plates. After 48 h, the cells were pretreated with 20, 10, or 5 μM D-512 for 1 h. Afterward, the cells were cotreated with 300 μM sodium nitroprusside (SNP, Across Organics) for 8 h. The cells were harvested using a cell scraper, washed with 1× PBS, and resuspended in 120 μL of 1× PBS. The cells were sonicated three times for 5 s each using a Branson ultrasonicator at 30% amplitude, and the whole cell lysate was used to determine the abundance of thiobarbituric acid (TBA) reactive species (TBARS). One-hundred microliters of 1% SDS and 100 μL of whole cell lysate were mixed with 4 mL of the color reagent. The color reagent was prepared by mixing 320 mg of TBA (Sigma-Aldrich) dissolved in 30 mL of 0.1 M NaOH and 30 mL of diluted acetic acid. The mixture was then boiled for 1 h at 100 °C, and the fluorescence was read using 520 nM excitation and 550 nM emission wavelengths (BioTek Synergy H-1 fluorescence plate reader). Fluorescence data was plotted after subtracting the blank that was prepared using 100 μL of 1× PBS, 100 μL of 1% SDS, and 4 mL of the color reagent. The control consisted of 100 μL of the cell lysate without any treatment in 100 μL of 1% SDS and 4 mL of the color reagent. The data were analyzed using GraphPad software (version 4).
Fluorometric Caspase-3/7 Assay
The caspase 3/7 activity was measured by the fluorometric Apo-ONE Homogeneous Caspase 3/7 assay kit following the specification provided by the manufacturer (Promega, Madison, WI) with slight modifications. Briefly, 1 × 104 MN9D cells/well were seeded in 90 μL of DMEM (with phenol red, containing 10% FetalClone III FBS and Pen/Strep) in a cell-bind 96-well plate (PLL coated) and incubated for 48 h at 37 °C under a 5% CO2 atmosphere. Ten microliters of fresh medium (as mentioned above) without DMSO was added to the control cells. Five microliters of fresh medium (as mentioned above) was added to the cells that would be treated with 6-OHDA alone. The required amount of D-512 was added to the cells that would be cotreated with 6-OHDA to maintain a final concentration of 20 and 1 uM, respectively. The plate was shaken at 400 rpm for 3 min on a Eppendorf Thermomix R shaker to ensure the homogeneous mixing of the drug. The plate was then incubated for 1 h at 37 °C under a 5% CO2 atmosphere. Next, the required amount of 6-OHDA solution (prepared from a 1 M stock solution in DMSO) was added very quickly into the cotreated cells to maintain a final concentration of 75 μM. The plate was incubated for 24 h at 37 °C under a 5% CO2 atmosphere. Following the incubation, the old medium was taken out from each well, and 75 μL of fresh DMEM medium (high glucose without serum and phenol red) was quickly added to each well. Seventy-five microliters of freshly prepared caspase-3/7 reagent (substrate + lysis buffer at a 1:100 dilution) was added to each well. The addition of the caspase 3/7 reagent was performed in the dark. The plate was then covered with aluminum foil and shaken at 400 rpm on a Eppendorf Thermopmix R shaker over a 16 h time period. A fluorescence reading was taken each hour at an excitation wavelength of 489 nM and an emission wavelength of 535 nM on a BioTek Synergy H-1 fluorescence plate reader with the sensitivity set to 65. The optimized result was obtained at 7 h after the addition of the caspase 3/7 reagent. The value from the blank well was subtracted from the control and the treated wells. The data were analyzed using GraphPad software (version 4).
Western Blot Analysis
A standard protocol was followed to perform the Western blotting experiment to evaluate TH activity. In brief, MN9D cells were grown in 6-well plates coated with 1 mg/mL of PLL. After the cells were seeded for 24 h, the cells were pretreated with 20 μM D-512 followed by cotreatment with 75 μM 6-OHDA for 24 h. The cells were then harvested by scraping and lysed in RIPA lysis buffer on ice for 30 min. The supernatant was collected, and the protein concentration was determined using the BCA protein assay reagents. Samples containing 30 μg of total protein were electrophoresed on 12% SDS-polyacrylamide gels and transferred to a polyvinylidene fluoride (PVDF) membrane for TH protein immunostaining. The membrane was blocked in 5% nonfat milk in tris-buffered saline with Tween 20 (TBST) for 1 h followed by probing with the TH primary antibody (1:1000) overnight at 4 °C. The membrane was treated with an HRP-conjugated rabbit secondary antibody for 1 h at rt.
For the determination of ERK and Akt activities, following the pretreatment with 10 μM D512 for 1 h MN9D cells were cotreated with 75 μM 6-OHDA for indicated time periods, as shown in Figure 10. Fifty micrograms of protein was separated on a 10% tris-glycine gel by SDS polyacrylamide gel electrophoresis (SDS-PAGE). Afterward, the proteins were transferred to a PVDF membrane (Bio-Rad). The membrane was blocked with 5% bovine serum albumin (BSA) in TBST for 1 h. The blocked membrane was incubated overnight with primary antibodies recognizing p-AKT, p-ERK1/2, total Akt, and total ERK1/2 at 1:1000 dilutions in 5% BSA in TBST. Afterward, the membrane was incubated with the appropriate HRP-conjugated secondary antibody (anti-mouse or anti-rabbit) at a 1:4000 dilution in 1% BSA in TBST. The chemiluminescence signal was developed using enhanced chemiluminescence reagents. The image was visualized and analyzed using ECL-Plus (PerkinElmer, Waltham, MA) and ImageQuant LAS 4000 (GE Healthcare Biosciences, Pittsburgh, PA).
Hoescht Staining
MN9D cells were seeded onto poly-l-lysine coated 12-well plates at a density of 1.4 × 105 cells/well in 1.5 mL of DMEM. After the plate was equilibrated for 40 h, the old medium was taken out from each well, and 1.5 mL of fresh medium containing 10 μM D-512 was added. Following 1 h of pretreatment with the drug, 75 μM of 6-OHDA was added into the well, and the cotreatment was continued for 16 h. The medium was removed, and the cells were washed twice with PBS. The cells were fixed with 1 mL of a 4% paraformaldehyde solution (EM Sciences) for 15 min at room temperature followed by washing twice with PBS. The cells were stained with 1 mL of a 0.5 mM Hoechst 33342/0.15% triton-100 solution for 15 min at room temperature followed by washing twice with PBS. The cells were observed with a fluorescence microscope at 40× (EVOS FL digital inverted microscope, AMG) with 357 nm excitation and 447 nm emission wavelengths.
Statistical Analysis
The data were expressed as the mean value ± standard error mean (SEM). For multiple groups, statistical significance was determined using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison posthoc test. In all cases, p < 0.05 was considered to be statistically significant.
Acknowledgments
We thank Dr. Michael Zigmond (University of Pittsburgh) for the kind gift of the MN9D cell lines. This work is supported by the National Institute of Neurological Disorders and Stroke/National Institute of Health (NS047198, AKD).
The authors declare no competing financial interest.
This paper was published ASAP on August 20, 2013, with incorrect concentration units for D-512 and 6-OHDA in the Figure 9 caption. The corrected version was reposted on August 27, 2013.
Funding Statement
National Institutes of Health, United States
References
- Hornykiewicz O. (1989) Ageing and neurotoxins as causative factors in idiopathic Parkinson’s disease--a critical analysis of the neurochemical evidence. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 13, 319–328. [DOI] [PubMed] [Google Scholar]
- Hornykiewicz O.; Kish S. J. (1987) Biochemical pathophysiology of Parkinson’s disease. Adv. Neurol. 45, 19–34. [PubMed] [Google Scholar]
- Hornykiewicz O. (1998) Biochemical aspects of Parkinson’s disease. Neurology 51, 2–9. [DOI] [PubMed] [Google Scholar]
- Braak H.; Del Tredici K.; Rub U.; de Vos R. A.; Jansen Steur E. N.; Braak E. (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Di Monte D. A. (2003) The environment and Parkinson’s disease: Is the nigrostriatal system preferentially targeted by neurotoxins?. Lancet Neurol. 2, 531–538. [DOI] [PubMed] [Google Scholar]
- Jenner P. (2003) Oxidative stress in Parkinson’s disease. Ann. Neurol. 53, 26–36. [DOI] [PubMed] [Google Scholar]
- Beal M. F. (2009) Therapeutic approaches to mitochondrial dysfunction in Parkinson’s disease. Parkinsonism Relat. Disord. 15, 189–194. [DOI] [PubMed] [Google Scholar]
- Linert W.; Jameson G. N. (2000) Redox reactions of neurotransmitters possibly involved in the progression of Parkinson’s disease. J. Inorg. Biochem. 79, 319–326. [DOI] [PubMed] [Google Scholar]
- Linert W.; Herlinger E.; Jameson R. F.; Kienzl E.; Jellinger K.; Youdim M. B. (1996) Dopamine, 6-hydroxydopamine, iron, and dioxygen--their mutual interactions and possible implication in the development of Parkinson’s disease. Biochim. Biophys. Acta 1316, 160–168. [DOI] [PubMed] [Google Scholar]
- Exner N.; Lutz A. K.; Haass C.; Winklhofer K. F. (2012) Mitochondrial dysfunction in Parkinson’s disease: Molecular mechanisms and pathophysiological consequences. EMBO J. 31, 3038–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M. T.; Beal M. F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795. [DOI] [PubMed] [Google Scholar]
- Birkmayer W.; Hornykiewicz O. (2001) The effect of l-3,4-dihydroxyphenylalanine (= DOPA) on akinesia in Parkinsonism. 1961. Wien. Klin. Wochenschr. 113, 851–4. [PubMed] [Google Scholar]
- Cotzias G. C.; Papavasiliou P. S.; Gellene R. (1969) Modification of Parkinsonism--chronic treatment with L-dopa. N. Engl. J. Med. 280, 337–345. [DOI] [PubMed] [Google Scholar]
- Marsden C. D.; Parkes J. D. (1976) “On-off” effects in patients with Parkinson’s disease on chronic levodopa therapy. Lancet 1, 292–296. [DOI] [PubMed] [Google Scholar]
- Fahn S. (1996) Is levodopa toxic?. Neurology 47, 184–195. [DOI] [PubMed] [Google Scholar]
- Olanow C. W. (1990) Oxidation reactions in Parkinson’s disease. Neurology 40, 37–39. [PubMed] [Google Scholar]
- Dutta A. K. L., W. (2006) Existing dopaminergic therapies for Parkinson’s disease. Expert Opin. Ther. Pat. 16, 1613–1625. [Google Scholar]
- Zou L.; Xu J.; Jankovic J.; He Y.; Appel S. H.; Le W. (2000) Pramipexole inhibits lipid peroxidation and reduces injury in the substantia nigra induced by the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in C57BL/6 mice. Neurosci. Lett. 281, 167–170. [DOI] [PubMed] [Google Scholar]
- Ramirez A. D.; Wong S. K.; Menniti F. S. (2003) Pramipexole inhibits MPTP toxicity in mice by dopamine D3 receptor dependent and independent mechanisms. Eur. J. Pharmacol. 475, 29–35. [DOI] [PubMed] [Google Scholar]
- Iida M.; Miyazaki I.; Tanaka K.; Kabuto H.; Iwata-Ichikawa E.; Ogawa N. (1999) Dopamine D2 receptor-mediated antioxidant and neuroprotective effects of ropinirole, a dopamine agonist. Brain Res. 838, 51–59. [DOI] [PubMed] [Google Scholar]
- Li C.; Biswas S.; Li X.; Dutta A. K.; Le W. (2010) Novel D3 dopamine receptor-preferring agonist D-264: Evidence of neuroprotective property in Parkinson’s disease animal models induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and lactacystin. J. Neurosci. Res. 88, 2513–2523. [DOI] [PubMed] [Google Scholar]
- Youdim M. B.; Buccafusco J. J. (2005) CNS targets for multi-functional drugs in the treatment of Alzheimer’s and Parkinson’s diseases. J. Neural Transm. 112, 519–537. [DOI] [PubMed] [Google Scholar]
- Youdim M. B.; Buccafusco J. J. (2005) Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol. Sci. 26, 27–35. [DOI] [PubMed] [Google Scholar]
- Cavalli A.; Bolognesi M. L.; Minarini A.; Rosini M.; Tumiatti V.; Recanatini M.; Melchiorre C. (2008) Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 51, 347–372. [DOI] [PubMed] [Google Scholar]
- Koller W. C.; Cersosimo M. G. (2004) Neuroprotection in Parkinson’s disease: An elusive goal. Curr. Neurol. Neurosci. Rep. 4, 277–283. [DOI] [PubMed] [Google Scholar]
- Dutta A. K.; Venkataraman S. K.; Fei X. S.; Kolhatkar R.; Zhang S.; Reith M. E. (2004) Synthesis and biological characterization of novel hybrid 7-[[2-(4-phenyl-piperazin-1-yl)-ethyl]-propyl-amino]-5,6,7,8-tetrahydro-naphthalen-2-ol and their heterocyclic bioisosteric analogues for dopamine D2 and D3 receptors. Bioorg. Med. Chem. 12, 4361–4373. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Hazeldine S.; Ghosh B.; Parrington I.; Kuzhikandathil E.; Reith M. E.; Dutta A. K. (2008) Bioisosteric heterocyclic versions of 7-{[2-(4-phenyl-piperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphthalen-2-ol: Identification of highly potent and selective agonists for dopamine D3 receptor with potent in vivo activity. J. Med. Chem. 51, 3005–3019. [DOI] [PubMed] [Google Scholar]
- Biswas S.; Zhang S.; Fernandez F.; Ghosh B.; Zhen J.; Kuzhikandathil E.; Reith M. E.; Dutta A. K. (2008) Further structure-activity relationships study of hybrid 7-{[2-(4-phenylpiperazin-1-yl)ethyl]propylamino}-5,6,7,8-tetrahydronaphthalen-2-ol analogues: Identification of a high-affinity D3-preferring agonist with potent in vivo activity with long duration of action. J. Med. Chem. 51, 101–117. [DOI] [PubMed] [Google Scholar]
- Gogoi S.; Antonio T.; Rajagopalan S.; Reith M.; Andersen J.; Dutta A. K. (2011) Dopamine D(2)/D(3) agonists with potent iron chelation, antioxidant and neuroprotective properties: Potential implication in symptomatic and neuroprotective treatment of Parkinson’s disease. ChemMedChem 6, 991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.; Antonio T.; Reith M. E.; Dutta A. K. (2012) Structure-activity relationship study of N(6)-(2-(4-(1H-indol-5-yl)piperazin-1-yl)ethyl)-N(6)-propyl-4,5,6,7-tetrahydroben zo[d]thiazole-2,6-diamine analogues: Development of highly selective D3 dopamine receptor agonists along with a highly potent D2/D3 agonist and their pharmacological characterization. J. Med. Chem. 55, 5826–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerstedt U. (1968) 6-Hydroxy-dopamine induced degeneration of central monoamine neurons. Eur. J. Pharmacol. 5, 107–10. [DOI] [PubMed] [Google Scholar]
- Xia Z.; Dickens M.; Raingeaud J.; Davis R. J.; Greenberg M. E. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- Yan C. Y.; Greene L. A. (1998) Prevention of PC12 cell death by N-acetylcysteine requires activation of the Ras pathway. J. Neurosci. 18, 4042–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H.; Datta S. R.; Franke T. F.; Birnbaum M. J.; Yao R.; Cooper G. M.; Segal R. A.; Kaplan D. R.; Greenberg M. E. (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275, 661–665. [DOI] [PubMed] [Google Scholar]
- Kauffmann-Zeh A.; Rodriguez-Viciana P.; Ulrich E.; Gilbert C.; Coffer P.; Downward J.; Evan G. (1997) Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 385, 544–548. [DOI] [PubMed] [Google Scholar]
- Choi H. K.; Won L.; Roback J. D.; Wainer B. H.; Heller A. (1992) Specific modulation of dopamine expression in neuronal hybrid cells by primary cells from different brain regions. Proc. Natl. Acad. Sci. U.S.A. 89, 8943–8947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese G. R.; Traylor T. D. (1970) Effect of 6-hydroxydopamine on brain norepinephrine and dopamine evidence for selective degeneration of catecholamine neurons. J. Pharmacol. Exp. Ther. 174, 413–420. [PMC free article] [PubMed] [Google Scholar]
- Bove J.; Prou D.; Perier C.; Przedborski S. (2005) Toxin-induced models of Parkinson’s disease. NeuroRx 2, 484–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston J. W.; Ballard P. A. Jr. (1983) Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 309, 310. [DOI] [PubMed] [Google Scholar]
- Przedborski S.; Jackson-Lewis V.; Djaldetti R.; Liberatore G.; Vila M.; Vukosavic S.; Almer G. (2000) The parkinsonian toxin MPTP: Action and mechanism. Restor. Neurol. Neurosci. 16, 135–142. [PubMed] [Google Scholar]
- Jackson-Lewis V.; Przedborski S. (2007) Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2, 141–151. [DOI] [PubMed] [Google Scholar]
- Signore A. P.; Weng Z.; Hastings T.; Van Laar A. D.; Liang Q.; Lee Y. J.; Chen J. (2006) Erythropoietin protects against 6-hydroxydopamine-induced dopaminergic cell death. J. Neurochem. 96, 428–443. [DOI] [PubMed] [Google Scholar]
- Prigol M.; Wilhelm E. A.; Schneider C. C.; Nogueira C. W. (2008) Protective effect of unsymmetrical dichalcogenide, a novel antioxidant agent, in vitro and an in vivo model of brain oxidative damage. Chem.–Biol. Interact. 176, 129–136. [DOI] [PubMed] [Google Scholar]
- Degterev A.; Boyce M.; Yuan J. (2003) A decade of caspases. Oncogene 22, 8543–8567. [DOI] [PubMed] [Google Scholar]
- Cryns V.; Yuan J. (1998) Proteases to die for. Genes Dev. 12, 1551–1570. [DOI] [PubMed] [Google Scholar]
- Choi W. S.; Yoon S. Y.; Oh T. H.; Choi E. J.; O’Malley K. L.; Oh Y. J. (1999) Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: Role of caspases, ROS, and JNK. J. Neurosci. Res. 57, 86–94. [DOI] [PubMed] [Google Scholar]
- Han B. S.; Hong H. S.; Choi W. S.; Markelonis G. J.; Oh T. H.; Oh Y. J. (2003) Caspase-dependent and -independent cell death pathways in primary cultures of mesencephalic dopaminergic neurons after neurotoxin treatment. J. Neurosci. 23, 5069–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huot P.; Levesque M.; Parent A. (2007) The fate of striatal dopaminergic neurons in Parkinson’s disease and Huntington’s chorea. Brain 130, 222–232. [DOI] [PubMed] [Google Scholar]
- Margolis E. B.; Coker A. R.; Driscoll J. R.; Lemaitre A. I.; Fields H. L. (2010) Reliability in the identification of midbrain dopamine neurons. PLoS One 5, e15222-1–e15222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taravini I. R.; Chertoff M.; Cafferata E. G.; Courty J.; Murer M. G.; Pitossi F. J.; Gershanik O. S. (2011) Pleiotrophin over-expression provides trophic support to dopaminergic neurons in parkinsonian rats. Mol. Neurodegener. 6, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin E.; Cavanaugh J. E.; Leak R. K.; Perez R. G.; Zigmond M. J. (2008) Rapid activation of ERK by 6-hydroxydopamine promotes survival of dopaminergic cells. J. Neurosci. Res. 86, 108–117. [DOI] [PubMed] [Google Scholar]
- Burgering B. M.; Coffer P. J. (1995) Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 376, 599–602. [DOI] [PubMed] [Google Scholar]