

Abstract
Colorectal cancer (CRC) is the third most common cancer disease in the Western world, and about 40% of the patients die from this disease. The cancer cells are commonly genetically unstable, but only a few low-frequency recurrent fusion genes have so far been reported for this disease. In this study, we present a thorough search for novel fusion transcripts in CRC using high-throughput RNA sequencing. From altogether 220 million paired-end sequence reads from seven CRC cell lines, we identified 3391 candidate fused transcripts. By stringent requirements, we nominated 11 candidate fusion transcripts for further experimental validation, of which 10 were positive by reverse transcription-polymerase chain reaction and Sanger sequencing. Six were intrachromosomal fusion transcripts, and interestingly, three of these, AKAP13-PDE8A, COMMD10-AP3S1, and CTB-35F21.1-PSD2, were present in, respectively, 18, 18, and 20 of 21 analyzed cell lines and in, respectively, 18, 61, and 48 (17%-58%) of 106 primary cancer tissues. These three fusion transcripts were also detected in 2 to 4 of 14 normal colonic mucosa samples (14%–28%). Whole-genome sequencing identified a specific genomic breakpoint in COMMD10-AP3S1 and further indicates that both the COMMD10-AP3S1 and AKAP13-PDE8A fusion transcripts are due to genomic duplications in specific cell lines. In conclusion, we have identified AKAP13-PDE8A, COMMD10-AP3S1, and CTB-35F21.1-PSD2 as novel intrachromosomal fusion transcripts and the most highly recurring chimeric transcripts described for CRC to date. The functional and clinical relevance of these chimeric RNA molecules remains to be elucidated.
Introduction
Colorectal cancer (CRC) is a global health problem with a high incidence and mortality. It is the second most common cancer type in Europe, and only lung cancer causes more cancer deaths per year [1]. The management of CRC is therefore in need of improved biomarkers for detection, monitoring, and prognostication, as well as prediction of treatment response [2]. Furthermore, effective targeted therapies are warranted for this disease [3]. A promising strategy to meet these demands is to identify highly cancer-specific molecules.
Gene expression profiling has been used to identify genes with ectopic expression in cancer. However, the efforts so far have not been sufficient for development of any differentially expressed genes into clinically useful biomarkers, probably because the gene expression is not specific enough for the malignant cells. Differential pre-mRNA processing adds an additional layer of complexity, and dysregulation of alternative splicing and promoter switches may yield cancer-specific transcripts and protein isoforms [4]. Chimeric fusion transcripts represent another source of common cancer-specific RNA and proteins and have, for other cancer types, been useful in both cancer detection and monitoring of patients with cancer, as well as being direct targets for treatment. Most recurrent fusion genes previously identified have been found from studies of hematological malignancies and sarcomas. Among the four most common carcinomas, the surprising discovery of the TMPRSS2-ERG fusion gene present in about half of all prostate cancers was reported in 2005 [5]. Later, several additional recurrent fusion genes in prostate cancer have been reported [6–10]. Furthermore, but with lower frequencies, fusion genes involving the targetable ALK, ROS1, and RET partners have been found in lung cancer [11–14]. In breast cancer, a recurrent fusion with a low frequency, SEC16A-NOTCH1, has been reported [15]. For CRC, there are three recent reports of recurrent fusion transcripts [13,16,17], but all of them occur in low frequencies.
Fusion transcripts are often produced after chromosomal rearrangements but can also be generated by RNA polymerase read-throughs and trans-splicing of pre-mRNA. Recently, there have been reports that chimeric fusion transcripts, generated by polymerase read-throughs and trans-splicing, are common within prostate cancers [18,19]. Low, but detectable, levels of SLC45A3-ELK4 mRNA was found in both benign and malignant prostate tissues, with higher expression of the fusion transcript in the malignant tissues [9]. Interestingly, there is also evidence for chimeric transcripts to be frequent and nonrandom within nonmalignant cells [20], and protein products from chimeric transcripts were recently reported to be commonly present in human cells [21]. Chimeric transcripts were recently also shown to have a regulatory role of growth in cancer cells [22]. The recent ENCODE transcriptome study suggests that the definition of a gene should be redefined on the basis of their findings of widespread overlapping of neighboring gene regions [23]. Furthermore, presence of fusion transcripts in nonmalignant cells, generated by trans-splicing, has been demonstrated to guide chromosomal rearrangements involving the same fusion partners in endometrial stromal tumors [24]. Regardless of the mechanisms, fusion transcripts may encode cancer-specific chimeric proteins, which are promising as biomarkers and also as targets for therapy.
Identification of recurrent fusion transcripts in CRC may aid the development of improved diagnostics and tailored treatment. In this study, we have identified novel fusion transcripts from colon cancer cell lines by use of paired-end RNA sequencing and shown their presence also in malignant, and sometimes nonmalignant, tissue from the large bowel.
Methods
Colon Cancer Cell Lines and Clinical Tissue Samples
Seven colon cancer cell lines were included in the RNA sequencing analyses. HCT15, SW48, HCT116, and RKO are known to be of the microsatellite instability (MSI) phenotype, and HT29, SW480, and LS1034 are microsatellite stable (MSS) [25]. Fourteen additional colon cancer cell lines were added to the validation panel (SW620, LoVo, Co115, Colo320, IS1, IS2, IS3, TC7, TC71, FRI, V9P, LS174T, EB, and NCI-H508). The cell lines have been obtained from Dr Richard Hamelin (INSERM, Paris, France) and American Type Culture Collection (Manassas, VA). Culturing conditions for the individual cell lines will be given on request. The cell lines have previously been karyotyped, and copy number was assessed by comparative genomic hybridization (CGH). Identities of the cell lines were verified by the AmpFlSTR Identifiler PCR Amplification Kit (Applied Biosystems by Life Technologies, Carlsbad, CA). Cell lines were harvested at a time point shortly before confluence was reached, and RNA was isolated using TRIzol (Life Technologies Inc, Rockville, MD). Quantity was measured using NanoDrop ND-1000 (Thermo Fisher Scientific, Waltham, MA), and quality was evaluated with Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA).
Tissue samples were collected from 106 patients treated surgically for CRC in hospitals in the Oslo region, Norway. The CRCs were enriched for clinical stages II and III (52 stage II, 53 stage III, and 1 stage IV CRCs) and included both MSI and MSS types [n = 20 and 85 (one sample not scored), respectively]. A summary of clinical data for the patients can be found in Table W1 (Supplementary Materials). For 14 patients, corresponding normal colonic mucosa was taken from visually disease-free areas. Tumors were staged according to the American Joint Cancer Committee/Union for International Cancer Control. Status for MSI, gene mutations (within KRAS, BRAF, PIK3CA, PTEN, and TP53), and transcriptome instability were obtained from previous publications [26–28]. The research biobanks have been registered according to national legislation, and the study has been approved by the Regional Committee for Medical Research Ethics (Biobank 2781; REK South-East S-09282c2009/4958). Informed consent was obtained from patients enrolled to the study. RNA from the tissue samples was isolated by using AllPrep DNA/RNA Mini Kit (Qiagen, Valencia, CA). Quantity and quality were measured and evaluated as described above. Further, a panel of 20 normal tissues from different organs and tissue types was included (FirstChoice Human Normal Tissue Total RNA, each has a pool of RNA from at least three individuals, with the exception of an individual sample from the stomach; Ambion, Applied Biosystems by Life Technologies, Carlsbad, CA).
High-Throughput Paired-End RNA Sequencing
Library construction followed the standard Illumina mRNA library preparation (icom.illumina.com, 2009; Illumina Inc, San Diego, CA), including poly-A mRNA isolation, fragmentation, and gel-based size selection. Shearing to about 250-bp fragments was achieved using the Covaris S2 focused-ultrasonicator (Covaris Inc, Woburn, MA). Sequencing was performed according to the paired-end RNA sequencing protocols from Illumina for Solexa sequencing on a Genome Analyzer IIx with paired-end module (Illumina Inc). For all seven cell lines, 23 to 38 million clusters were generated (Table W2; Supplementary Materials). Seventy-six bps were sequenced, from each side of a fragment about 250 bp long.
Gene Fusion Prediction and Gene Expression
Only reads marked by the Illumina pipeline (Bustard.py, OLB 1.6.0 and 1.8.0) as passed filtering were used in the analysis. We used the fusion discovery software tool deFuse [29], version 0.2.1, with hg19 sequence reference and Ensembl release 58 annotation databases, to assist in locating potential gene fusions. Several filtering steps of the fusions were performed. The first step included filtering against fusions identified in normal cells and tissues. These were identified from analysis of the Illumina Human Body Map v2 data set, including paired-end RNA sequencing data from 16 nonmalignant sample types, including normal colonic mucosa (ArrayExpress accession ID E-MTAB-513 and European Nucleotide Archive study accession ID ERP000546). The deFuse software was applied with the same settings as for analyzing the seven colon cancer cell lines. Fusions from the 16 cell and tissue types were pooled together and defined as normal tissue fusions. The second filtering step removed fusions where at least one of the fusion partners was annotated as a ribosomal gene (as listed in Biomart Ensembl release 60, GO term 0005840). The third step removed promiscuous fusions where one of the genes had multiple partners within the same cell line. The fourth step removed intrachromosomal fusions where the gene partners were located less than 100 kbp apart. The fifth, and final, step removed genes where the chimeric breakpoint sequences were introns or intraexonic, leaving only fusion transcripts with intact exon-exon boundaries, using predominantly consensus splice sites. Gene expression levels were computed by using Cufflinks v1.1.0 [30], with the Illumina iGenomes Ensembl GRCh37 data set (2011-06-20) as reference, on reads aligned with TopHat v1.3.3 [31] and Bowtie v0.12.5 [32]. Coverage and annotation plots were created using R [33] and the GenomeGraphs [34] package in Bioconductor [35].
Reverse Transcription-Polymerase Chain Reaction and cDNA Sequencing
First-strand cDNA synthesis was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA), and reverse transcription-polymerase chain reaction (RT-PCR) with HotStart Taq DNA Polymerase Kit (Qiagen) was performed to validate the existence of the nominated fusion transcripts. Primers were designed to span the fusion breakpoints using the Primer3 software [36] with default parameters (primer sequences are shown in Table W3; Supplementary Materials). The PCR products were run on a 2% agarose gel containing ethidium bromide. When only one band was present after electrophoresis, the PCR product was cleaned using ExoSAP-IT (GE Healthcare, Little Chalfont, United Kingdom) before sequencing. When several bands were present, each was cut and eluted using the MinElute Gel Extraction Kit (Qiagen). All samples were further sequenced (BigDye with ABI 3730 DNA analyzer; Applied Biosystems).
Whole-Genome Paired-End DNA Sequencing
Whole-genome paired-end sequencing was performed by BGI Hong Kong (Hong Kong, China) on the four cell lines with confirmed fusion transcripts (HCT15, HCT116, HT29, and SW480) to an average coverage of x30. Around 850 million 100-bp paired-end reads were produced for each cell line. The sequence reads were aligned by BWA version 0.6.1 [37] against hg19, and the loci of the validated fusion transcripts were visualized in the Integrative Genomics Viewer [38]. Genomic breakpoints were identified by nFuse version 0.2.0 [39].
Fluorescence in situ Hybridization (FISH)
To detect chromosomal rearrangements involving AKAP13 and PDE8A, we used a triple color probe FISH strategy flanking the aforementioned genes. Bacterial artificial chromosome (BAC) clones targeting the 5′ region of PDE8A (CTD-2253L13), the 3′ region of AKAP13 (RP11-296P8 and CTD-3247B18), and the 200-kb region between the two genes (CTD-2222G4) were selected using the UCSC Human Genome Browser and obtained from the BACPAC Resources Center (Oakland, CA). BAC DNA was extracted using the Plasmid DNA Purification Kit (MACHEREY-NAGEL GmbH KG, Duren, Germany) and amplified using the GenomiPhi V2 DNA Amplification Kit (WGA kit; GE Healthcare) according to the manufacturer's instructions. BAC DNA was labeled with SpectrumGreen (CTD-2222G4)-, SpectrumRed (CTD-2253L13)-, and SpectrumAqua (CTD-3247B18, RP11-296P8)-conjugated nucleotides by nick translation according to the manufacturer's instructions (Nick Translation DNA Labelling System; Enzo Life Sciences, Farmingdale, NY). Adequate mapping and probe specificity of all BAC clones was confirmed by hybridization onto normal human metaphases. SW480 metaphase spreads were obtained according to standard procedures.
Results
Identification of Fusion Transcripts and Gene Expression from Paired-End RNA Sequencing Data
Altogether 220 million paired-end sequence reads were generated from seven colon cancer cell lines (Table W2; Supplementary Materials; European Nucleotide Archive study accession ID ERP002049). From these, 3391 candidate fusion transcripts were identified with at least five-fold coverage of sequence pairs across the fusion partners and at least three individual sequence reads spanning the actual breakpoint. Subsequent filtering resulted in a set of 11 fusion transcripts reliably nominated for experimental validation (Figure 1 and Table W4, A and B; Supplementary Materials). Briefly, first, 644 fusions were removed, because they were also identified in a set of 16 miscellaneous types of normal cells and tissues by use of the same data processing algorithm as for the cancer cell lines. The second filter removed 1419 fusions where at least one of the partner genes encoded ribosomal proteins, known to be frequent artifacts. The third filter removed 1116 fusions in which a common partner in a number of different fusions in the same sample was included. The fourth filter removed 39 of the remaining chimeric sequences where the gene partners were localized within less than 100 kb on the same chromosome, likely to be read-throughs. The fifth filter removed 162 additional chimeric sequences and ensured that only chimeric sequences with exact whole exons at either side of the breakpoints were included, preserving consensus splice sites.
Figure 1.

The identified fused sequences were filtered in a stepwise manner for nomination of fusion transcripts for further experimental validation.
Experimental Verification and Exploration within Additional Colon Cancer Cell Lines
The presence of the 11 CRC fusion transcripts selected for experimental validation were verified by RT-PCR of RNA from the same cell lines in which they were identified, and the junction was confirmed by Sanger sequencing (Table 1). Four of these were interchromosomal, whereas six were intrachromosomal. For all experimentally verified fusions, RT-PCR spanning the same exon-exon boundaries as initially identified was performed on a set of 19 colon cancer cell lines (including the seven analyzed by RNA sequencing). For three of the 10 fusion transcripts multiple positive bands were observed (86%–95% of cell lines; Table 2), and for these, an additional nested PCR primer pair was designed to ensure specificity of the analysis. The three recurrent fusion transcripts were AKAP13-PDE8A (86%; Figure 2), COMMD10-AP3S1 (95%; Figure 3), and CTB-35F21.1-PSD2 (86%; Figure W1; Supplementary Materials). The other fusion transcripts were positive in one or two cell lines each (Table W5; Supplementary Materials).
Table 1.
Fusion Transcripts Experimentally Validated by RT-PCR and Sanger Sequencing.
| Fusion Gene | Chromosome | Bands | Cell Line | Distance (kb)* | Genomic Breakpoint† |
| Interchromosomal | |||||
| SLC39A14-TSPAN15 | 8p21.3 | 10q22.1 | HT29 | N | |
| NCOA3-SPINT1 | 20q12 | 15q13.3 | HCT15 | N | |
| GRIN2B-CYP4F3 | 12p12 | 19p12.12 | SW480 | Y | |
| FAM96A-STIM1 | 15q22.31 | 11p15.5 | SW480 | Y | |
| Intrachromosomal | |||||
| PRMT1-FLT3LG | 19q13.33 | 19q13.33 | HCT15 | 214 | N |
| MGRN1-C16orf96 | 16p13.3 | 16p13.3 | HCT15 | 134 | Y |
| COMMD10-AP3S1 | 5q23.1 | 5q22.3 | HCT116 | 198 | Y |
| SPAG9-MBTD1 | 17q21.33 | 17q21.33 | HCT116 | 298 | Y |
| AKAP13-PDE8A | 15q25.3 | 15q25.3 | SW480 | 556 | N |
| CTB-35F21.1-PSD2 | 5q31.2 | 5q31.2 | SW480 | 160 | N |
Distance is the outer distance between the two genes.
Predicted genomic breakpoint by nFuse.
Table 2.
Nested PCR Performed on Two Patient Sample Series, One of Them Paired with Normals, and Cell Lines.
| Fusion | CRC Series 1 | CRC Series 2 | Normal Mucosa Series 2 | Series 2, Pairs (Positive in Both Tumor and Normal) | Tumors, Series 1 + 2 | Cell Lines |
| n | 92 | 14 | 14 | 14 | 106 | 21 |
| AKAP13-PDE8A | 15 (16%) | 4 (29%) | 4 (29%) | 2 (14%) | 19 (18%) | 18 (86%) |
| CTB35F21.1-PSD2 | 41 (45%) | 8 (57%) | 2 (14%) | 1 (7%) | 49 (46%) | 18 (86%) |
| COMMD10-AP3S1 | 49 (53%) | 12 (86%) | 4 (29%) | 4 (29%) | 61 (58%) | 20 (95%) |
Figure 2.
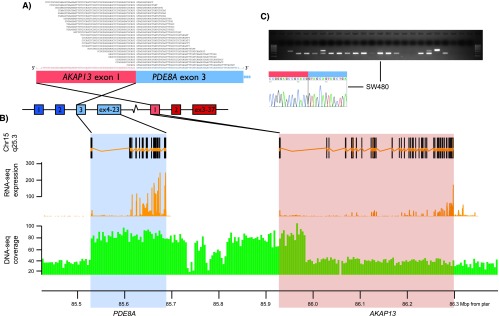
The AKAP13-PDE8A fusion transcript is recurrent in CRC, and the genomic locus is rearranged in the SW480 cell line. (A) Twenty-four RNA sequence reads spanned the chimeric transcript breakpoint, passing from exon 1 of AKAP13 (ENST00000361243) to exon 3 of PDE8A (ENST00000310298). Dark colors indicate exons that are not part of the fusion transcript. (B) Genomic view of the rearranged locus, from the top showing annotated exons of the fused genes (exons belonging to genes located between and within PDE8A and AKAP13 were removed for clarification), relative RNA expression levels, and DNA copy numbers. The two latter are based on coverage data from high-throughput sequencing of RNA and DNA from the SW480 cell line. (C) The AKAP13-PDE8A fusion transcript was initially detected from the SW480 cell line, but nested RT-PCR demonstrated detectable levels from 17 additional colon cancer cell lines (see Table 2 for cell line identities). The specific breakpoints of the AKAP13-PDE8A fusion transcripts were verified by Sanger sequencing and shown to follow the consensus splicing sites of the fusion partner genes.
Figure 3.
The COMMD10-AP3S1 fusion transcript is recurrent in CRC, and the genomic locus is rearranged in the HCT116 cell line. (A) Twelve RNA sequence reads spanned the chimeric transcript breakpoint, passing from exon 4 of COMMD10 (ENST00000274458) to exon 4 of AP3S1 (ENST00000316788). Dark colors indicate exons that are not part of the fusion transcript. (B) Genomic view of the rearranged locus, from the top showing annotated exons of the fused genes (exons belonging to genes located between and within COMMD10 and AP3S1 were removed for clarification), relative RNA expression levels, and DNA copy numbers. The two latter are based on coverage data from high-throughput sequencing of RNA and DNA from the HCT116 cell line. (C) The COMMD10-AP3S1 fusion transcript was initially detected from the HCT116 cell line, but nested RT-PCR demonstrated detectable levels in 19 additional colon cancer cell lines (see Table 2 for cell line identities). The specific breakpoints of the COMMD10-AP3S1 fusion transcripts were verified by Sanger sequencing and shown to follow the consensus splicing sites of the fusion partner genes.
The AKAP13-PDE8A locus was analyzed by three-color interphase and metaphase FISH on the SW480 cell line, showing predominantly five signals per cell (two in seemingly normal chromosome 15 and three in aberrant chromosomes), but no evidence of chromosome rearrangement splitting signals from within or between AKAP13 and PDE8A. However, from whole-genome sequencing data of SW480, we found an increased coverage in the particular genomic segment from intron 2 in PDE8A to intron 1 in AKAP13, involving the exact set of exons corresponding to the observed AKAP13-PDE8A fusion transcript (Figure 2B), indicating that there is a duplication of that small genomic segment (below the resolution level of FISH analysis) that juxtaposes exon 1 of AKAP13 5′ to exon 3 of PDE8A (Figure 2). Theoretically, this fusion gene could also be originated by translocation or insertion between the two chromosome 15 regions, but these two mechanisms do not generate copy number changes and they would have been detectable by the FISH strategy we used.
Out of the additional experimentally verified fusion transcripts, a similar increased coverage from the whole-genome sequence data for the corresponding cell line was found for the COMMD10-AP3S1 region in the HCT116 cell line (Figure 3B).
By analyzing the whole-genome sequencing data using nFuse [39], we identified a genomic breakpoint between the genes COMMD10 and AP3S1 and also of four additional fusion transcripts (Table 1). We performed genomic PCR of the predicted breakpoint of the COMMD10-AP3S1 fusion on the set of 19 colon cancer cell lines. Only HCT116, from which both the fusion transcript and the genomic breakpoint were originally identified, harbored this exact breakpoint.
The sequenced cell lines have also previously been karyotyped [25]. From this, we do not find cytogenetic evidence of genomic breakpoints at the loci of the fusion partner genes. However, the reported intrachromosomal fusion transcripts have also proximal loci that are visible at the cytogenetic level.
Validation in Clinical Specimens
Altogether 106 clinical CRC specimens, 14 with corresponding normal colonic mucosa, were analyzed for the presence of any of the 10 fusion transcripts. To ensure specificity of the products, nested PCR primers were generated for the three with multiple bands in the cell lines (AKAP13-PDE8A, COMMD10-AP3S1, and CTB-35F21.1-PSD2). A PCR-on-PCR protocol was applied for the remaining seven to detect the small amount of expressed fusion transcripts. The AKAP13-PDE8A fusion transcript, originally identified in the SW480 cell line, was positive in 19 of the 106 CRCs (18%) and as well in 4 of the 14 normal colonic mucosa samples (29%; Tables 2 and W6). Of the four positive normal samples, one was also positive for the COMMD10-AP3S1 fusion transcript, and another sample was also positive for the CTB-35F21.1-PSD2 fusion transcript. The COMMD10-AP3S1 fusion transcript, originally identified in the HCT116 cell line, was positive in 61 of the 106 CRCs (58%) and in 4 of the 14 normal colonic mucosa samples (29%). The CTB-35F21.1-PSD2 fusion transcript, originally identified in the SW480 cell line, was positive in 49 of the 106 CRCs (46%) and in 2 of the 14 normal colonic mucosa samples (14%). Furthermore, SPAG9-MBTD1 was positive in four cancers (4%) and one normal sample (7%), NCOA3-SPINT1 was positive in one normal sample (7%), and the remaining five fusion transcripts were negative in all clinical CRC samples (Table W5; Supplementary Materials). The identity of the fusion transcripts were further ensured by Sanger sequencing in 36 samples, confirming the sequence in all, and the chimeric sequences were shown to be located precisely at known exon boundaries in 31 of these (Table W7; Supplementary Materials). Nested PCR on the three fusion transcripts AKAP13-PDE8A, COMMD10-AP3S1, and CTB-35F21.1-PSD2 were performed on 20 additional normal tissues of miscellaneous sources. Of the 20 samples, 3, 10, and 17 were positive for the fusions AKAP13-PDE8A, COMMD10-AP3S1, and CTB-35F21.1-PSD2, respectively (tissue identities in Table W8).
For the three fusion transcripts with more than 10% positive CRCs, we tested for associations with clinical parameters (stage, MSI status, tumor location, gender, and age) and molecular data (MSI, mutations in BRAF, KRAS, PIK3CA, and PTEN, and transcriptome instability [26–28]). None of the associations were statistically significant (data not shown).
Discussion
Here, we report three novel and highly recurrent fusion transcripts in CRC, AKAP13-PDE8A, COMMD10-AP3S1, and CTB-35F21.1-PSD2. Their high prevalence in CRC, as well as their presence in a significant proportion of normal colonic mucosa samples, indicates that they are commonly produced and most often not the result of genomic rearrangement. All three recurrently detected fusion transcripts have partner genes from the same chromosome, suggesting a polymerase read-through mechanism. However, the switch of order of the AKAP13-PDE8A implies that this mechanism cannot be the sole explanation, and we see both genomic duplications, which is evident from the SW480 cell line, and trans-splicing as other likely mechanisms. A genomic breakpoint was identified in the COMMD10-AP3S1 fusion in HCT116, and together with the increased coverage between the breakpoints, this supports a genomic duplication as the mechanism for these fusion transcripts and cell line. Further, since the genomic PCR covering the breakpoint sequence was only positive for this sample, the COMMD10-AP3S1 fusion transcript is not likely to be due to a common copy number variant.
In the majority of the positive samples, the fusion transcripts were expressed at low levels, underscored by the need for nested PCR for detection. Originally, using regular one-step PCR, we detected the CTB-35F21.1-PSD2 fusion transcript in the cell lines SW480 and HCT15. Given also the precise joining of sequences at known exon boundaries, we reason that the measured fusion transcripts are not produced by artifacts of the laboratory protocol. However, the presence of genomic rearrangements of AKAP13-PDE8A and COMMD10-AP3S1 in one cancer cell line each indicates that the production of fusion transcripts may guide the generation of genomic rearrangements, similarly to a previous report on the fusion of JAZF1 and JJAZ1 in endometrial stromal tumors [24]. Recently, a study showed that fusion transcripts in healthy cells may also generate fusion proteins [21].
The predicted protein encoded by the AKAP13-PDE8A fusion transcript includes only coding parts from the PDE8A partner and with a truncation of 72 amino acid residues from its N terminus. PDE8A encodes a phosphodiesterase involved in regulation of cyclic adenosine monophosphate (cAMP) metabolism [40]. Five alternative splicing variants have been characterized, with a conserved catalytic domain located toward the C-terminal region, starting on amino acid number 555, located on exon 18 [41]. This suggests that the catalytic domain is still active in the predicted fusion protein encoded by the AKAP13-PDE8A fusion transcript. Interestingly, the protein encoded by AKAP13 is an A-kinase anchoring protein [42]. Although speculative, the regulation of PDE8A by the AKAP13 promoter may alter cAMP-mediated signaling in cells harboring this fusion. As FISH analysis of the fusion gene did not reveal large-scale structural rearrangements of the region containing both partner genes, the DNA copy number increase suggests some local rearrangement, such as duplication. Importantly, the increased coverage from the whole genome sequencing data in the SW480 cell line spans the exact region corresponding to the AKAP13-PDE8A fusion transcript.
The breakpoint in the COMMD10-AP3S1 fusion transcript is between exon 5 of COMMD10 and exon 4 of AP3S1. COMMD10 is predicted to encode a suppressor of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [43], whereas AP3S1 encodes a partner of the AP-3 complex, an adapter-related complex that is associated with the Golgi apparatus and more peripheral structures. AP3S1 facilitates the budding of vesicles from the Golgi membrane and may be directly involved in trafficking to lysosomes [44]. Increased coverage in the COMMD10-AP3S1 locus from the whole-genome sequence data on HCT116 suggests, as with the AKAP13-PDE8A fusion, that a duplication of a segment covering both genes may have caused the fusion and, hence, the DNA copy number change.
The predicted protein encoded by the CTB-35F21.1-PSD2 fusion transcript includes the first three exons of CTB-35F21.1, which is annotated as a lincRNA in the Havana database, and all coding exons of PSD2, beginning with the start codon in exon 2. PSD2 encodes a protein containing pleckstrin and Sec7 domains and is shown to interact with PMS2 in the DNA mismatch repair complex [45].
The prevalence of the identified fusion transcripts may be even higher than what is reported here, as we have only tested the breakpoint between two exons originally identified to be involved in the fusion, and other CRCs may have fusion transcripts between the same genes at another exon at one or both sides of the breakpoint. Furthermore, other CRCs may have one of the fusion partners exchanged for another gene.
Conclusions
We have identified three novel recurrent and seven novel private fusion transcripts in CRC. This adds significantly to the knowledge on chimeric transcripts in CRC and provides a new context for further studies targeted at the usage of fusions as biomarkers or drug targets.
Supplementary Material
Acknowledgments
We are grateful to Shujun Luo, Irina Khrebtukova, and Gary Schroth from Illumina for providing the RNA sequencing data for the 16 different human tissues ahead of release to the ArrayExpress public data repository.
Footnotes
This study was funded by grants from the Norwegian Cancer Society (PR-2006-0442 to R.A.L. and PR-2007-0166 to R.I.S.). Instrumentation for high-throughput sequencing has been financed by the Norwegian Radium Hospital Legacies. The authors declare that they have no competing interests.
This article refers to supplementary materials, which are designated by Tables W1 to W8 and Figure W1 and are available online at www.transonc.com.
References
- 1.Ferlay J, Shin H-R, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Bacolod MD, Barany F. Molecular profiling of colon tumors: the search for clinically relevant biomarkers of progression, prognosis, therapeutics, and predisposition. Ann Surg Oncol. 2011;18:3694–3700. doi: 10.1245/s10434-011-1615-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waldner MJ, Neurath MF. The molecular therapy of colorectal cancer. Mol Aspects Med. 2010;31:171–178. doi: 10.1016/j.mam.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Skotheim RI, Nees M. Alternative splicing in cancer: noise, functional, or systematic. Int J Biochem Cell Biol. 2007;39:1432–1449. doi: 10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 5.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun X-W, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 6.Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer. 2008;8:497–511. doi: 10.1038/nrc2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paulo P, Barros-Silva JD, Ribeiro FR, Ramalho-Carvalho J, Jerónimo C, Henrique R, Lind GE, Skotheim RI, Lothe RA, Teixeira MR. FLI1 is a novel ETS transcription factor involved in gene fusions in prostate cancer. Genes Chromosomes Cancer. 2012;51:240–249. doi: 10.1002/gcc.20948. [DOI] [PubMed] [Google Scholar]
- 8.Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, Han B, Cao Q, Cao X, Suleman K, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rickman DS, Pflueger D, Moss B, VanDoren VE, Chen CX, de la Taille A, Kuefer R, Tewari AK, Setlur SR, Demichelis F, et al. SLC45A3-ELK4 is a novel and frequent erythroblast transformation-specific fusion transcript in prostate cancer. Cancer Res. 2009;69:2734–2738. doi: 10.1158/0008-5472.CAN-08-4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barros-Silva JD, Paulo P, Bakken AC, Cerveira N, Lovf M, Henrique R, Jeronimo C, Lothe RA, Skotheim RI, Teixeira MR. Novel 5′ fusion partners of ETV1 and ETV4 in prostate cancer. Neoplasia. 2013;15:720–726. doi: 10.1593/neo.13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S-I, Watanabe H, Kurashina K, Hatanaka H, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 12.Takeuchi K, Choi YL, Togashi Y, Soda M, Hatano S, Inamura K, Takada S, Ueno T, Yamashita Y, Satoh Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15:3143–3149. doi: 10.1158/1078-0432.CCR-08-3248. [DOI] [PubMed] [Google Scholar]
- 13.Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 15.Robinson DR, Kalyana-Sundaram S, Wu Y-M, Shankar S, Cao X, Ateeq B, Asangani IA, Iyer M, Maher CA, Grasso CS, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med. 2011;17:1646–1651. doi: 10.1038/nm.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bass AJ, Lawrence MS, Brace LE, Ramos AH, Drier Y, Cibulskis K, Sougnez C, Voet D, Saksena G, Sivachenko A, et al. Genomic sequencing of colorectal adenocarcinomas identifies a recurrent VTI1A-TCF7L2 fusion. Nat Genet. 2011;43:964–968. doi: 10.1038/ng.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, et al. Recurrent R-spondin fusions in colon cancer. Nature. 2012;488:660–664. doi: 10.1038/nature11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nacu S, Yuan W, Kan Z, Bhatt D, Rivers CS, Stinson J, Peters BA, Modrusan Z, Jung K, Seshagiri S, et al. Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med Genomics. 2011;4:11. doi: 10.1186/1755-8794-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Gong M, Yuan H, Park HG, Frierson HF, Li H. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov. 2012;2:598–607. doi: 10.1158/2159-8290.CD-12-0042. [DOI] [PubMed] [Google Scholar]
- 20.Djebali S, Lagarde J, Kapranov P, Lacroix V, Borel C, Mudge JM, Howald C, Foissac S, Ucla C, Chrast J, et al. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS One. 2012;7:e28213. doi: 10.1371/journal.pone.0028213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frenkel-Morgenstern M, Lacroix V, Ezkurdia I, Levin Y, Gabashvili A, Prilusky J, del Pozo A, Tress M, Johnson R, Guigo R, et al. Chimeras taking shape: potential functions of proteins encoded by chimeric RNA transcripts. Genome Res. 2012;22:1231–1242. doi: 10.1101/gr.130062.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plebani R, Oliver GR, Trerotola M, Guerra E, Cantanelli P, Apicella L, Emerson A, Albiero A, Harkin PD, Kennedy RD, et al. Long-range transcriptome sequencing reveals cancer cell growth regulatory chimeric mRNA. Neoplasia. 2012;14:1087–1096. doi: 10.1593/neo.121342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H, Wang J, Mor G, Sklar J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science. 2008;321:1357–1361. doi: 10.1126/science.1156725. [DOI] [PubMed] [Google Scholar]
- 25.Kleivi K, Teixeira MR, Eknaes M, Diep CB, Jakobsen KS, Hamelin R, Lothe RA. Genome signatures of colon carcinoma cell lines. Cancer Genet Cytogenet. 2004;155:119–131. doi: 10.1016/j.cancergencyto.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 26.Diep CB, Thorstensen L, Meling GI, Skovlund E, Rognum TO, Lothe RA. Genetic tumor markers with prognostic impact in Dukes' stages B and C colorectal cancer patients. J Clin Oncol. 2003;21:820–829. doi: 10.1200/JCO.2003.05.190. [DOI] [PubMed] [Google Scholar]
- 27.Berg M, Danielsen SA, Ahlquist T, Merok MA, Agesen TH, Vatn MH, Mala T, Sjo OH, Bakka A, Moberg I, et al. DNA sequence profiles of the colorectal cancer critical gene set KRAS-BRAF-PIK3CA-PTEN-TP53 related to age at disease onset. PLoS One. 2010;5:e13978. doi: 10.1371/journal.pone.0013978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sveen A, Agesen TH, Nesbakken A, Rognum TO, Lothe RA, Skotheim RI. Transcriptome instability in colorectal cancer identified by exon microarray analyses: associations with splicing factor expression levels and patient survival. Genome Med. 2011;3:32. doi: 10.1186/gm248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McPherson A, Hormozdiari F, Zayed A, Giuliany R, Ha G, Sun MGF, Griffith M, Heravi Moussavi A, Senz J, Melnyk N, et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput Biol. 2011;7:e1001138. doi: 10.1371/journal.pcbi.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Development Core Team, author. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2009. [Google Scholar]
- 34.Durinck S, Bullard J, Spellman PT, Dudoit S. GenomeGraphs: integrated genomic data visualization with R. BMC Bioinformatics. 2009;10:2. doi: 10.1186/1471-2105-10-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 37.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McPherson A, Wu C, Wyatt AW, Shah S, Collins C, Sahinalp SC. nFuse: discovery of complex genomic rearrangements in cancer using high-throughput sequencing. Genome Res. 2012;22:2250–2261. doi: 10.1101/gr.136572.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fisher DA, Smith JF, Pillar JS, St Denis SH, Cheng JB. Isolation and characterization of PDE8A, a novel human cAMP-specific phosphodiesterase. Biochem Biophys Res Commun. 1998;246:570–577. doi: 10.1006/bbrc.1998.8684. [DOI] [PubMed] [Google Scholar]
- 41.Wang P, Wu P, Egan RW, Billah MM. Human phosphodiesterase 8A splice variants: cloning, gene organization, and tissue distribution. Gene. 2001;280:183–194. doi: 10.1016/s0378-1119(01)00783-1. [DOI] [PubMed] [Google Scholar]
- 42.Diviani D, Soderling J, Scott JD. AKAP-Lbc anchors protein kinase A and nucleates Gα12-selective Rho-mediated stress fiber formation. J Biol Chem. 2001;276:44247–44257. doi: 10.1074/jbc.M106629200. [DOI] [PubMed] [Google Scholar]
- 43.Burstein E, Hoberg JE, Wilkinson AS, Rumble JM, Csomos RA, Komarck CM, Maine GN, Wilkinson JC, Mayo MW, Duckett CS. COMMD proteins, a novel family of structural and functional homologs of MURR1. J Biol Chem. 2005;280:22222–22232. doi: 10.1074/jbc.M501928200. [DOI] [PubMed] [Google Scholar]
- 44.Simpson F, Peden AA, Christopoulou L, Robinson MS. Characterization of the adaptor-related protein complex, AP-3. J Cell Biol. 1997;137:835–845. doi: 10.1083/jcb.137.4.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cannavo E, Gerrits B, Marra G, Schlapbach R, Jiricny J. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J Biol Chem. 2007;282:2976–2986. doi: 10.1074/jbc.M609989200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.