

Abstract
A culture-independent genome sequencing approach was developed and used to examine genomic variability in Chlamydia trachomatis-positive specimens that were collected from patients in the Seattle, WA, USA, area. The procedure is based on an immunomagnetic separation approach with chlamydial LPS-specific mAbs, followed by DNA purification and total DNA amplification, and subsequent Illumina-based sequence analysis. Quality of genome sequencing was independent of the total number of inclusion-forming units determined for the sample and the amount of non-chlamydial DNA in the Illumina libraries. A geographically and temporally linked clade of isolates was identified with evidence of several different regions of recombination and variable ompA sequence types, suggesting that recombination is common within outbreaks. Culture-independent sequence analysis revealed a linkage pattern at two nucleotide positions that was unique to the genomes of isolates from patients, but not in C. trachomatis recombinants generated in vitro. These data demonstrated that culture-independent sequence analysis can be used to rapidly and inexpensively collect genome data from patients infected by C. trachomatis, and that this approach can be used to examine genomic variation within this species.
Introduction
Chlamydia trachomatis is an obligate intracellular bacterium and the most prevalent sexually transmitted bacterial infection of humans (Centers for Disease Control and Prevention, 2011). Studying these genetically difficult organisms has become less arduous, with the recent advent of high-throughput sequencing technologies and workable genetic systems (Wang et al., 2011). While acquisition by chlamydiae of genetic material from other species is considered rare (Dugan et al., 2004), recombination within C. trachomatis, both in vitro and in vivo, appears to be relatively common (Dugan et al., 2004; Gomes et al., 2004, 2007; Harris et al., 2012; Jeffrey et al., 2010; Joseph et al., 2011; Somboonna et al., 2011; Srinivasan et al., 2012). Recombination has been documented between the lymphogranuloma venereum and urogenital biovars, fusogenic and non-fusogenic strains (Jeffrey et al., 2013), and strains with tropism for different tissues (Harris et al., 2012; Jeffrey et al., 2010). The implications of these recombination events are only beginning to be elucidated.
The application of high-throughput sequencing for Chlamydia is challenging because of the extensive culturing process required for generating sufficient quantities of genomic DNA for sequencing. Moreover, extended in vitro culturing may lead to changes in the chlamydial genome due to the absence of host immune pressures (Borges et al., 2013). To address these issues, a system for sequencing chlamydial genomes without the need for growth in cell culture was developed. Immunomagnetic separation (IMS), utilizing antibodies specific to chlamydial LPS, was used to isolate Chlamydia directly from clinical samples. Multiple displacement amplification (MDA) was then employed to amplify the isolated chlamydial genomic DNA and produce sufficient quantities of DNA for high-throughput genomic sequencing. Utilizing this protocol, we sequenced and analysed the chlamydial genomes collected from 10 clinical endo-cervical swab specimens isolated from the Seattle, WA, USA, area collected between April 1993 and January 1998. The results revealed a geographically linked clade of similar chlamydial genomes with variable ompA sequences, distinct recombination blocks and evidence of in-patient mutation.
Methods
C. trachomatis collection, inclusion-forming unit (IFU) determination and serotyping.
De-identified patient materials used for this study were selected from frozen specimens in the University of Washington Chlamydia Repository. This resource contains over 15 000 patient samples including isolates from culture-documented patients attending Seattle–King County Health Department sexually transmitted disease clinics from 1988 to 2006 (Suchland et al., 2003). In the original diagnostic procedure, McCoy cells in 96-well trays were infected in a standardized dilution series with patient material and incubated for chlamydial growth. Cells were fixed with methanol, and inclusions were identified and quantified using genus-specific FITC-labelled anti-chlamydial LPS mAb (clone 2C1; Washington Research Foundation). Monolayers having 100 inclusions or less were individually counted to determine the inclusion number per swab. The average inclusion count per well was calculated by enumerating three fields on a Zeiss microscope and multiplying the average count by predetermined conversion factors to calculate the number of inclusions per well (Eckert et al., 2000). Serotypes of primary diagnostic cultures were determined using the methods of Suchland & Stamm (1991). After culture manipulations for diagnostic analysis, primary sample material was returned to the repository and stored until use in this study.
IMS of chlamydial elementary bodies (EBs).
Cervical swabs initially used for culture-based diagnostics were stored at −80 °C prior to processing. Specimens were selected for analysis on the basis of chlamydial IFU values (ranging from 4 to 15 000 IFU per swab) and serovar (Fig. 1). The identified specimens were removed from the freezer, thawed at 37 °C and placed on ice. Three Pyrex beads (2 mm diameter) were added to 1 ml of sample in PBS and vortexed [4 °C, 33 relative centrifugal force (RCF) for 3 min]. Samples were then split into two aliquots, with one being stored at −80 °C and the other being used for the enrichment of chlamydial EBs through IMS. Initial enrichments used primary mouse antibodies directed at LPS (EVI-HI; Suchland et al., 2008) or the major outer membrane protein (MOMP; Zhang et al., 1989). In each case, antibody concentrations were standardized to a final concentration of 3.1 ng µl−1. The primary antibody/cell suspension was incubated for 30 min at 4 °C on a rotating shaker (33 r.p.m.) and 1.0 ml of MACS buffer [Miltenyi; PBS (pH 7.2), EDTA (2 mM) and FBS (0.55 %)] was added. The sample was vortexed gently and centrifuged at 20 800 RCF for 10 min at room temperature. The supernatant was aspirated and pellet resuspended in 1.0 ml MACS buffer. IgG-specific secondary antibodies, conjugated to 50 nm paramagnetic particles (Miltenyi), were added and incubated using the same conditions as the primary antibody incubation. The labelled cells were loaded onto a disposable column and placed in a strong magnetic field (MACS Multi Stand). Bound material was washed twice with PBS, the columns were removed from the magnetic field and target material was collected with MACS buffer.
Fig. 1.
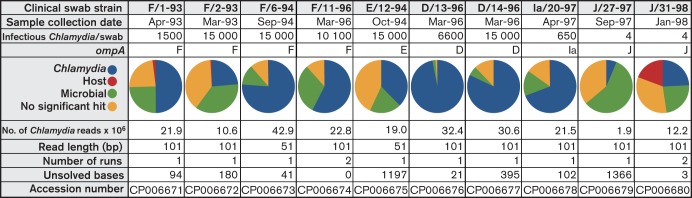
Clinical swab sample classification and sequencing output distribution.
DNase treatment for removal of host DNA.
Eluted material from the chlamydial cell separation was incubated (37 °C, 5 % CO2, 60 min) with RQ1 DNase (Promega). RQ1 stop buffer was added and the reaction was incubated at 65 °C for 10 min with gentle mixing. Total DNA was then extracted from the enriched EBs using the DNeasy Blood & Tissue kit (Qiagen) as described previously (Jeffrey et al., 2010). Real-time PCR analysis of this EB preparation was carried out using the Applied Biosystems Taqman Fast Universal PCR Master Mix with primers defining unique sequences of the second allele (groEL_2; CT604) of the C. trachomatis chaperone gene Hsp60 (primers: CTHsp60F GATTCTCTCTTCCTCGCTGTCTTC, CTHsp60R GAGGGTTTTCCCTGTCTGTGC). A plasmid containing the groEL_2 ORF was created, quantified and used as a standard curve in quantifying genome copy number from sample DNA.
MDA.
Column-purified EBs were removed from −80 °C storage, thawed quickly at 37 °C and placed on ice. Samples were centrifuged at 21 500 RCF for 10 min, the supernatant was aspirated and was the pellet resuspended in PBS. MDA was performed as described by the manufacturer (Qiagen Repli-g kit), using a 90 min reaction time at 30 °C. Amplified material was then stored at −20 °C.
Genome sequencing.
MDA-amplified genomic DNA samples from clinical swab samples were prepared for multiplex Illumina sequencing using the NEBNext DNA Library Prep Master Mix Set for Illumina kit and according to manufacturer-specified protocols (Illumina). Sequencing was performed on the Illumina HiSeq 2000 platform at the Center for Genome Research and Biocomputing Core Lab facility at Oregon State University. Multiplexing of samples was conducted using a commercial kit (Illumina Multiplexing Sample Preparation Oligonucleotide kit).
The clinical isolates were sequenced in two different groups; both were single-end, multiplexed runs with either 51 bp read-lengths or 101 bp read-lengths, as indicated (Fig. 1). For two samples (J/31-98 and F/11-96), single IMS preparations were divided in half, with each half being processed independently through all subsequent steps. These parallel runs were used to assemble and cross-check the sequence analysis. The initial 51-cycle run was completed with two multiplexed samples per lane, while the 101-cycle runs were performed with three and four multiplexed samples per lane.
Genome assembly and sequence analysis.
Genome sequence assemblies were done using the reference guided assembly software package maq (Li et al., 2008). Loci that could not be assembled using maq were resolved by assembling scaffolds from contiguous segments of sequence generated via the de novo assembly software package vcake (Jeck et al., 2007) or through the use of an ad hoc perl script pipeline. Any remaining ambiguous gaps in the assembled contiguous drafts were resolved through the use of traditional PCR and Sanger sequencing. macvector software (Macvector) was used for sequence manipulation and draft editing, and the mafft software package (Katoh et al., 2002, 2009) was used for whole-genome alignments and phylogenetic analysis. All ORFs were annotated based on the D/UW-3/CX genome as published by Stephens et al. (1998).
Regional recombination analyses.
A sliding window perl script described by Jeffrey et al. (2010) was initially used to compare compiled sequence information, for variation and recombination, against a database consisting of previously published chlamydial genome sequences: D/UW-3/CX (NC_000117; Stephens et al., 1998), J/6276 [National Center for Biotechnology Information (NCBI) Taxonomic ID 564416; Suchland et al., 2008], G/9768 (NC_017429; Jeffrey et al., 2010), E/11023 (NC_017431; Jeffrey et al., 2010) and/or F/70 (NCBI Taxonomic ID_564418 (Suchland et al., 2008). Genomes were aligned using mafft and custom perl scripts. To identify regions of recombination, the Recombination Identification Program (rip) available on the HIV sequence database website (http://www.hiv.lanl.gov/) was used. The software package phipack was used to assess overall probability of recombination in each region based on ascertaining the Pairwise Homoplasy Index statistic for each region (Bruen et al., 2006). The r statistical software package was used to generate data plots for analysis (R Development Core Team, 2008).
Results
Comparison of different mAbs for isolating C. trachomatis EBs from a clinical swab sample
Optimization of the culture-independent sequencing technology first required a comparison of available surface-reactive mAbs, to determine if antibody specificity affected EB harvesting efficiency. mAbs EVI-HI (specific to a genus common chlamydial LPS epitope), L2-I-V (specific to L2 MOMP) and HV-AV (specific to A, C, H, I and J MOMP) were each tested as possible primary antibodies for IMS. HV-AV was used as a negative control as it is non-reactive to L2 MOMP. The use of the anti-LPS primary mAb resulted in the elution of 95.5 % of the genome copies in test samples, while the use of the surface-epitope-targeting anti-MOMP antibody L2-I-V led to the recovery of far less material in these assays (Fig. 2a). Experiments in which either the primary or secondary antibodies were excluded demonstrated that the overall procedure was efficient and specific (Fig. 2b). Based on these results, we elected to use the anti-LPS mAb for all subsequent IMSs.
Fig. 2.
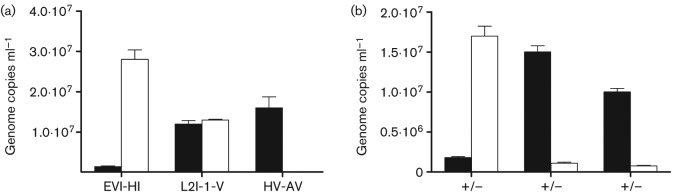
Enrichment efficiency of C. trachomatis serovar L2, as a factor of surface-reactive mAbs. qPCR of CT604 was used to infer bacterial enrichment efficiency based on chlamydial genome abundance. The white bars indicate genome copy numbers in the eluate of an IMS reaction, while the black bars indicate genome copies in the flow-through. (a) A laboratory-generated swab sample was harvested using antibodies to chlamydial LPS (EVI-HI), serovar L2 MOMP (L2-I-V) or to a serologically different MOMP (HV-AV) and processed through the IMS protocol. (b) Successful IMS only occurred in the presence of both primary and secondary antibodies. +/+, Recovery using both primary and secondary antibodies; +/−, no secondary antibody; –/+ indicates no primary antibody. This test was conducted using EVI-HI primary antibody.
Amplification of chlamydial genomes in samples with varying numbers of IFUs
The minimal necessary number of chlamydial genome copies required for culture-independent Illumina sequencing was determined by creating a set of standards with different amounts of genomic DNA and using these in independent MDA reactions. Amplification of samples containing fewer than 10 genome copies provided sufficient DNA for sequencing (data not shown). Sufficient genomic DNA for sequencing and complete assembly was collected and amplified from samples containing maximal numbers of infectious chlamydiae (i.e. 15 000 IFUs in specimen F/6-94) to as few as four IFUs per swab (i.e. specimens J/31-98 and J/27-98; Fig. 1).
Mitigating contamination from non-chlamydial DNA
The clinical samples used for amplification and sequence analysis were expected to contain non-chlamydial sources of DNA. For example, preliminary quantitative PCR (qPCR) on post-IMS samples revealed a substantial amount of human DNA that was carried through the separation process. A DNase incubation step was included post-IMS to help minimize contaminating host DNA. Subsequent qPCR performed after the DNase treatment demonstrated that this was successful at reducing host DNA (not shown).
To assess the abundance of DNA from non-chlamydial sources in our final Illumina-generated sequence data, a total of 1000 randomly selected reads from each of the clinical swabs were used in a blast-base search against the non-redundant nucleotide sequence database. The mentioned DNase reaction led to minimal contribution of host DNA in sequence reads (Fig. 1). The percentage of non-chlamydial reads from all sources ranged from around 4 % in the case of D/13-96, to around 75 % and 95 % in J/31-98 and J/27-97, respectively. Levels of each of the individual contaminants were variable across all the sequenced samples (Fig. 1).
Comparative analysis of generated genome sequences
Ten specimens were subjected to IMS purification, MDA whole-genome amplification and Illumina-based sequencing. For eight of these specimens, unambiguous sequence data were collected for greater than 99.98 % of the genome. The range of ambiguous bases per genome ranged from 0 to 1197. In two specimens (F/11-96 and J/31-98), a parallel sequencing approach was used, leading to the generation of two independently assembled genome sequences that were compared against each other. This approach resulted in the lowest numbers of ambiguous bases in the final assembled sequences (Fig. 1).
The assembled unambiguous sequence data were compared to a set of chlamydial genomes published by our group and other investigators (Fig. 3) (Harris et al., 2012; Jeffrey et al., 2010; Stephens et al., 1998). In all specimens, the chlamydial plasmid was present and displayed over 99 % identity with that of strain D/UW-3/CX. None of the changes present led to truncations in any of the plasmid coding sequences. In every specimen, CT135, a gene recently identified as necessary for in vivo pathogenicity, was intact and identical (Borges et al., 2013; Sturdevant et al., 2010). CT119, the ORF encoding IncA, was also intact in each of the genomes collected from these swabs.
Fig. 3.

mafft-generated whole-genome phylogeny aligning all 10 sequenced clinical swab samples with nine of the already published chlamydial genomes. rRNA sequences were removed to eliminate the repeat-associated error associated with highly homologous and non-informative sequence regions. The colour and the first letter of each strain indicate ompA genotype. The branch lengths are proportional to the genetic distance between specimens, as indicated by the scale bar.
Six assembled genomes carried a D, F or J ompA genotype, but otherwise shared a highly similar genomic background (F/1-93, F/2-93, F/6-94, F/11-96, D/13-96 and J/27-97; Fig. 4). While phylogenetic analysis of the complete genome sequence of specimen J/27-97 placed it outside of this grouping (Fig. 3), removal of the region surrounding ompA (Fig. 4) increased the similarity of this strain to other members of the clade (not shown). These sequences were each closely related to strain D(s)/2923, an IncA-negative isolate collected during the same time period and geographical area as the specimens analysed in this study (Jeffrey et al., 2010; Suchland et al., 2000) (Fig. 4). These genomes will be referred to as the D/13-like clade and D(s)/2923 will be included in all subsequent comparisons.
Fig. 4.

Genome maps of clinical isolates examined in this study. Each horizontal bar represents the complete genome sequence of the indicated strain or specimen, beginning with CT001. Sequence origin was identified by plotting informative sites using rip. Regions coloured grey indicate areas of apparent recombination that, in some strains, have margins that indicate independent recombination events. Black regions indicate conserved sequence between informative sites of sequences of different origin. The region showing the expanded sequences surrounding CT681 contains colour-coded sequence that identifies the ompA sequence origin (red, F; yellow, D; purple, J). J/27-97 is included in the ompA comparisons because the sequences shown were solved to a high degree of confidence, while the overall genome structure contained a relatively high level of ambiguous bases.
While the D/13-like clade was common in this population of patients, this was not the only sequence group identified in this set of specimens. Four other specimens (D/14-96, Ia/20-97, J/31-98 and E/12-94) contained C. trachomatis with genome sequences that were significantly different than the D/13-like strains and aligned more closely with other published chlamydial genomes (Fig. 3).
Several different regions of apparent recombination were identified in the D/13-like clade. The clearest example involved sequences surrounding ompA (CT681), the gene that codes for the MOMP (Figs 4 and S1, available with the online version of this paper in Microbiology Online). ompA from the D/13-96 genome is identical to that found in D(s)/2923, while F/2-93, F/6-94 and F/11-96 had similar flanking sequence but contained an ompA sequence of a different serovar. Specimen J/27-97 was included in the ompA comparative analysis because sequences surrounding ompA were fully resolved in this strain, against the background of an otherwise incomplete genome (Fig. 1). Analysis of the J/27-97 ompA region showed upstream flanking sequence highly similar to the D/13-like clade, with an ompA genotype representative of serovar J strains (Fig. 4). The location of the apparent integration sites for these different ompA regions varied with the different genotypes and there was no evidence that ompA variability was a function of sequence-specific recombination hot spots.
Two other regions of apparent recombination within this group include ORFs CT050-055 and CT193-218 (Fig. 4). These regions showed differences at their margins within the different specimens, indicating that they likely originated from distinct recombination events and were not the product of clonal expansion of a single recombinant lineage.
The genome sequence of F/6-94 is similar to D/13-96 in sharing several patterns of recombination. However, F/6-94 contains sequence representing a different integration event, leading to a large region of the chromosome (around 7 %, CT833-872) containing sequence with low similarity to the D/13-like clade (Fig. 4). All of the examined regions (CT050-55, CT193-218, CT681 and CT833-872) had significant support for recombination based on grouped patterns of homoplasic SNPs (P<0.05).
The genome sequence carried in specimen J/31-98 was most similar to a previously published strain collected from a patient in the Seattle area, J/6276 (Suchland et al., 2008). This strain has a short region of the genome that is less related to J/6276, including ORFs CT660 through CT662 (Fig. S3). This approximately 12-kb region is most similar to the sequences within the D/13-like clade and likely represents a short region of recombination into a background similar to strain J/6276.
Analysis of mutations within C. trachomatis from a single specimen
For most of the specimens, a single sequencing run was completed following IMS enrichment of samples. This resulted in many instances of unsolvable bases, based on ambiguity among the collected reads at that position (Fig. 1). While in some cases these might represent a non-clonal population within a single patient, it was not possible to separate genuine polymorphisms of sequence from errors generated in the generation of sequence data. This problem was addressed in two specimens (F/11-96 and J/31-98) by independently sequencing the genomes twice and cross-checking the results with an ad hoc perl script. Using this approach, sequences that represented genuine polymorphisms were represented similarly in both sequencing runs. For specimen F/11-96, this approach led to a complete genome sequence with no detectable ambiguities, leading to the assessment that this patient was infected with a clonal C. trachomatis population. In contrast, examination of sequence reads for specimen J/31-98 identified a total of three variable bases, demonstrating a low level of clonal variation in the patient (Fig. 1). Each variable base is associated with amino acid changes in the respective coding sequences (CT042, glycogen hydrolase; CT308, ATP synthase subunit A; CT449, hypothetical; Table S1).
The coding sequences for ORFs CT308 and CT449 are separated by approximately 150 000 bp. As a result of the recently established paradigm that these organisms commonly recombine in vitro and in vivo (Gomes et al., 2007; Harris et al., 2012; Jeffrey et al., 2010; Suchland et al., 2008), we hypothesized that these two mutations would segregate independently in published C. trachomatis genomes. However, comparison of these variable loci across all sequenced chlamydial genomes demonstrated that the different nucleotide polymorphisms are absolutely linked in published genomes from clinical strains. In this set of 47 published genomes collected from patients, 21 of the genomes carried an ‘A’ at position 2 in CT308 and a ‘C’ at position 320 in CT449 (Fig. S2a). In contrast, the remaining 26 genomes had a ‘G’ at position 2 in CT308 and a ‘T’ at position 320 in CT449. Our sequence data for J/31-98 contains consistent mixtures of these two polymorphic nucleotides in each of the two independently assembled sequences. Therefore, complete and published genome sequences from patients had specific combinations of nucleotides, each leading to a different amino acid in the encoded protein, while our culture-independent clinical specimen had variability at that position. In contrast, our group recently generated complete genome sequences from 10 recombinant clones generated in vitro, from parents that were members of the 47 strains discussed above (Jeffrey et al., 2013). Examination of these polymorphic nucleotide positions in these recombinant progeny demonstrated that three of 10 independent in vitro generated recombinants had nucleotide combinations that were not consistent with that seen in the clinical strains (Fig. S2b). Therefore, the absolute linkage found at these physically distant nucleotide positions is only found in chlamydiae growing under undetermined selective pressures in vivo and not in recombinant clones generated in vitro.
Discussion
Culture and propagation of chlamydiae can be time and reagent intensive, and opens the door for attenuations in vitro that might alter the genome sequence actually present in the patient. To address these issues, we have developed a rapid, inexpensive method for sequencing chlamydial genomes using material collected directly from clinical swab samples, which can be used even in the presence of large amounts of non-chlamydial DNA contamination. While we have applied our method directly to uncultured patient specimens, a similar approach should also be useful for sequencing hard to grow specimens or low-passage in vitro generated mutant strains. For the latter approach, we consider the use of parallel independent amplifications and sequence analysis to be a relatively inexpensive and ultimately cost-saving approach to generating unambiguous DNA sequence. Contamination is a significant challenge faced when dealing with samples directly collected from patients. Enrichment of chlamydial EBs with the LPS-specific antibody, DNase treatment of samples to reduce the level of non-chlamydial DNA and the strength of high-throughput sequencing technologies collectively led to high numbers of sequence reads for each position in the target genome. This was accomplished against a background of variable levels of contamination from the host, other microbes collected in the specimen and template-independent products that can occur artefactually in MDA procedures (Fig. 1). Regardless of these challenges, non-chlamydial reads from all sources did not reduce the overall success of sequencing, or assembly, of any of the samples processed.
A very recent report by Seth-Smith et al. (2013) discussed a similar technology for creating culture-independent C. trachomatis sequences from clinical specimens. Our approach is very similar to that described by these investigators, with the exception that we have included a RQ1 DNase treatment that leads to near total elimination of host DNA (Fig. 1). While this treatment adds another step to the process of generating a complete genome sequence, the strength of this approach lies in the much higher percentage of Chlamydia-specific reads within each Illumina sequencing run, allowing a higher level of multiplexing during the sequencing process.
Sequence ambiguities still existed in several of our assembled genomes, as is true with many published genomes available in the databases. However, we greatly reduced the numbers of ambiguities through the use of a parallel sequencing strategy, in which a single IMS-harvested Chlamydia preparation is subjected to two independent MDA reactions and sequencing runs. This approach is recommended for two reasons: it yields a much clearer overall dataset and greater confidence can be attributed to remaining polymorphisms, because these polymorphisms will remain variable in independent sequencing runs. In our data, sequence ambiguities ranged from 21 to 1366 nt in genomes sequenced a single time, but were reduced to zero and three in the two genomes that were sequenced twice. Variations at the three remaining positions in specimen J/31-98 were consistent both in the identities of the variable bases and in the percentage of a specific base at that position. Additionally, the success of a particular sequence analysis was not directly associated with the percentage of Chlamydia-specific reads, as samples with as little as 25 % Chlamydia-specific reads were successfully sequenced (strain J/31-98). The most significant factor in generating dependable and unambiguous sequence was the use of two parallel, independent amplifications and sequencing runs within a single clinical specimen.
Using this method we elucidated a geographically and temporally linked clade of C. trachomatis, termed the D/13-like clade, that displayed shared apparent recombination blocks. While each of these specimens was collected in the Seattle area, a similar genome sequence was identified in the genomic survey by Harris et al. (2012); their strain, D/SotonD1, was collected from the UK. Chlamydiae in these specimens display a highly similar overall genomic content to that of the previously sequenced isolate D(s)/2923 (Jeffrey et al., 2010). Sequence variability within the clade can be attributed to both recombination and mutation. Evidence of mutation is most apparent when comparing two highly related genomes, D/13-96 and D(s)/2923, which have 126 sequence differences and no apparent blocks of recombination that distinguish the genomes. One polymorphism between these genomes led to inactivation of incA in D(s)/2923, while the coding sequence of D/13-96 is intact and expresses a properly localized IncA polypeptide (not shown). These data are consistent with previous results from our group, in which highly related strains that were either IncA-positive or -negative can exist within single patients (Suchland et al., 2000). Collectively these results demonstrate that variability at incA is common and is found in similar specimens both in single patients and in related specimens being circulated in a geographically linked population.
The use of multiple sequence runs was critical to the solving of complete genomes and for the identification of apparently genuine nucleotide polymorphisms within a selected genome. This was evident during analysis of J/31-98, in which three nucleotide positions were variable at similar rates in each of the independent Illumina runs. These analyses identified polymorphic nucleotides that are uniquely matched in each sequenced clinical strain, but not in in vitro-generated recombinant strains. Such results led to the hypothesis that there are functional linkages between [V-type ATP synthase subunit A (CT308) and the hypothetical inclusion protein (CT449)] that are important in vivo, but it is challenging to design experiments to test this hypothesis. Further, it is not likely that uncovering of such a linkage between these two nucleotide positions could have been accomplished using any technology besides a culture-independent sequencing approach.
Recombination is also evident within clade members. Recombination-driven ompA variability is evident in specimens D/13-96, F/2-93, F/6-94, F/11-96 and J/27-98, where unique exchange events were identified leading to a phenotypic change of serovar (Figs 4 and S1). Changes were also evident in other regions of the chromosome, in events that appear to have occurred independent of one another (CT050-055, CT193-218 and CT826-874) (Fig. 4). For example, the region containing CT050-055 (Fig. 4) is very similar within the clade, but the margins of apparent integration differ for each sequence. The data from each of the D/13-like clade members suggest that variation within a common genetic background, similar to that described by Harris et al. (2012), was supplemented with independent recombination events involving common regions of the genome. These results demonstrate that recombination and mutation shape the overall mosaicism that is evident in the published C. trachomatis genome sequences, and support a hypothesis that the described mosaicism is the rule, not the exception, in chlamydial genome composition. The multi-locus sequence-typing (MLST) scheme designed by Dean et al. (2009) supported grouping these genomes by their similar mosaic structure. However, while serotyping and MLST analyses remain useful in understanding variation among chlamydial strains, the underlying mosaicism, against the background of overall genomic synteny and sequence identity, can only be assessed via more extensive whole-genome sequence analysis.
We have developed a rapid system for sequencing chlamydial genomes directly from clinical swabs without the need for growth in cell culture. This method could theoretically be used to isolate and expand the genome of any microbe with a surface antigen that is targetable by antibodies, directly from a clinical or mixed sample. Through its use we have elucidated a geographically and temporally linked clade of C. trachomatis that has undergone widespread recombination and is variable in the ompA sequence type carried by its members.
The use of the recently developed transformation system, next-generation sequencing and culture-independent methods allowing the sequencing of genomes directly from clinical samples represents a powerful combination of tools that will allow us to address issues in chlamydial biology that were previously not possible. Future efforts will continue in the investigation of the mechanisms and significance of recombination in chlamydial populations, and how recombination and genomic variation pertain to disease outcome.
Acknowledgements
Will Valiant is acknowledged for his participation in sequence assembly and verification. We thank Mark Dasenko, of the Center for Genomic Research and Biocomputing at Oregon State University, for his help and expertise with the high-throughput sequencing. We thank Harlan Caldwell of the Rocky Mountain Laboratories, NIH, for antibodies to C. trachomatis. We thank Dr Jeff Chang of Oregon State University for editorial assistance. This research was supported by NIH grants AI088540-02 and AI086469-01.
Abbreviations:
- EB
elementary body
- IFU
inclusion-forming unit
- IMS
immunomagnetic separation
- MDA
multiple displacement amplification
- MLST
multi locus sequence typing
- MOMP
major outer membrane protein
- NCBI
National Center for Biotechnology Information
- qPCR
quantitative PCR
- RCF
relative centrifugal force
- rip
Recombination Identification Program
Footnotes
Three supplementary figures and one supplementary table are available with the online version of this paper.
References
- Borges V., Ferreira R., Nunes A., Sousa-Uva M., Abreu M., Borrego M. J., Gomes J. P. (2013). Effect of long-term laboratory propagation on Chlamydia trachomatis genome dynamics. Infect Genet Evol 17, 23–32. 10.1016/j.meegid.2013.03.035 [DOI] [PubMed] [Google Scholar]
- Bruen T. C., Philippe H., Bryant D. (2006). A simple and robust statistical test for detecting the presence of recombination. Genetics 172, 2665–2681. 10.1534/genetics.105.048975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2011). CDC Grand Rounds: Chlamydia prevention: challenges and strategies for reducing disease burden and sequelae. MMWR Morb Mortal Wkly Rep 60, 370–373. [PubMed] [Google Scholar]
- Dean D., Bruno W. J., Wan R., Gomes J. P., Devignot S., Mehari T., de Vries H. J., Morré S. A., Myers G. & other authors (2009). Predicting phenotype and emerging strains among Chlamydia trachomatis infections. Emerg Infect Dis 15, 1385–1394. 10.3201/eid1509.090272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan J., Rockey D. D., Jones L., Andersen A. A. (2004). Tetracycline resistance in Chlamydia suis mediated by genomic islands inserted into the chlamydial inv-like gene. Antimicrob Agents Chemother 48, 3989–3995. 10.1128/AAC.48.10.3989-3995.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert L. O., Suchland R. J., Hawes S. E., Stamm W. E. (2000). Quantitative Chlamydia trachomatis cultures: correlation of chlamydial inclusion-forming units with serovar, age, sex, and race. J Infect Dis 182, 540–544. 10.1086/315738 [DOI] [PubMed] [Google Scholar]
- Gomes J. P., Bruno W. J., Borrego M. J., Dean D. (2004). Recombination in the genome of Chlamydia trachomatis involving the polymorphic membrane protein C gene relative to ompA and evidence for horizontal gene transfer. J Bacteriol 186, 4295–4306. 10.1128/JB.186.13.4295-4306.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes J. P., Bruno W. J., Nunes A., Santos N., Florindo C., Borrego M. J., Dean D. (2007). Evolution of Chlamydia trachomatis diversity occurs by widespread interstrain recombination involving hotspots. Genome Res 17, 50–60. 10.1101/gr.5674706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S. R., Clarke I. N., Seth-Smith H. M., Solomon A. W., Cutcliffe L. T., Marsh P., Skilton R. J., Holland M. J., Mabey D. & other authors (2012). Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat Genet 44, 413–419, S1. 10.1038/ng.2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck W. R., Reinhardt J. A., Baltrus D. A., Hickenbotham M. T., Magrini V., Mardis E. R., Dangl J. L., Jones C. D. (2007). Extending assembly of short DNA sequences to handle error. Bioinformatics 23, 2942–2944. 10.1093/bioinformatics/btm451 [DOI] [PubMed] [Google Scholar]
- Jeffrey B. M., Suchland R. J., Quinn K. L., Davidson J. R., Stamm W. E., Rockey D. D. (2010). Genome sequencing of recent clinical Chlamydia trachomatis strains identifies loci associated with tissue tropism and regions of apparent recombination. Infect Immun 78, 2544–2553. 10.1128/IAI.01324-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey B. M., Suchland R. J., Eriksen S. G., Sandoz K. M., Rockey D. D. (2013). Genomic and phenotypic characterization of in vitro-generated Chlamydia trachomatis recombinants. BMC Microbiol 13, 142. 10.1186/1471-2180-13-142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph S. J., Didelot X., Gandhi K., Dean D., Read T. D. (2011). Interplay of recombination and selection in the genomes of Chlamydia trachomatis. Biol Direct 6, 28. 10.1186/1745-6150-6-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Misawa K., Kuma K., Miyata T. (2002). mafft: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30, 3059–3066. 10.1093/nar/gkf436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Asimenos G., Toh H. (2009). Multiple alignment of DNA sequences with mafft. Methods Mol Biol 537, 39–64. 10.1007/978-1-59745-251-9_3 [DOI] [PubMed] [Google Scholar]
- Li H., Ruan J., Durbin R. (2008). Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18, 1851–1858. 10.1101/gr.078212.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2008). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Seth-Smith H. M., Harris S. R., Skilton R. J., Radebe F. M., Golparian D., Shipitsyna E., Duy P. T., Scott P., Cutcliffe L. T. & other authors (2013). Whole-genome sequences of Chlamydia trachomatis directly from clinical samples without culture. Genome Res 23, 855–866. 10.1101/gr.150037.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somboonna N., Wan R., Ojcius D. M., Pettengill M. A., Joseph S. J., Chang A., Hsu R., Read T. D., Dean D. (2011). Hypervirulent Chlamydia trachomatis clinical strain is a recombinant between lymphogranuloma venereum (L(2)) and D lineages. MBio 2, e00045-11. 10.1128/mBio.00045-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan T., Bruno W. J., Wan R., Yen A., Duong J., Dean D. (2012). In vitro recombinants of antibiotic-resistant Chlamydia trachomatis strains have statistically more breakpoints than clinical recombinants for the same sequenced loci and exhibit selection at unexpected loci. J Bacteriol 194, 617–626. 10.1128/JB.06268-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens R. S., Kalman S., Lammel C., Fan J., Marathe R., Aravind L., Mitchell W., Olinger L., Tatusov R. L. & other authors (1998). Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science 282, 754–759. 10.1126/science.282.5389.754 [DOI] [PubMed] [Google Scholar]
- Sturdevant G. L., Kari L., Gardner D. J., Olivares-Zavaleta N., Randall L. B., Whitmire W. M., Carlson J. H., Goheen M. M., Selleck E. M. & other authors (2010). Frameshift mutations in a single novel virulence factor alter the in vivo pathogenicity of Chlamydia trachomatis for the female murine genital tract. Infect Immun 78, 3660–3668. 10.1128/IAI.00386-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchland R. J., Stamm W. E. (1991). Simplified microtiter cell culture method for rapid immunotyping of Chlamydia trachomatis. J Clin Microbiol 29, 1333–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchland R. J., Rockey D. D., Bannantine J. P., Stamm W. E. (2000). Isolates of Chlamydia trachomatis that occupy nonfusogenic inclusions lack IncA, a protein localized to the inclusion membrane. Infect Immun 68, 360–367. 10.1128/IAI.68.1.360-367.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchland R. J., Eckert L. O., Hawes S. E., Stamm W. E. (2003). Longitudinal assessment of infecting serovars of Chlamydia trachomatis in Seattle public health clinics: 1988–1996. Sex Transm Dis 30, 357–361. 10.1097/00007435-200304000-00016 [DOI] [PubMed] [Google Scholar]
- Suchland R. J., Jeffrey B. M., Xia M., Bhatia A., Chu H. G., Rockey D. D., Stamm W. E. (2008). Identification of concomitant infection with Chlamydia trachomatis IncA-negative mutant and wild-type strains by genomic, transcriptional, and biological characterizations. Infect Immun 76, 5438–5446. 10.1128/IAI.00984-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Kahane S., Cutcliffe L. T., Skilton R. J., Lambden P. R., Clarke I. N. (2011). Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 7, e1002258. 10.1371/journal.ppat.1002258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. X., Stewart S. J., Caldwell H. D. (1989). Protective monoclonal antibodies to Chlamydia trachomatis serovar- and serogroup-specific major outer membrane protein determinants. Infect Immun 57, 636–638. [DOI] [PMC free article] [PubMed] [Google Scholar]