

Significance
The role of protein dynamics in enzyme catalysis remains a topic of considerable debate. Here, we use a combination of experimental and computational methods to identify the origins of the observed changes in reactivity on isotopic substitution of dihydrofolate reductase from Escherichia coli. Isotopic substitution causes differences in environmental coupling to the hydride transfer step and protein dynamics have therefore a small but measurable effect on the chemical reaction rate.
Keywords: kinetics, computational chemistry, biological chemistry, biophysics, quantum biology
Abstract
Protein dynamics have controversially been proposed to be at the heart of enzyme catalysis, but identification and analysis of dynamical effects in enzyme-catalyzed reactions have proved very challenging. Here, we tackle this question by comparing an enzyme with its heavy (15N, 13C, 2H substituted) counterpart, providing a subtle probe of dynamics. The crucial hydride transfer step of the reaction (the chemical step) occurs more slowly in the heavy enzyme. A combination of experimental results, quantum mechanics/molecular mechanics simulations, and theoretical analyses identify the origins of the observed differences in reactivity. The generally slightly slower reaction in the heavy enzyme reflects differences in environmental coupling to the hydride transfer step. Importantly, the barrier and contribution of quantum tunneling are not affected, indicating no significant role for “promoting motions” in driving tunneling or modulating the barrier. The chemical step is slower in the heavy enzyme because protein motions coupled to the reaction coordinate are slower. The fact that the heavy enzyme is only slightly less active than its light counterpart shows that protein dynamics have a small, but measurable, effect on the chemical reaction rate.
There is heated debate about the role of protein dynamics in enzyme catalysis, especially for reactions that involve transfer of hydrogen (H+, H·, H–), in which quantum tunneling is significant. It has been suggested that “promoting protein motions”, i.e., specific fluctuations that might reduce the barrier height or promote tunneling by reducing donor–acceptor distances, can drive enzymatic reactions (1, 2). Such models include promoting vibrations (3), environmentally coupled tunneling (1), and vibrationally enhanced ground-state tunneling (4). Several of these proposals suggest that the anomalous temperature and pressure dependences of experimentally observed reaction rates and kinetic isotope effects are the consequence of protein motions on the pico- to femtosecond timescale that reduce the width and/or height of the potential energy barrier along the chemical reaction coordinate. However, a connection between promoting motions and potential energy barrier modulation has never been demonstrated directly, and recent work has shown that the temperature dependence of kinetic isotope effects can be accounted for by conformational effects for a number of enzymes (5). Whereas some authors postulate dynamics as a key driving force in catalysis (1–4), others have performed analyses showing activation free-energy reduction, which is an equilibrium property, to be the source of catalysis (6–14). Enzyme reactions, and particularly their dynamics, present formidable challenges for study, and progress requires a combination of theoretical, experimental, and computational approaches (5, 15–18).
Dihydrofolate reductase (DHFR) has been at the heart of the debates about the relationship between enzyme dynamics and catalysis. DHFR catalyses the NAPDH-dependent reduction of 7,8-dihydrofolate (H2F) to 5,6,7,8-tetrahydrofolate (H4F) by hydride transfer from C4 of NADPH and protonation of N5 of H2F. The enzyme from Escherichia coli (EcDHFR) cycles through five reaction intermediates, namely E·NADPH, E·NADPH·H2F, E·NADP+·H4F, E·H4F, and E·NADPH·H4F (19), and adopts two major conformations, the closed conformation in the reactant complexes E·NADPH and E·NADPH·H2F and the occluded conformation in the three product complexes E·NADP+·H4F, E·H4F, and E·NADPH·H4F (20). The physical steps of ligand association and dissociation have been shown to depend on movements between these two conformations (20, 21). The actual chemical step of hydride transfer from NADPH to H2F occurs with a reaction-ready configuration of the closed complex (Fig. 1), where the M20 loop (residues 8–23) closes over the active site to shield the reactants from solvent and provide an optimal geometry and electrostatic environment of the active site for the reaction (6, 20). Results for mutants of DHFR (22–25) have been interpreted as showing a central role for protein dynamics in catalysis. However, mutations that affect protein dynamics may actually influence the chemical reaction in other ways (7), such as through changing conformational preferences of the enzyme (26). Strong evidence exists against a direct coupling of large-scale millisecond protein motions to the reaction coordinate during hydride transfer from NADPH to H2F (6, 7, 27–29), but the coupling of short-range promoting enzyme motions to the reaction coordinate in DHFR cannot be excluded experimentally (6, 22, 27). The effects of protein dynamics on chemical reactions in enzymes have previously been investigated directly only by simulations. These have found that the effects of mutation on reaction in DHFR are not dynamical; rather, the free-energy barrier for reaction is affected (7, 30, 31). Given the lack of clear evidence of dynamical effects on the reaction per se, more direct probes are required.
Fig. 1.

Active site of EcDHFR in the reaction-ready configuration. Substrate, cofactor, and key amino acid residues are shown as sticks. The portion of the reactants treated quantum mechanically in the QM/MM simulations (SI Text) is shown in an overlaid surface representation. The figure was created from the crystal structure with PDB code 1RX2, using UCSF Chimera (60).
Dynamical effects can be rigorously defined as deviations of phenomenological rate constants, k(T), from the predictions of transition-state theory (TST) (32–34). In a canonical ensemble, phenomenological rate coefficients are typically represented as
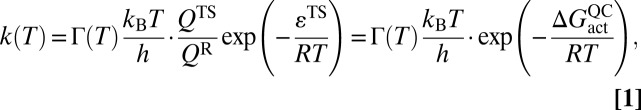 |
where R is the ideal gas constant, kB is the Boltzmann constant, h is Planck’s constant, QTS and QR are the respective transition-state (TS) and reactant (R) partition functions, εTS is the classical transition-state barrier height, 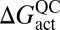 is the quasiclassical activation free energy (for more detail see SI Text) (35), and Γ(T) is the temperature-dependent transmission coefficient, which generally lumps together the so-called “dynamical” corrections to the classical TST expression. In the limit of classical TST, Γ(T) in Eq. 1 is equal to unity. In such circumstances, an Arrhenius plot of ln(k(T)) vs. 1/T should be nearly linear, as long as the temperature range is small enough that the preexponential factor is approximately constant.
is the quasiclassical activation free energy (for more detail see SI Text) (35), and Γ(T) is the temperature-dependent transmission coefficient, which generally lumps together the so-called “dynamical” corrections to the classical TST expression. In the limit of classical TST, Γ(T) in Eq. 1 is equal to unity. In such circumstances, an Arrhenius plot of ln(k(T)) vs. 1/T should be nearly linear, as long as the temperature range is small enough that the preexponential factor is approximately constant.
Several enzymes show nonlinear Arrhenius plots for H-transfer reactions (5, 36–40). However, the microscopic origin of these nonlinearities remains an open question. The most common explanations invoke recrossing and tunneling, both of which are folded into Γ(T),
where the recrossing transmission coefficient, γ, corrects the rate coefficient for trajectories that recross the dividing surface back to the reactant valley, and the tunneling coefficient, κ, accounts for reactive trajectories that do not reach the classical threshold energy. In general, 0 ≤ γ(T) ≤ 1, with values less than unity arising from the coupling of the reaction coordinate to other coordinates (discussed in further detail below). γ(T) can be estimated from molecular dynamics (MD) trajectories starting from the TS with a thermal distribution of velocities. Recent studies on several enzyme-catalyzed reactions (11–14) suggest that recrossing coefficients tend to be somewhat closer to unity than the corresponding counterpart reactions in solution. In general, κ(T) ≥ 1, with values larger than unity when quantum tunneling is important (41, 42).
Isotopic substitution of substrates or cofactors has provided strong evidence for quantum tunneling in enzyme reactions. The temperature and pressure dependences of experimentally observed reaction rates and kinetic isotope effects have been interpreted to be a consequence of protein motions on the pico- to femtosecond timescale that reduce the width and/or height of the potential energy barrier along the chemical reaction coordinate (1–4, 43). Others have postulated that millisecond conformational fluctuations may also be involved in driving the chemical step of the reaction (22). To focus more directly on protein dynamics, rather than dynamics of the reactants, entire enzymes can be isotopically substituted, with all nonexchangeable atoms of a particular type (e.g., N, C, H) replaced by a heavier isotope; the “heavy” enzyme can then be compared with its natural, lighter counterpart. Within the Born–Oppenheimer approximation, the electronic potential energy surface, V, governing atomic motion is identical in the light and heavy enzymes. The forces acting on the atoms are also identical, being the negative gradient of the potential with respect to atomic coordinates (i.e., –dV/dq = F, where F is the force acting on an atom and q is a vector of atomic coordinates). Consequently, any differences in reaction rate between the light and heavy enzymes must arise from mass-induced differences in atomic motions, ranging from fast bond vibrations on the femtosecond timescale to conformational changes on the millisecond timescale.
Isotopic substitution of HIV protease, purine nucleoside phosphorylase, alanine racemase, and pentaerythritol tetranitrate reductase has been proposed to affect catalysis by changing ultrafast vibrations that couple to the reaction coordinate (44–47). However, the precise manner in which such mass-dependent effects impact the different terms of the preexponential factor in Eq. 1 remains uncertain. Exactly how γ(T) and κ(T) contribute to Γ(T), and in particular how these are affected by protein dynamics, remains a fundamental and hotly debated question with important consequences for understanding enzyme catalysis. Using a combination of experiment, theory, and computation, we analyze dynamical effects by comparing the rate coefficients for hydride transfer in NADPH catalyzed by both “heavy” (15N, 13C, 2H isotopically substituted) and “light” (natural isotopic abundance) EcDHFR. We have measured, analyzed, and simulated the temperature dependence of the EcDHFR-catalyzed hydride transfer from NADPH to H2F in the heavy and light enzymes. A key component of these experiments is the fact that we isotopically modified only the protein, leaving the substrate unchanged. This universal isotopic substitution of the protein provides an exquisitely sensitive means of probing dynamical effects.
Results and Discussion
Creation of Heavy EcDHFR.
Heavy EcDHFR was produced in M9 medium containing exclusively 15NH4Cl, U-13C,2H-glucose, and 2H2O. All exchangeable 2H atoms were replaced by 1H during enzyme purification and storage in buffers made of 1H2O. The observed 10.76% increase in molecular mass for purified heavy EcDHFR (SI Text) showed that 98.6% of the 14N, 12C, and nonexchangeable 1H atoms (76% of total 1H atoms) had been replaced by their heavier isotopes. The circular dichroism spectra of light and heavy EcDHFR were indistinguishable, indicating that the isotope substitution did not alter the overall structure of the protein (Fig. S1).
Experimental Results.
The experimentally measured kinetics (Fig. 2 and Tables S1–S3) show intriguing differences in reactivity between the light and heavy enzymes under otherwise identical conditions (SI Text). The EcDHFR reaction is strongly dependent on pH; at pH 7, hydride transfer from the reduced cofactor NADPH to (mostly) protonated dihydrofolate is a fast step in the overall turnover of H2F. At pH values of 9.5 and above, hydride transfer to (mostly) unprotonated substrate is rate limiting (19). At elevated values of pH, the hydride transfer can therefore be monitored in multiple-turnover steady-state experiments. The steady-state rate constants at pH 9.5 for light and heavy EcDHFR, kcatLE and kcatHE, are similar at low temperatures, but notably diverge with increasing temperature because the rate constants of the light enzyme increase more rapidly (Table S1 and Fig. 2). At 40 °C, kcatLE is 27% larger than kcatHE (Table S1). Michaelis constants for NADPH and dihydrofolate are identical within error for the heavy and light enzymes at both 20 °C and 35 °C (Table S2), suggesting that binding interactions are unaltered in the heavy enzyme. The difference between kcatLE and kcatHE therefore reflects a difference in reactivity between the light and heavy enzymes after formation of the respective Michaelis complexes.
Fig. 2.

Experimental EcDHFR data for hydride transfer rate constants and corresponding fits using a tunneling model (5) (VTS = 15 kcal⋅mol−1) at pH 7 and pH 9.5 (main text). Upper Left shows the pH 7.0 pre–steady-state kinetic data (ln kHLE as red circles and ln kHHE as blue circles); Upper Right shows the pH 9.5 steady-state kinetic data (ln kcatLE as red circles and ln kcatHE as blue circles). Fits to the light and heavy enzyme data are shown using red and blue lines, respectively. Lower Left and Lower Right show the KIE (ratio of light to heavy enzyme rate constants, kLE/kHE), at pH 7.0 and pH 9.5, respectively, with red circles showing experimental data and the line indicating the fit from the tunneling TST model.
At pH 7.0, the overall turnover rate is determined by release of tetrahydrofolate from the EcDHFR·NADPH·H4F mixed ternary complex (19). Crystal structures and NMR spectroscopy have revealed that this is accompanied by movement of the M20 and βFG loops with rates similar to those for product release and therefore kcat (20, 21, 48). The enzyme kinetic isotope effects on kcat (KIEcat = kcatLE/kcatHE) measured here at pH 7 are in agreement with these observations. Whereas the Michaelis constants were not sensitive to enzyme isotopic substitution, the steady-state rate constants of the light and heavy enzymes (Table S1) showed KIEcat of 1.04 ± 0.03 and 1.16 ± 0.01 at 20 °C and 35 °C, respectively.
To determine the rates of the fast hydride transfer from reduced NADPH to (mostly) protonated dihydrofolate at physiological pH, pre–steady-state stopped-flow experiments that follow the fluorescence resonance energy transfer from the protein to reduced NADPH were conducted. We have shown previously that these are the most physiologically relevant conditions for hydride transfer measurements (29). The rate constants, kHLE and kHHE, for hydride transfer catalyzed by the light and heavy enzymes show a similar dependence on temperature to that observed in the steady-state measurements at elevated pH (Table S1 and Fig. 2). The (enzyme) kinetic isotope effect (KIEH = kHLE/kHHE) increased from 0.93 ± 0.02 at 10 °C to 1.18 ± 0.06 at 40 °C (Fig. 2). Measurements of the pH dependence of the pre–steady-state rate coefficients for hydride transfer indicated that the apparent pKa value of the reaction was not affected by isotopic substitutions (Table S3). The apparent pKa values were 6.26 ± 0.15 and 6.67 ± 0.31 for the light and heavy EcDHFR-catalyzed reactions at 20 °C and 6.40 ± 0.11 and 6.55 ± 0.37 at 35 °C.
Data Fitting.
Curvature in the Arrhenius plots (Fig. 2), especially in the pH 7.0 data, hints at microscopic effects beyond those described by simple classical TST. Recently, we have shown that the temperature dependence of rate constants and KIEs in several enzymes can be described using physically reasonable kinetic models that include tunneling corrections (5, 49). For some enzymes, such as aromatic amine dehydrogenase and methylamine dehydrogenase, two conformations with different reactivity are required to reproduce observed behavior, whereas for others like soybean lipoxygenase-1, a single conformation is sufficient. The experimental data in Fig. 2 can be fitted well, using a one-conformation tunneling model of the form
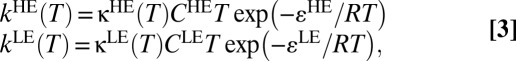 |
where kHE and kLE are the respective temperature-dependent rate constants for hydride transfer in heavy and light EcDHFR, κHE and κLE are the tunneling transmission coefficients in the heavy and light enzymes (calculated from an analytical expression for tunneling through a one-dimensional barrier as discussed in SI Text), CHET and CLET are prefactors that fold in the effect of both recrossing and temperature-dependent contributions of the reaction entropy to the total rate coefficient, and εHE and εLE are the enthalpic activation barriers for hydride transfer. The fitting procedure gives excellent agreement with experiment over the entire temperature range (Table S4, Fig. 2, and Fig. S2).
Molecular Dynamics Simulations.
Separately, we carried out quantum mechanics/molecular mechanics (QM/MM) molecular dynamics simulations at 300 K with the substrate dihydrofolate fully protonated, to investigate the intricate molecular details of the reaction (Fig. S3). QM/MM ensemble-averaged variational TST (EA-VTST) calculations with multidimensional tunneling corrections have provided useful insight into many enzyme-catalyzed reactions (9, 12, 14, 41, 50). For the molecular dynamics simulations, the reaction coordinate was defined as the difference of distances between the transferred hydride and the donor and acceptor atoms. Simulations of the heavy enzyme were performed using the masses of 15N, 13C, and 2H for nitrogen, carbon, and nonexchangeable hydrogen atoms. Structures from the QM/MM molecular dynamics simulations were used for EA-VTST calculations, which work out Γ(T) by calculating both γ and κ (Eqs. 1 and 2) (further details in SI Text).
The extent of dynamical coupling between the reaction coordinate and other motions within the protein–substrate complex is indicated by the magnitude of the recrossing coefficient, γ. It is important to point out that the calculated value of γ is related to the free-energy profile (51). The free-energy profile in turn depends on the choice of the reaction coordinate. Here, we confine our discussion to the difference in the distances between the transferred hydride and its donor/acceptor. The reasons for this are twofold: (i) The extent to which our recrossing coefficients deviate from unity is comparable to that in previous studies (11, 12, 30), suggesting that we have chosen a reasonable reaction coordinate, and (ii) a reaction coordinate based on bond distances conforms to the local mode picture that chemists typically use to rationalize whether or not a reaction has occurred. In SI Text, we describe more sophisticated quantized variational transition state searches carried out on the mass weighted coordinates of larger atomic subsets surrounding the hydride transfer region. The important point of these additional tests is that the free-energy profiles for hydride transfer in both the light and the heavy systems are statistically identical for the reaction coordinates that we investigated. As shown in SI Text (e.g., Fig. S4B), changes in the reaction coordinate significantly affect neither the height nor the position of the free-energy maximum, in agreement with the results of the experimental fits described above.
Discussion
The 300-K classical potentials of mean force (PMF), obtained using the semiempirical Austin Model 1 Hamiltonian with specific reaction parameters and molecular mechanics (AM1-SRP/MM), give free-energy barriers that are statistically identical in the heavy and light enzymes and close to the value of 15 kcal⋅mol−1 obtained above for the classical barrier height (VTS) through fits to the experimental data (SI Text). This value is considerably different from the activation energy of ∼6.3 kcal⋅mol−1 obtained using an Arrhenius-type fit (Table S4), providing an excellent cautionary example of how Arrhenius-type fits can be misleading when Γ(T) is significant. The quantized vibrational corrections to the reactant PMF are small and statistically indistinguishable in the heavy and light enzymes (SI Text). The findings from the QM/MM MD simulations are consistent with the kinetic fits, which found εLE and εHE to be very similar at each pH and within 1.2 kcal⋅mol−1 of VTS (Table S4). The magnitude of these corrections is similar to those found in previous studies on hydride transfer reactions in other NAD(P)H-dependent enzymes (52–54). The 300-K rate constants obtained from MD simulations, which result directly from the simulations without any fitting to the experimental data, are in excellent agreement with the experimental values (which are of course themselves subject to some uncertainty) (Table 1). It is important to note that such good agreement (apparently within the errors of all of the various methods) is, to some extent, fortuitous given the complexity of simulating enzyme reactions. Nevertheless, these results (taken alongside the kinetic modeling) suggest that the computational approach is reasonable.
Table 1.
Transmission coefficient components due to recrossing (γ) and tunneling (κ) for hydride transfer in light and heavy EcDHFR, determined by QM/MM calculations
| EcDHFR | γ | κ |
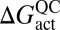 , kcal⋅mol−1 , kcal⋅mol−1
|
ΔGeff, kcal⋅mol−1 | ktheor, s−1 | kexp, s−1 |
| Light | 0.57 ± 0.02 | 2.61 ± 0.49 | 14.59 ± 0.41 | 14.35 ± 0.54 | 219 | 209.1 ± 5.0 |
| Heavy | 0.49 ± 0.02 | 14.46 ± 0.54 | 188 | 190.1 ± 8.5 |
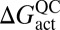 is the quasiclassical (QC) free energy of activation (Eq. 1); ΔGeff is the effective phenomenological free energy of activation, into which the effects of tunneling and recrossing are folded; ktheor is the predicted hydrogen transfer rate coefficient at 300 K; and kexp is the experimentally determined hydrogen transfer rate coefficient at 303 K.
is the quasiclassical (QC) free energy of activation (Eq. 1); ΔGeff is the effective phenomenological free energy of activation, into which the effects of tunneling and recrossing are folded; ktheor is the predicted hydrogen transfer rate coefficient at 300 K; and kexp is the experimentally determined hydrogen transfer rate coefficient at 303 K.
The EA-VTST tunneling coefficients, κ, are statistically identical for the heavy and light enzymes (Table 1), with an effective contribution to the phenomenological barrier less than 1 kcal⋅mol−1. Qualitatively, this result is identical to that found from fitting the kinetic data: The best-fit values for ωLE and ωHE (Table S4) suggest a barrier that is rather broad and smooth. The tunneling coefficients obtained from EA-VTST and independently from the fitting model agree within errors (Table S4), both methods suggesting that contributions from tunneling in the heavy and light enzymes are small and effectively identical. The EA-VTST values of the barrier reduction due to tunneling are also very close to those calculated for DHFR from Thermotoga maritima (ca. 0.7 kcal⋅mol−1, computed for the monomer at 298 K) (16) and for lactate dehydrogenase (ca. 0.8 kcal⋅mol−1) (55), but slightly smaller than those found for thymidylate synthase (1.4 kcal⋅mol−1 for hydride transfer at 303 K) (14) or morphinone reductase (1.5 kcal⋅mol−1 for hydride transfer at 298 K) (56). In both light and heavy enzymes, the time-dependent flux–flux correlation functions used to obtain the transmission coefficients show a fast decay during the first 20 fs and a subsequent plateau after 40–60 fs (Fig. S4C), giving 0.57 for γLE and 0.49 for γHE. This corresponds to a γLE:γHE ratio of 1.16, in good agreement with the results of the fitting, in which the preexponential factors CLE and CHE have a ratio of 1.14 at pH 9.5 and 1.08 at pH 7.
The fits and the QM/MM results both point to a scenario in which the difference in phenomenological rate constants between the heavy and light enzymes arises in part from the different participation of protein motions in the reaction coordinate. In the EA-VTST model, this is captured through differences in the recrossing coefficients γ, whereas in the fitting model differences in γ are folded into differences in CLE and CHE. The origin of the difference in the recrossing coefficients lies in the coupling between the reaction coordinate and the environmental motions. In general, the coupling of environmental modes to motion along the reaction coordinate depends on the relative values of each mode’s frequencies. The speed of passage over the TS is largely determined by the curvature of the energy surface around the TS. In general, environmental motions characterized by vibrational frequencies that are greater than or equal to the equivalent characteristic time for passage through the transition state region quickly adapt to geometrical changes in the reaction coordinate, and the fast equilibrium assumption (implicit in TST) holds. Environmental motions characterized by vibrational frequencies less than the characteristic time for passage over the reaction coordinate reaction frequency adapt to geometrical changes along the reaction coordinate more slowly and the fast equilibrium assumption becomes less valid. In ergodic systems, faster environmental response is therefore often linked to higher-energy frequency distributions. The friction spectrum (Fig. S4D) shows the distribution of frequencies that couple to the reaction coordinate in both the heavy and the light enzymes. Qualitatively, the most significant differences between the friction spectra in both systems occur below frequencies of ∼1,000 cm−1 (with many of the most intense peaks in the heavy enzymes red-shifted by ∼30 cm–1 compared with the light enzyme, owing to the greater masses). The lower frequency distribution in the heavy enzyme system is consistent with a slower environmental response time in the heavy protein and a transmission coefficient that has a correspondingly larger departure from unity.
The MD simulations suggest that differences in the environmental response time between the heavy and light proteins translate to an effective free-energy difference in the barrier heights for hydride transfer, with the barrier in the light enzyme 0.11 kcal⋅mol–1 lower than that in the heavy enzyme at 300 K (Table 1). Within the error limits at 300 K, the recrossing factor γ captures most of the difference between the experimentally observed k(T) values in the heavy and light enzymes.
To investigate possible dynamical differences between the light and heavy enzymes, we examined isolated dynamical observables either side of the transition state calculated from the QM/MM simulations of hydride transfer in each enzyme (Table S5). These reveal that the light and heavy enzymes are very similar, with little or no significant difference in many dynamical observables. For example, the donor–acceptor distance (Fig. S4E) and the angle between the donor, hydride, and acceptor atoms (Fig. S4F) in the heavy and light systems show very similar time-dependent profiles. Other distances between atoms in the substrate and the active site during reaction also show similar behavior in the heavy and light enzymes: The approach of Met20 to the substrate and the amide group of the cofactor nicotinamide ring (Fig. S4G), which precedes the formation of the TS [and has been suggested to stabilize the hydride transfer TS (57)], has very similar time profiles in the light and heavy enzymes. Compelling evidence for a faster environmental response in the light enzyme is clearly seen only with a global analysis that accounts for all atomic positions within both the light and the heavy enzyme. The root mean squared deviation (RMSD) along reactive trajectories between the average geometry at time t and the average TS geometry (Fig. S4H) shows that the global environmental response is slightly faster in the light enzyme, as deduced from the exponential decays of the RMSD on both enzymes. The relaxation rate constants obtained from a least-squares fit are 9.0 ± 0.1 ps−1 and 8.7 ± 0.1 ps−1 for the light and heavy enzymes, respectively.
Most significantly, analysis of the RMSD reveals that the light enzyme environment—taken as a global aggregate—responds more quickly to motion along the reaction coordinate (Fig. S4H). It is also interesting to consider the converse: namely, how the reaction coordinate responds to motions in the environment. Thermodynamic detailed balance requires that a faster response in one direction must be linked to a faster response in the reverse direction—i.e., the chemical reaction rate in the light enzyme must be more responsive to environmental fluctuations and perturbations than that in the heavy enzyme. For DHFR, protein motion couples to the reaction coordinate in a rather subtle way that is apparent only via a global description of all atomic positions. This makes it difficult to specifically identify any “promoting motions” that couple EcDHFR motions to progress along the reaction coordinate. Clearly any dynamical effects on the chemical step are small, subtle, and not localized, but apparently play a role in making the heavy enzyme less active than its natural, light counterpart. Unraveling the microscopic mechanisms responsible for this sort of global dynamical coupling offers interesting and fertile territory for future investigations into the microscopic mechanisms that underlie enzyme function. Work is currently underway to investigate the effect of isotopically labeling segments of EcDHFR to determine whether certain portions of the enzyme play a greater role in the dynamical effects.
Conclusions
Our experimental results show that hydride transfer from NADPH to dihydrofolate is generally somewhat faster in light EcDHFR than in its heavy counterpart, over the temperature range 280–313 K. Fitting this temperature-dependence data to a recently developed model based on TST suggests that both the tunneling contributions and the barrier heights in the heavy and light enzymes are identical; the fitting indicates that the differences in the rate coefficients arise from variations in the respective preexponential factors. This conclusion is supported by QM/MM MD simulations and EA-VTST calculations carried out at 300 K, which suggest that (i) the tunneling probabilities and barrier heights are statistically indistinguishable in the light and heavy enzymes and (ii) the differences in the phenomenological rate coefficients are mostly accounted for by differences in the recrossing coefficient. Thus, the difference in reactivity is due neither to differences in quantum tunneling nor to differences in barrier height, but rather to differences in the extent to which the protein environment of the light and heavy enzymes globally couples to the reaction coordinate. These findings run counter to proposals that invoke enhancement of tunneling or barrier modulation by specific protein (“promoting”) motions or claims that protein dynamics “drive” tunneling.
Although TST with tunneling corrections broadly accounts for the observed hydride transfer rate coefficients, more detailed quantitative analysis requires dynamical recrossing corrections. Specifically, our simulations and modeling indicate that the main cause of the difference in the rate constants for the reactions catalyzed by light and heavy EcDHFR is different coupling of global motions with the protein environment along the reaction coordinate. In a TST treatment this can be translated into a slightly different reaction coordinate (e.g., a more global coordinate allowing more of the protein to participate directly) or, more conveniently in this case, into a different value of the recrossing transmission coefficient. Irrespective of this procedural choice, our experimental and theoretical results agree that the small differences in reactivity between the light and heavy enzymes most probably arise from differences in the extent to which the protein environment is coupled to the chemical step. In the light enzyme, where atomic motion is characterized by higher frequencies, the environment responds more rapidly to changes along the reaction coordinate, resulting in fewer trajectory recrossings. This study, which compares kinetics in the light and heavy enzymes, provides important insight into the nature of enzyme reaction dynamics. Although protein dynamics have a measurable effect on the chemical reaction, the effect is relatively small and is not related to differences in quantum tunneling.
Methods
Experimental Methods.
EcDHFR and 15N-, 13C-, 2H-labeled (heavy) EcDHFR were produced in M9 medium and purified as previously described (58). Electrospray ionization mass spectrometry was used to determine the degree of isotopic substitution in the heavy enzyme, and structural integrity was confirmed by circular dichroism spectroscopy. Steady-state and stopped-flow kinetic measurements were performed as previously described (38, 59).
Fitting Methodology.
The temperature-dependent experimental hydride transfer data at different values of pH were fitted to Eq. 3, using a nonlinear least-squares minimization algorithm. Fitting the data to a more sophisticated multiconformation model did not give improved nonlinear least-squares fits compared with single-conformer models.
QM/MM EA-VTST Calculations and Molecular Dynamics Simulations.
Protein Data Bank entry 3QL3 (22) was used as the starting structure for simulations. Heavy EcDHFR was prepared by modifying the masses of all 14N, 12C, and nonexchangeable 1H atoms to those of 15N, 13C, and 2H. QM/MM EA-VTST calculations and molecular dynamics simulations were performed to determine reactive trajectories and to extract contributions to the transmission coefficient (Eq. 2) and activation parameters.
A full description of experimental, fitting, and simulation methods is provided in SI Text.
Supplementary Material
Acknowledgments
We acknowledge the computational facilities of Universitat Jaume I and Universitat de València (Tirant Supercomputer). This work was supported by Grants BB/E008380/1 and BB/J005266/1 (to R.K.A.) from the UK Biotechnology and Biological Sciences Research Council, by Doctoral Training Account funding from the UK Engineering and Physical Sciences Research Council (EPSRC), by the Vice Chancellor Fund of Cardiff University, by the Spanish Ministerio de Economía y Competitividad (Project CTQ2012-36253-C03), by Generalitat Valenciana (Projects ACOMP/2012/119, GV/2012/044, GV/2012/053, and Prometeo/2009/053), and by Universitat Jaume I-Bancaixa (Projects P1·1A2010-08 and P1·1B2011-23). J.J.R.-P. thanks the Spanish Ministerio de Ciencia e Innovación for a “Juan de la Cierva” contract. A.J.M. is an EPSRC Leadership Fellow (Grant EP/G007705/1). D.R.G. has support under EPSRC Programme Grant EP/G00224X/1.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
1L.Y.P.L. and J.J.R.-P. contributed equally to this work.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1312437110/-/DCSupplemental.
References
- 1.Nagel ZD, Klinman JP. A 21st century revisionist’s view at a turning point in enzymology. Nat Chem Biol. 2009;5(8):543–550. doi: 10.1038/nchembio.204. [DOI] [PubMed] [Google Scholar]
- 2.Limbach H-H, Schowen KB, Schowen RL. Heavy atom motions and tunneling in hydrogen transfer reactions: The importance of the pre-tunneling state. J Phys Org Chem. 2010;23:586–605. [Google Scholar]
- 3.Antoniou D, Caratzoulas S, Kalyanaraman C, Mincer JS, Schwartz SD. Barrier passage and protein dynamics in enzymatically catalyzed reactions. Eur J Biochem. 2002;269(13):3103–3112. doi: 10.1046/j.1432-1033.2002.03021.x. [DOI] [PubMed] [Google Scholar]
- 4.Scrutton NS, Basran J, Sutcliffe MJ. New insights into enzyme catalysis. Ground state tunnelling driven by protein dynamics. Eur J Biochem. 1999;264(3):666–671. doi: 10.1046/j.1432-1327.1999.00645.x. [DOI] [PubMed] [Google Scholar]
- 5.Glowacki DR, Harvey JN, Mulholland AJ. Taking Ockham’s razor to enzyme dynamics and catalysis. Nat Chem. 2012;4(3):169–176. doi: 10.1038/nchem.1244. [DOI] [PubMed] [Google Scholar]
- 6.Loveridge EJ, Behiry EM, Guo J, Allemann RK. Evidence that a ‘dynamic knockout’ in Escherichia coli dihydrofolate reductase does not affect the chemical step of catalysis. Nat Chem. 2012;4(4):292–297. doi: 10.1038/nchem.1296. [DOI] [PubMed] [Google Scholar]
- 7.Adamczyk AJ, Cao J, Kamerlin SCL, Warshel A. Catalysis by dihydrofolate reductase and other enzymes arises from electrostatic preorganization, not conformational motions. Proc Natl Acad Sci USA. 2011;108(34):14115–14120. doi: 10.1073/pnas.1111252108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olsson MHM, Parson WW, Warshel A. Dynamical contributions to enzyme catalysis: Critical tests of a popular hypothesis. Chem Rev. 2006;106(5):1737–1756. doi: 10.1021/cr040427e. [DOI] [PubMed] [Google Scholar]
- 9.Garcia-Viloca M, Gao J, Karplus M, Truhlar DG. How enzymes work: Analysis by modern rate theory and computer simulations. Science. 2004;303(5655):186–195. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- 10.Gao J, et al. Mechanisms and free energies of enzymatic reactions. Chem Rev. 2006;106(8):3188–3209. doi: 10.1021/cr050293k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roca M, Moliner V, Tuñón I, Hynes JT. Coupling between protein and reaction dynamics in enzymatic processes: Application of Grote-Hynes Theory to catechol O-methyltransferase. J Am Chem Soc. 2006;128(18):6186–6193. doi: 10.1021/ja058826u. [DOI] [PubMed] [Google Scholar]
- 12.Ruiz-Pernía JJ, Tuñón I, Moliner V, Hynes JT, Roca M. Dynamic effects on reaction rates in a Michael addition catalyzed by chalcone isomerase. Beyond the frozen environment approach. J Am Chem Soc. 2008;130(23):7477–7488. doi: 10.1021/ja801156y. [DOI] [PubMed] [Google Scholar]
- 13.Roca M, Oliva M, Castillo R, Moliner V, Tuñón I. Do dynamic effects play a significant role in enzymatic catalysis? A theoretical analysis of formate dehydrogenase. Chemistry. 2010;16(37):11399–11411. doi: 10.1002/chem.201000635. [DOI] [PubMed] [Google Scholar]
- 14.Kanaan N, et al. Temperature dependence of the kinetic isotope effects in thymidylate synthase. A theoretical study. J Am Chem Soc. 2011;133(17):6692–6702. doi: 10.1021/ja1114369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roston D, Cheatum CM, Kohen A. Hydrogen donor-acceptor fluctuations from kinetic isotope effects: A phenomenological model. Biochemistry. 2012;51(34):6860–6870. doi: 10.1021/bi300613e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pang JY, Pu JZ, Gao JL, Truhlar DG, Allemann RK. Hydride transfer reaction catalyzed by hyperthermophilic dihydrofolate reductase is dominated by quantum mechanical tunneling and is promoted by both inter- and intramonomeric correlated motions. J Am Chem Soc. 2006;128(24):8015–8023. doi: 10.1021/ja061585l. [DOI] [PubMed] [Google Scholar]
- 17.Pu J, Ma S, Gao J, Truhlar DG. Small temperature dependence of the kinetic isotope effect for the hydride transfer reaction catalyzed by Escherichia coli dihydrofolate reductase. J Phys Chem B. 2005;109(18):8551–8556. doi: 10.1021/jp051184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamerlin SCL, Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78(6):1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fierke CA, Johnson KA, Benkovic SJ. Construction and evaluation of the kinetic scheme associated with dihydrofolate reductase from Escherichia coli. Biochemistry. 1987;26(13):4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- 20.Sawaya MR, Kraut J. Loop and subdomain movements in the mechanism of Escherichia coli dihydrofolate reductase: Crystallographic evidence. Biochemistry. 1997;36(3):586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- 21.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313(5793):1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 22.Bhabha G, et al. A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science. 2011;332(6026):234–238. doi: 10.1126/science.1198542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Tharp S, Selzer T, Benkovic SJ, Kohen A. Effects of a distal mutation on active site chemistry. Biochemistry. 2006;45(5):1383–1392. doi: 10.1021/bi0518242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agarwal PK, Billeter SR, Rajagopalan PTR, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci USA. 2002;99(5):2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cameron CE, Benkovic SJ. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry. 1997;36(50):15792–15800. doi: 10.1021/bi9716231. [DOI] [PubMed] [Google Scholar]
- 26.Swanwick RS, Shrimpton PJ, Allemann RK. Pivotal role of Gly 121 in dihydrofolate reductase from Escherichia coli: The altered structure of a mutant enzyme may form the basis of its diminished catalytic performance. Biochemistry. 2004;43(14):4119–4127. doi: 10.1021/bi036164k. [DOI] [PubMed] [Google Scholar]
- 27.Loveridge EJ, et al. The role of large-scale motions in catalysis by dihydrofolate reductase. J Am Chem Soc. 2011;133(50):20561–20570. doi: 10.1021/ja208844j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loveridge EJ, Tey LH, Allemann RK. Solvent effects on catalysis by Escherichia coli dihydrofolate reductase. J Am Chem Soc. 2010;132(3):1137–1143. doi: 10.1021/ja909353c. [DOI] [PubMed] [Google Scholar]
- 29.Loveridge EJ, Allemann RK. Effect of pH on hydride transfer by Escherichia coli dihydrofolate reductase. ChemBioChem. 2011;12(8):1258–1262. doi: 10.1002/cbic.201000794. [DOI] [PubMed] [Google Scholar]
- 30.Boekelheide N, Salomón-Ferrer R, Miller TF., 3rd Dynamics and dissipation in enzyme catalysis. Proc Natl Acad Sci USA. 2011;108(39):16159–16163. doi: 10.1073/pnas.1106397108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan Y, Cembran A, Ma S, Gao J. Connecting protein conformational dynamics with catalytic function as illustrated in dihydrofolate reductase. Biochemistry. 2013;52:2036–2049. doi: 10.1021/bi301559q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glasstone S, Laidler KJ, Eyring H. The Theory of Rate Processes: The Kinetics of Chemical Reactions, Viscosity, Diffusion and Electrochemical Phenomena. New York: McGraw-Hill; 1941. [Google Scholar]
- 33.Keck JC. Variational theory of reaction rates. Adv Chem Phys. 1967;13:85–121. [Google Scholar]
- 34.Truhlar DG, Garrett BC, Klippenstein SJ. Current status of transition-state theory. J Phys Chem. 1996;100:12771–12800. [Google Scholar]
- 35.Alhambra C, et al. Canonical variational theory for enzyme kinetics with the protein mean force and multidimensional quantum mechanical tunneling dynamics. Theory and application to liver alcohol dehydrogenase. J Phys Chem B. 2001;105:11326–11340. [Google Scholar]
- 36.Kohen A, Cannio R, Bartolucci S, Klinman JP. Enzyme dynamics and hydrogen tunnelling in a thermophilic alcohol dehydrogenase. Nature. 1999;399(6735):496–499. doi: 10.1038/20981. [DOI] [PubMed] [Google Scholar]
- 37.Truhlar DG, Kohen A. Convex Arrhenius plots and their interpretation. Proc Natl Acad Sci USA. 2001;98(3):848–851. doi: 10.1073/pnas.98.3.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maglia G, Allemann RK. Evidence for environmentally coupled hydrogen tunneling during dihydrofolate reductase catalysis. J Am Chem Soc. 2003;125(44):13372–13373. doi: 10.1021/ja035692g. [DOI] [PubMed] [Google Scholar]
- 39.Anandarajah K, Schowen KB, Schowen RL. Hydrogen tunneling in glucose oxidation by the archaeon Thermoplasma acidophilum. Z Phys Chem. 2008;222:1333–1347. [Google Scholar]
- 40.Tsai SC, Klinman JP. Probes of hydrogen tunneling with horse liver alcohol dehydrogenase at subzero temperatures. Biochemistry. 2001;40(7):2303–2311. doi: 10.1021/bi002075l. [DOI] [PubMed] [Google Scholar]
- 41.Masgrau L, et al. Atomic description of an enzyme reaction dominated by proton tunneling. Science. 2006;312(5771):237–241. doi: 10.1126/science.1126002. [DOI] [PubMed] [Google Scholar]
- 42.Knapp MJ, Rickert K, Klinman JP. Temperature-dependent isotope effects in soybean lipoxygenase-1: Correlating hydrogen tunneling with protein dynamics. J Am Chem Soc. 2002;124(15):3865–3874. doi: 10.1021/ja012205t. [DOI] [PubMed] [Google Scholar]
- 43.Major DT, et al. Differential quantum tunneling contributions in nitroalkane oxidase catalyzed and the uncatalyzed proton transfer reaction. Proc Natl Acad Sci USA. 2009;106(49):20734–20739. doi: 10.1073/pnas.0911416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kipp DR, Silva RG, Schramm VL. Mass-dependent bond vibrational dynamics influence catalysis by HIV-1 protease. J Am Chem Soc. 2011;133(48):19358–19361. doi: 10.1021/ja209391n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva RG, Murkin AS, Schramm VL. Femtosecond dynamics coupled to chemical barrier crossing in a Born-Oppenheimer enzyme. Proc Natl Acad Sci USA. 2011;108(46):18661–18665. doi: 10.1073/pnas.1114900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pudney CR, et al. Fast protein motions are coupled to enzyme H-transfer reactions. J Am Chem Soc. 2013;135(7):2512–2517. doi: 10.1021/ja311277k. [DOI] [PubMed] [Google Scholar]
- 47.Toney MD, Castro JN, Addington TA. Heavy-enzyme kinetic isotope effects on proton transfer in alanine racemase. J Am Chem Soc. 2013;135(7):2509–2511. doi: 10.1021/ja3101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Falzone CJ, Wright PE, Benkovic SJ. Dynamics of a flexible loop in dihydrofolate reductase from Escherichia coli and its implication for catalysis. Biochemistry. 1994;33(2):439–442. doi: 10.1021/bi00168a007. [DOI] [PubMed] [Google Scholar]
- 49.Glowacki DR, Harvey JN, Mulholland AJ. Protein dynamics and enzyme catalysis: The ghost in the machine? Biochem Soc Trans. 2012;40(3):515–521. doi: 10.1042/BST20120047. [DOI] [PubMed] [Google Scholar]
- 50.Pu J, Gao J, Truhlar DG. Multidimensional tunneling, recrossing, and the transmission coefficient for enzymatic reactions. Chem Rev. 2006;106(8):3140–3169. doi: 10.1021/cr050308e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Truhlar DG, et al. Ensemble-averaged variational transition state theory with optimized multidimensional tunneling for enzyme kinetics and other condensed-phase reactions. Int J Quantum Chem. 2004;100:1136–1152. [Google Scholar]
- 52.Garcia-Viloca M, Alhambra C, Truhlar DG, Gao J. Inclusion of quantum-mechanical vibrational energy in reactive potentials of mean force. J Chem Phys. 2001;114:9953–9958. [Google Scholar]
- 53.Alhambra C, Corchado JC, Sanchez ML, Gao JL, Truhlar DG. Quantum dynamics of hydride transfer in enzyme catalysis. J Am Chem Soc. 2000;122:8197–8203. [Google Scholar]
- 54.Webb SP, Agarwal PK, Hammes-Schiffer S. Combining electronic structure methods with the calculation of hydrogen vibrational wavefunctions: Application to hydride transfer in liver alcohol dehydrogenase. J Phys Chem B. 2000;104:8884–8894. [Google Scholar]
- 55.Ferrer S, et al. A theoretical analysis of rate constants and kinetic isotope effects corresponding to different reactant valleys in lactate dehydrogenase. J Am Chem Soc. 2006;128(51):16851–16863. doi: 10.1021/ja0653977. [DOI] [PubMed] [Google Scholar]
- 56.Pang J, Hay S, Scrutton NS, Sutcliffe MJ. Deep tunneling dominates the biologically important hydride transfer reaction from NADH to FMN in morphinone reductase. J Am Chem Soc. 2008;130(22):7092–7097. doi: 10.1021/ja800471f. [DOI] [PubMed] [Google Scholar]
- 57.Garcia-Viloca M, Truhlar DG, Gao J. Reaction-path energetics and kinetics of the hydride transfer reaction catalyzed by dihydrofolate reductase. Biochemistry. 2003;42(46):13558–13575. doi: 10.1021/bi034824f. [DOI] [PubMed] [Google Scholar]
- 58.Hay S, et al. Are the catalytic properties of enzymes from piezophilic organisms pressure adapted? ChemBioChem. 2009;10(14):2348–2353. doi: 10.1002/cbic.200900367. [DOI] [PubMed] [Google Scholar]
- 59.Evans RM, et al. Catalysis by dihydrofolate reductase from the psychropiezophile Moritella profunda. ChemBioChem. 2010;11(14):2010–2017. doi: 10.1002/cbic.201000341. [DOI] [PubMed] [Google Scholar]
- 60.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.