

Significance
There is an unmet need for factors that can protect pancreatic islet beta cells from apoptosis and improve insulin secretion in the context of diabetes. There are many candidate factors produced locally in islets. We investigated the role of axon guidance factors and found that the SLIT–Roundabout receptors system is present, where it responds to stress. Full expression of SLIT ligands is essential for optimal beta-cell survival. Recombinant SLIT promotes survival and increases insulin secretion via mechanisms involving Ca2+ and actin.
Keywords: programmed cell death, glucose metabolism, intracellular calcium signaling
Abstract
We previously cataloged putative autocrine/paracrine signaling loops in pancreatic islets, including factors best known for their roles in axon guidance. Emerging evidence points to nonneuronal roles for these factors, including the Slit–Roundabout receptor (Robo) family, in cell growth, migration, and survival. We found SLIT1 and SLIT3 in both beta cells and alpha cells, whereas SLIT2 was predominantly expressed in beta cells. ROBO1 and ROBO2 receptors were detected in beta and alpha cells. Remarkably, even modest knockdown of Slit production resulted in significant beta-cell death, demonstrating a critical autocrine/paracrine survival role for this pathway. Indeed, recombinant SLIT1, SLIT2, and SLIT3 decreased serum deprivation, cytokine, and thapsigargin-induced cell death under hyperglycemic conditions. SLIT treatment also induced a gradual release of endoplasmic reticulum luminal Ca2+, suggesting a unique molecular mechanism capable of protecting beta cells from endoplasmic reticulum stress-induced apoptosis. SLIT treatment was also associated with rapid actin remodeling. SLITs potentiated glucose-stimulated insulin secretion and increased the frequency of glucose-induced Ca2+ oscillations. These observations point to unexpected roles for local Slit secretion in the survival and function of pancreatic beta cells. Because diabetes results from a deficiency in functional beta-cell mass, these studies may contribute to therapeutic approaches for improving beta-cell survival and function.
Emerging evidence highlights the important role of locally released pancreatic islet peptide factors on beta-cell mass growth, maintenance, and survival (1–12). We have published a list of 233 ligands and 234 receptors expressed in islets and/or beta cells (12). Although our list is undoubtedly not comprehensive, it provides a starting point for the investigation of factors in adult islets that had previously only been reported in other cell types or in fetal pancreas (12). We identified a group of secreted molecules known to provide axonal guidance cues during neuronal development, comprising members of the netrin, slit, semaphorin, and ephrin families (13). The parallels between cell fate decisions in neurons and the endocrine pancreas prompted us to examine some factors in detail and discover that netrin treatment modulates beta-cell survival signaling (12).
The Slit ligands and their Roundabout receptors (Robo) were discovered in Drosophila as regulators of axon guidance during development (14–17). Mammalian homologs of Slit and Robo with functions outside of axon guidance have since been identified (18, 19). Slit ligands have been implicated in liver, kidney, lung, and mammary development by modulating cell adhesion, migration, differentiation, and death (18, 20, 21). It was not known whether Slit–Robo signaling functions in beta cells. Here, we report that Slit expression can be regulated by stress and that local Slit production is required for beta-cell survival and optimal function via a mechanism involving endoplasmic reticulum (ER) Ca2+ homeostasis and actin remodeling. Our work provides examples of local guidance factors that are required for beta-cell survival and suggests avenues for protecting functional beta-cell mass.
Results
Slits Are Expressed in Adult Mouse and Human Islets.
The mammalian genome contains three Slit ligands and four Robo receptors. Our bioinformatic studies identified the expression of several Slit and Robo family members in adult human and rodent pancreatic islet cells (12), and Robo1 was identified by others as a transcript enriched in pancreatic endocrine cells during development (22). Nevertheless, no in-depth studies of these proteins have been reported. We detected Slit1, Slit2, and Slit3 transcripts in 6- and 30 wk-old mouse islets, with higher expression of Slit2 and Slit3 (Fig. 1A). In human islets, SLIT1, SLIT2, and SLIT3 expression was similar. Robo1 and Robo2 were expressed in MIN6 cells, mouse islets, and human islets (Fig. 1A). SLIT1, SLIT2, SLIT3, ROBO1, and ROBO2 were confirmed at the protein level (Fig. 1 B–F). Secretion of SLIT2 and SLIT3 from mouse islets was detected following incubation under 3 mM and 15 mM glucose conditions (Fig. 1B). Although SLIT1 secretion from islets and SLIT1 protein content within islets fell below the detection threshold of the ELISA kit, the protein could be detected by immunoblotting and immunostaining (Fig. 1 B–F). SLIT2 immunoreactivity was more predominant in beta cells, whereas SLIT1 and SLIT3 were detected in both beta and alpha cells with the same intensity (Fig. 1 E and F). Deconvolution microscopy illustrated that SLIT2 colocalized with insulin-positive granules, whereas SLIT1 and SLIT3 were present in distinct granules (Fig. 1F). These data suggest that SLIT2 may act in an autocrine manner on beta cells, whereas SLIT1 and SLIT3 may play both autocrine and paracrine roles.
Fig. 1.

Slit–Robo autocrine/paracrine network in pancreatic islet cells. (A) qRT-PCR of Slit and Robo expression in mouse islets, human islets, and MIN6 cells (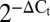 relative to actin). (B) SLIT1–3 secretion from mouse islets under 3 mM and 15 mM glucose (n = 11) (ND, not detected). (C) SLIT1–3 protein content in mouse islets (n = 11). (D) SLIT and ROBO immunoblot in MIN6, mouse islet, and human islet lysates. (E and F) SLIT and ROBO staining in mouse pancreas sections and mouse islet cells. (G) Slit and Robo expression in mouse islet cells treated with cytokine mixture (25 ng/mL TNF-α, 10 ng/mL IL-1β, 10 ng/mL IFN-γ), 1 μM thapsigargin (Tg), 1.5 mM palmitate, or 5 mM or 20 mM glucose containing medium supplemented with 10% FBS or under serum-free (SF) conditions (
relative to actin). (B) SLIT1–3 secretion from mouse islets under 3 mM and 15 mM glucose (n = 11) (ND, not detected). (C) SLIT1–3 protein content in mouse islets (n = 11). (D) SLIT and ROBO immunoblot in MIN6, mouse islet, and human islet lysates. (E and F) SLIT and ROBO staining in mouse pancreas sections and mouse islet cells. (G) Slit and Robo expression in mouse islet cells treated with cytokine mixture (25 ng/mL TNF-α, 10 ng/mL IL-1β, 10 ng/mL IFN-γ), 1 μM thapsigargin (Tg), 1.5 mM palmitate, or 5 mM or 20 mM glucose containing medium supplemented with 10% FBS or under serum-free (SF) conditions (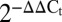 ; n = 4, mean ± SEM, *P < 0.05 compared with 20 mM glucose, 10% FBS; †P < 0.05 compared with 5 mM glucose, 10% FBS).
; n = 4, mean ± SEM, *P < 0.05 compared with 20 mM glucose, 10% FBS; †P < 0.05 compared with 5 mM glucose, 10% FBS).
Next, we assessed the mechanism of local Slit signaling by investigating the expression and localization of Slit receptors. ROBO1 and ROBO2 staining was detected in both beta and alpha cells (Fig. 1E). However, although ROBO1 could be found in the plasma membrane and cytosolic compartments typical for receptors of soluble ligands, ROBO2 displayed nuclear localization (Fig. 1F). Further biochemical assays, such as nuclear fractionation, will be required to support this observation. Although ROBO1 has been reported to be nuclear in some cell types (23), we are unaware of reports of nuclear ROBO2. Our data suggest that SLIT ligands act via ROBO1 receptors on islet cell plasma membranes.
It is not well understood in any cell type whether the Slit–Robo system can be dynamically regulated, for example by stresses (24, 25). Quantitative RT-PCR (qRT-PCR) revealed that cytokines, thapsigargin, and palmitate down-regulated Slit3, and serum deprivation down-regulated Slit2 (Fig. 1G). Slit1 expression could not be consistently detected under all of the treatment conditions or up-regulated under stress. In contrast to the situation in primary islets, thapsigargin and palmitate up-regulated Slit1 and Slit2 in MIN6 cells (Fig. S1). Slit3, which is typically absent in MIN6 cells, was robustly induced by ER stress (Fig. S1). These data suggest that the production of Slit ligands can be regulated in response to specific cellular stresses, with differential effects observed between primary islet cells and MIN6 insulinoma cells. We also examined the effects of stress on the Robo genes, and observed significant regulation (Fig. 1G).
Knockdown of Endogenous Slits Decreases Beta-Cell Survival.
The regulation of Slit expression by stress suggests that Slit–Robo signaling play a role in beta-cell survival. A loss-of-function approach was used to determine the role of endogenous Slit ligands in beta-cell survival. In mouse islet cells, simultaneous siRNA targeting of all Slit ligands (to circumvent isoform compensation) resulted in a 48% knockdown of Slit1, 46% of Slit2, and 49% of Slit3 mRNA (Fig. 2A). Remarkably, even this incomplete reduction in endogenous Slit production significantly reduced beta-cell survival in serum-free conditions (Fig. 2B). Similarly, knockdown of Slit1/2/3 in MIN6 cells had significant negative effects on beta-cell survival (Fig. S2). These studies demonstrate that the local production of Slit ligands is required for optimal beta-cell survival.
Fig. 2.

Knockdown of endogenous Slits increases cell death. (A) Dispersed mouse islet cells were transfected with siRNA for Slit1, Slit2, and Slit3 or scramble siRNA as control, and examined by qRT-PCR after 72 h (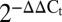 ; n = 6, *P < 0.05 compared with control). (B) Mouse islet cells transfected with Slit siRNAs were stained with 0.05 µg/mL Hoechst and 0.5 µg/mL propidium iodide (PI) 48 h following transfection and imaged. Cells were in SF conditions with 20 mM and 5 mM glucose. Percent PI-positive cells and area under the curve (AUC) were calculated for indicated time intervals (n = 12, *P < 0.05 compared with control). (C) Mouse islet cells transfected with Slit siRNAs were incubated in 5 mM glucose SF conditions supplemented with 10 nM SLIT1-3 (n = 10, *P < 0.05 compared with scramble control, †P < 0.05 compared with Slit knockdown without SLIT supplement). (D) Mouse islet cells transfected with Slit siRNAs were stained and treated with cytokine mixture (25 ng/mL TNF-α, 10 ng/mL IL-1β, 10 ng/mL IFN-γ), 1 μM thapsigargin, or 1.5 mM palmitate in 20 mM glucose. (Inset) AUC from 0 to 60 h (n = 8–10).
; n = 6, *P < 0.05 compared with control). (B) Mouse islet cells transfected with Slit siRNAs were stained with 0.05 µg/mL Hoechst and 0.5 µg/mL propidium iodide (PI) 48 h following transfection and imaged. Cells were in SF conditions with 20 mM and 5 mM glucose. Percent PI-positive cells and area under the curve (AUC) were calculated for indicated time intervals (n = 12, *P < 0.05 compared with control). (C) Mouse islet cells transfected with Slit siRNAs were incubated in 5 mM glucose SF conditions supplemented with 10 nM SLIT1-3 (n = 10, *P < 0.05 compared with scramble control, †P < 0.05 compared with Slit knockdown without SLIT supplement). (D) Mouse islet cells transfected with Slit siRNAs were stained and treated with cytokine mixture (25 ng/mL TNF-α, 10 ng/mL IL-1β, 10 ng/mL IFN-γ), 1 μM thapsigargin, or 1.5 mM palmitate in 20 mM glucose. (Inset) AUC from 0 to 60 h (n = 8–10).
We asked whether supplementing islet cell cultures with recombinant SLIT would be sufficient to rescue the effects of Slit1/2/3 knockdown. Indeed, although SLIT1 and SLIT2 alone could not rescue the elevated level of cell death observed under 5 mM glucose serum-free condition (Fig. 2C), SLIT3 and a combination of all SLITs reversed the effects of SLIT knockdown on islet cell death (Fig. 2C). We did not observe significant differences in cell survival between control and Slit knockdown in primary islet cells and MIN6 treated with cytokines, thapsigargin, or palmitate (Fig. 2D and Fig. S3). Collectively, our data indicate that SLIT ligands have acute protective effects on islet cells.
Exogenous SLITs Increase Beta-Cell Survival During Stress and Hyperglycemia.
Next, we tested whether exogenous SLIT1, SLIT2, and SLIT3 could protect beta cells from multiple forms of death. We first sought to determine whether the glucose milieu altered the protective effects of Slit treatment, as we have observed with netrin and Notch signaling (12, 26). Indeed, treatment with SLIT1 and SLIT2 recombinant proteins significantly reduced thapsigargin-induced death in MIN6 cells under high-, but not low-glucose conditions (Fig. S4 A and B). In mouse islet cells, SLIT1, SLIT2, and SLIT3 also reduced islet cell death in response to serum deprivation alone and in combination with cytokines (Fig. 3 A and B). These effects were only seen in the context of high-glucose and never in low-glucose conditions, suggesting a context-dependent switch. Cell death induced by exposure to thapsigargin under serum deprivation was rescued with combination treatment of SLIT1-3 (Fig. 3C). SLIT1 and SLIT2 also reduced the level of cell death induced by thapsigargin treatment alone (Fig. S4C). Palmitate-induced cell death was not significantly reduced with SLITs (Fig. 3D). These data show that exogenous SLIT ligands, especially in combination, exert significant protection against several harsh stresses.
Fig. 3.

Slits reduce stress-induced islet cell death under high glucose conditions. Dispersed mouse islet cells were stained with Hoechst and PI and imaged. The percentage of PI-positive cells was determined following treatments with 10 nM SLIT1, SLIT2, and/or SLIT3 in 20 mM glucose SF (A), 20 mM glucose SF with cytokine mixture (B), 20 mM glucose SF with 1 µM thapsigargin (C), and 20 mM glucose SF with 1.5 mM palmitate (D). (Inset) Area under the curve from 0 to 50 h (n = 8, *P < 0.05 compared with untreated control).
Slits Protect Beta Cells by Suppressing Apoptosis and ER Stress.
To assess the molecular mechanisms associated with Slit action in beta cells, we examined markers of ER stress and apoptosis. Significant increases in annexin V-positive cells were observed in Slit1/2/3 knockdown cells compared with control (Fig. 4 A–C), pointing to an effect on apoptotic cell death. Treatment of mouse islets with exogenous SLIT decreased expression of Chop, Gadd34, and sXbp1 mRNA, but only in high glucose (Fig. 4D). In low glucose, SLIT increased the expression of Bip, a protective chaperone (Fig. 4D and Fig. S5). Consistent with the down-regulation of Chop observed in mouse islet cells, we also found a decrease in thapsigargin-induced CHOP protein upon treatment with SLIT2 in high glucose (Fig. S6A). Treating mouse islets with SLITs reduced cleaved caspase-3 and cleaved PARP (Fig. 4E).
Fig. 4.

Slits down-regulate proapoptotic and ER-stress signaling. MIN6 cells transfected with Slit1, Slit2, and Slit3 siRNAs were stained with Hoechst, PI, and Alexa Fluor 647-conjugated Annexin V 48 h following transfection. Cells were cultured in 22 mM (A and B) and 5 mM (C) glucose in the presence or absence of FBS and imaged. (Inset) Area under the curve from 20 to 40 h (n = 10, *P < 0.05 compared with scramble siRNA control). (D) Mouse islet cells were treated with SLIT1 or SLIT2 for 4 h before RNA isolation and qRT-PCR analysis (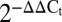 ; n = 8, *P < 0.05). (E) Mouse islet cells were treated with SLIT1-3 under 20 mM glucose SF condition. Immunoassay for cleaved caspase-3, cleaved PARP, phospho-JNK, phospho-p38, phospho-p53, and phospho-HSP27 (n = 6–8, *P < 0.05).
; n = 8, *P < 0.05). (E) Mouse islet cells were treated with SLIT1-3 under 20 mM glucose SF condition. Immunoassay for cleaved caspase-3, cleaved PARP, phospho-JNK, phospho-p38, phospho-p53, and phospho-HSP27 (n = 6–8, *P < 0.05).
Upon induction of ER stress, IRE1 activation can lead to the downstream activation of NF-κB and ASK1-p38MAPK/JNK signaling cascades. Treatment with SLITs significantly reduced phospho-JNK and phospho-p38MAPK, indicative of the down-regulation of these signaling cascades (Fig. 4E). Downstream mediators of p38MAPK pathway, p53 and HSP27, were also down-regulated. Treatment of MIN6 cells with SLIT2 following ER-stress induction reduced ASK1 activation (Fig. S6B). Thapsigargin-induced cleaved caspase-3 and cleaved caspase-12 were also down-regulated by SLIT2 (Fig. S6 C–E). Serum deprivation-induced cleaved caspase-3 levels were also down-regulated upon SLIT1 and SLIT2 treatments under high-glucose, but not low-glucose conditions (Fig. S6 F and G). Cleaved caspase-7 and cleaved caspase-12 levels were also significantly decreased in cells treated with SLIT1 under hyperglycemic serum-free conditions (Fig. S6 H and I). Together, these experiments indicate that SLIT protects beta cells by broad suppression of the ER stress-induced apoptosis pathway. Because we have previously shown that the CHOP–caspase axis in beta cells can be controlled by luminal Ca2+ (24, 25), we predicted that the effects of SLIT–ROBO signaling on cell death signaling were downstream of the modulation of ER Ca2+.
SLITs Accelerate Ca2+ Oscillations and Modulate ER Luminal Ca2+.
Ca2+ homeostasis, especially within the ER, plays a key role in both beta-cell survival (24, 25, 27, 28) and glucose responsiveness (27, 29, 30). Given that SLIT2 signaling in neurons involves Ca2+ release from the ER (31–33), we examined this mechanism in beta cells. Interestingly, SLIT treatment increased the frequency of glucose-stimulated cytosolic Ca2+ oscillations (Fig. 5 A and B), a phenomenon that is known to be modulated by the ER Ca2+ filling state and associated with increased insulin secretion (27, 29, 30, 34). At basal glucose, SLIT treatment had little or no effect on cytosolic Ca2+ (Fig. 5C). However, in cells transfected with luminal ER Ca2+ sensor, D1ER (25, 35), we observed that SLIT induced a gradual release of Ca2+ from the ER filled after exposure to high glucose (Fig. 5D and Fig. S7). These effects correlate well with the conditions under which SLIT proteins protect beta cells from ER stress induced by cytokines and by thapsigargin, a drug that blocks ER Ca2+ refilling. This result fits with a model whereby ER stress-induced cell death is dependent on the rate at which Ca2+ is depleted and the level of depletion (25). SLITs only partially depleted ER Ca2+ because thapsigargin treatment led to further depletion of ER Ca2+ (Fig. 5D and Fig. S7). The partial depletion of ER Ca2+ was maintained throughout a 6-h treatment with SLITs (Fig. S7, Bottom). These experiments demonstrate that Slit signaling has direct effects on ER luminal Ca2+, a parameter known to modulate ER stress and insulin secretion (24, 25, 28).
Fig. 5.

Slits modulate cytosolic Ca2+ and ER Ca2+ signaling and actin remodeling. Dispersed mouse islet cells loaded with Fura-2-AM. Representative single-cell traces following 10 nM SLIT1, SLIT2, or SLIT3 treatment in 15 mM glucose (A). Single-cell oscillation frequency of cytosolic Ca2+ (n = 91–104, *P < 0.05) (B). Representative single-cell traces following exposure to 10 nM SLIT2 in 3 mM glucose (C). (D) Dispersed mouse islet cells were transfected with D1ER cameleon to image ER Ca2+. Cells were exposed to 1 µM Tg 15 min following treatment with 10 nM SLIT1, SLIT2, or SLIT3 (colored lines) or untreated (black line) in 15 mM glucose (n = 13–14). (Inset) D1ER FRET/CFP ratios normalized to the time point following Tg addition. (Right) AUC for the 15-min preincubation with 15 mM glucose and 15-min treatment with SLIT1-3 (n = 13–14, *P < 0.05 compared with untreated). (E) Mouse islet cells treated with 10 nM SLIT1, SLIT2, or SLIT3 in 11 mM glucose-containing RPMI. Beta cells stained for insulin and phalloidin and peak intensity of cortical F-actin staining was normalized to untreated cells at the same time point (n = 39–60 cells).
Ca2+-dependent actin remodeling is induced by SLIT proteins during axon guidance (31, 32). Treatment of mouse beta cells with recombinant SLIT1, SLIT2, or SLIT3 decreased cortical F-actin (Fig. 5E), an event expected to promote glucose-stimulated insulin secretion (36–38). Temporal analysis of F-actin levels revealed a biphasic response following SLIT1 and SLIT3 treatments (Fig. 5E).
SLITs Potentiate Glucose-Stimulated Insulin Secretion.
Given the roles of Ca2+ and actin on insulin secretion, we investigated whether SLITs affect insulin release. Static incubation showed that mouse islets cultured in 15 mM glucose secreted more insulin in the presence of SLIT (control: 9.6 ± 2.8 ng/mL, SLIT1: 21.6 ± 8.2 ng/mL, SLIT2: 19.1 ± 8.4 ng/mL). This observation led us to conduct islet perifusions. Indeed, glucose-stimulated insulin secretion was significantly potentiated in the presence of SLIT1, SLIT2, or SLIT3 (Fig. 6A). SLIT treatment did not potentiate insulin secretion when beta cells were depolarized with 30 mM KCl (Fig. 6A), ruling out effects distal to the opening of voltage-gated Ca2+ channels and pointing to effects on glucose sensing/signaling. SLITs did not affect Ins1 and Ins2 transcription (Fig. S8). Thus, Slit proteins can both protect beta cells and increase insulin secretion, which itself is antiapoptotic (8, 9, 39).
Fig. 6.

Slits modulate insulin secretion. (A) Mouse islets perifused with Krebs–Ringer buffer containing 3 or 15 mM glucose in combination with 10 nM SLIT1, SLIT2, or SLIT3 (n = 5–6). (Right) Baseline subtracted AUC for the 15 mM glucose- and KCl-stimulated insulin secretion, along with first- and second-phase insulin secretion (n = 5–6, *P < 0.05 compared with control). (B) Model of Slit–Robo signaling in beta cells. TCA, tricarboxylic acid.
Discussion
The present study was conducted to determine the expression pattern, regulation, and roles of the Slit family of secreted factors and their Robo receptors in pancreatic islet cells. Slit and Robo transcripts and protein were detected in adult mouse and human islets and regulated in stress conditions. We identified an important role for this local autocrine/paracrine network in beta-cell survival and function. We identified a unique anti-ER stress and antiapoptotic mechanism of action involving the controlled release of Ca2+ from the ER lumen (Fig. 6B).
The roles of Slit–Robo signaling in cell survival remain poorly understood. Knockout mouse studies, along with the observed loss of expression of ROBO1 in some human cancers, provide evidence that the loss of ROBO is tumorigenic. SLIT1 and SLIT3 are candidate tumor suppressor genes (40), although SLIT1 is up-regulated in prostate tumors (41). In islet cells, a modest knockdown of Slits significantly increased cell death, suggesting that endogenous SLIT secretion plays an important role in cell survival. Conversely, SLIT1, SLIT2, and/or SLIT3 supplementation reduced stress-induced cell death. We observed significant decreases in both ER stress- and serum starvation-induced cell death, but only under hyperglycemic conditions. Our data suggest that Ca2+-dependent mechanisms are important for the protective effects of Slit, which is in line with a role for Ca2+ in Slit–Robo signaling in other cell types (24, 25, 27, 28, 31–33). In particular, our results implicate a controlled depletion in ER Ca2+ and an increase in the frequency of Ca2+ oscillations.
It is possible that Slit may protect beta cells via interaction with receptors for other guidance factors. DCC, UNC5, and Neogenin are dependence receptors that can increase cell survival in the presence of netrins and induce cell death in the absence of netrins (42, 43). Interaction between ROBO1 and DCC in a Slit-dependent manner decreases netrin-induced chemoattraction in neurons (44, 45). Perhaps Slit–Robo signaling regulates beta-cell survival through down-regulation of netrin receptor-induced apoptosis. Additionally, because Slit–Robo signaling induces tumor angiogenesis by attracting endothelial cells, perhaps local production of SLITs could improve islet engraftment following transplantation (46). The stress-induced up-regulation of Slit expression, the survival effects of exogenous SLITs under high-glucose conditions, and the protective effects of endogenous SLITs suggest an autocrine/paracrine compensatory survival network.
In addition to the protective effects on beta cells, we uncovered effects of SLIT ligands on proximal glucose sensing/signaling, which were associated with changes in Ca2+ and actin. It was not surprising to find that SLITs potentiate glucose-stimulated insulin secretion given that Cdc42, Rac1, and RhoA play important roles in Slit-induced cellular migration (33, 47, 48). The down-regulation of cortical F-actin staining observed upon SLIT treatment is in agreement with the requirement for Cdc42 and RhoA inactivation via increasing Rho GTPase-activating proteins for the repulsive effects of SLITs in neurons and olfactory ensheathing cells (33, 47). Our current data do not allow us to distinguish whether the biphasic F-actin depletion following SLIT1 and SLIT3 treatments directly or indirectly mediates the potentiation of insulin secretion, but this could be a fruitful area for future investigation. In addition to the release of antiapoptotic insulin (8, 9, 39, 49–51), modulation of actin polymerization may directly promote survival (52–55). We have not determined whether the survival effects of SLITs are due to direct down-regulation of apoptotic pathways or indirect effects through up-regulation of insulin secretion (39, 56).
In conclusion, we provide detailed evidence that Slit ligands and their Robo receptors are present in pancreatic islet cells and define multiple roles for the Slit-secreted factors in beta-cell physiology. Our results reveal that Slit signaling depletes ER Ca2+ and protects beta cells from ER stress. Together, our results point to the Slit–Robo pathway and a novel area for investigations around beta-cell survival and function.
Materials and Methods
Detailed methods can be found in SI Materials and Methods.
Primary Islet Isolation, Cell Culture, and Perifusion.
Islets were isolated from C57BL/6J mice using collagenase and filtration (12, 56). Human islets (>80% pure) were provided by Garth Warnock (University of British Columbia, Vancouver), collected via protocols approved by the University of British Columbia Research Ethics Board. Islets and MIN6 cells were cultured as described (8, 12, 57). Insulin secretion was measured by perifusion and RIA (26).
siRNA, RT-PCR, and Immunoblotting.
MIN6 and dispersed islet cells were Neon (Invitrogen)-transfected with 100 nM (MIN6) or 200 nM (islet cell) Silencer Select siRNA (Ambion) targeting Slit1, Slit2, and Slit3, with scramble siRNA (Ambion) as a negative control. Total RNA was isolated and qRT-PCR was performed. Immunoblotting was performed as described (12).
Imaging.
MIN6 and dispersed islet cells stained as described (12). For cell death assays, cells were seeded into 96-well plates and exposed to 0.05 µg/mL Hoechst 33342 (Invitrogen), 0.5 µg/mL propidium iodide (Sigma), and AlexaFluor647-conjugated Annexin V (1:250; Invitrogen) (25). Cells were imaged with ImageXpressMICRO (Molecular Devices) every 1 or 2 h at 37 °C and 5% (vol/vol) CO2 (12). Cytosolic Ca2+ was imaged using Fura-2-AM (Invitrogen) (57). To measure ER Ca2+, mouse islet and MIN6 cells were transfected with the D1ER sensor (24, 25). To detect F-actin, mouse islet cells were fixed with Z-FIX (Anatech Ltd.) and stained with Alexa Fluor 488-conjugated phalloidin (Invitrogen). Beta cells were identified by insulin staining (Santa Cruz Biotechnology). Line scans were performed using ImageJ (version 1.41o; National Institutes of Health) and mean peak-intensity calculated.
Statistics.
Data are expressed as mean ± SEM unless otherwise indicated. Results were considered significant when P < 0.05 using Student t test or ANOVA, where appropriate (Prism; GraphPad).
Supplementary Material
Acknowledgments
We thank Keshika Nanda, Xiaoke Hu, and Dr. Kwan Yi Chu for technical assistance. These studies were supported by grants from the JDRF–Johnson & Johnson (to J.D.J.) and Canadian Diabetes Association (to P.E.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1214312110/-/DCSupplemental.
References
- 1.Rosenbaum T, Vidaltamayo R, Sánchez-Soto MC, Zentella A, Hiriart M. Pancreatic beta cells synthesize and secrete nerve growth factor. Proc Natl Acad Sci USA. 1998;95(13):7784–7788. doi: 10.1073/pnas.95.13.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsuda H, et al. Immunohistochemical localization of hepatocyte growth factor protein in pancreas islet A-cells of man and rats. Jpn J Cancer Res. 1992;83(12):1262–1266. doi: 10.1111/j.1349-7006.1992.tb02756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dezaki K, Kageyama H, Seki M, Shioda S, Yada T. Neuropeptide W in the rat pancreas: Potentiation of glucose-induced insulin release and Ca2+ influx through L-type Ca2+ channels in beta-cells and localization in islets. Regul Pept. 2008;145(1-3):153–158. doi: 10.1016/j.regpep.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 4.Yada T, et al. Pituitary adenylate cyclase-activating polypeptide (PACAP) is an islet substance serving as an intra-islet amplifier of glucose-induced insulin secretion in rats. J Physiol. 1997;505(Pt 2):319–328. doi: 10.1111/j.1469-7793.1997.319bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamamoto K, et al. Overexpression of PACAP in transgenic mouse pancreatic beta-cells enhances insulin secretion and ameliorates streptozotocin-induced diabetes. Diabetes. 2003;52(5):1155–1162. doi: 10.2337/diabetes.52.5.1155. [DOI] [PubMed] [Google Scholar]
- 6.Fujita Y, et al. Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet alpha-cells and promotes insulin secretion. Gastroenterology. 2010;138(5):1966–1975. doi: 10.1053/j.gastro.2010.01.049. [DOI] [PubMed] [Google Scholar]
- 7.Szabat M, Johnson JD, Piret JM. Reciprocal modulation of adult beta cell maturity by activin A and follistatin. Diabetologia. 2010;53(8):1680–1689. doi: 10.1007/s00125-010-1758-0. [DOI] [PubMed] [Google Scholar]
- 8.Alejandro EU, et al. Acute insulin signaling in pancreatic beta-cells is mediated by multiple Raf-1 dependent pathways. Endocrinology. 2010;151(2):502–512. doi: 10.1210/en.2009-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson JD, Alejandro EU. Control of pancreatic beta-cell fate by insulin signaling: The sweet spot hypothesis. Cell Cycle. 2008;7(10):1343–1347. doi: 10.4161/cc.7.10.5865. [DOI] [PubMed] [Google Scholar]
- 10.Beith JL, Alejandro EU, Johnson JD. Insulin stimulates primary beta-cell proliferation via Raf-1 kinase. Endocrinology. 2008;149(5):2251–2260. doi: 10.1210/en.2007-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szabat M, et al. Maintenance of β-cell maturity and plasticity in the adult pancreas: Developmental biology concepts in adult physiology. Diabetes. 2012;61(6):1365–1371. doi: 10.2337/db11-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang YH, et al. Paracrine signalling loops in adult human and mouse pancreatic islets: Netrins modulate beta cell apoptosis signalling via dependence receptors. Diabetologia. 2011;54(4):828–842. doi: 10.1007/s00125-010-2012-5. [DOI] [PubMed] [Google Scholar]
- 13.Chilton JK. Molecular mechanisms of axon guidance. Dev Biol. 2006;292(1):13–24. doi: 10.1016/j.ydbio.2005.12.048. [DOI] [PubMed] [Google Scholar]
- 14.Long H, et al. Conserved roles for Slit and Robo proteins in midline commissural axon guidance. Neuron. 2004;42(2):213–223. doi: 10.1016/s0896-6273(04)00179-5. [DOI] [PubMed] [Google Scholar]
- 15.Seeger M, Tear G, Ferres-Marco D, Goodman CS. Mutations affecting growth cone guidance in Drosophila: Genes necessary for guidance toward or away from the midline. Neuron. 1993;10(3):409–426. doi: 10.1016/0896-6273(93)90330-t. [DOI] [PubMed] [Google Scholar]
- 16.Kidd T, et al. Roundabout controls axon crossing of the CNS midline and defines a novel subfamily of evolutionarily conserved guidance receptors. Cell. 1998;92(2):205–215. doi: 10.1016/s0092-8674(00)80915-0. [DOI] [PubMed] [Google Scholar]
- 17.Kidd T, Bland KS, Goodman CS. Slit is the midline repellent for the robo receptor in Drosophila. Cell. 1999;96(6):785–794. doi: 10.1016/s0092-8674(00)80589-9. [DOI] [PubMed] [Google Scholar]
- 18.Ypsilanti AR, Zagar Y, Chédotal A. Moving away from the midline: New developments for Slit and Robo. Development. 2010;137(12):1939–1952. doi: 10.1242/dev.044511. [DOI] [PubMed] [Google Scholar]
- 19.Brose K, et al. Slit proteins bind Robo receptors and have an evolutionarily conserved role in repulsive axon guidance. Cell. 1999;96(6):795–806. doi: 10.1016/s0092-8674(00)80590-5. [DOI] [PubMed] [Google Scholar]
- 20.Fujiwara M, Ghazizadeh M, Kawanami O. Potential role of the Slit/Robo signal pathway in angiogenesis. Vasc Med. 2006;11(2):115–121. doi: 10.1191/1358863x06vm658ra. [DOI] [PubMed] [Google Scholar]
- 21.Strickland P, Shin GC, Plump A, Tessier-Lavigne M, Hinck L. Slit2 and netrin 1 act synergistically as adhesive cues to generate tubular bi-layers during ductal morphogenesis. Development. 2006;133(5):823–832. doi: 10.1242/dev.02261. [DOI] [PubMed] [Google Scholar]
- 22.Juhl K, Sarkar SA, Wong R, Jensen J, Hutton JC. Mouse pancreatic endocrine cell transcriptome defined in the embryonic Ngn3-null mouse. Diabetes. 2008;57(10):2755–2761. doi: 10.2337/db07-1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seki M, et al. Human ROBO1 is cleaved by metalloproteinases and gamma-secretase and migrates to the nucleus in cancer cells. FEBS Lett. 2010;584(13):2909–2915. doi: 10.1016/j.febslet.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Gwiazda KS, Yang TL, Lin Y, Johnson JD. Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am J Physiol Endocrinol Metab. 2009;296(4):E690–E701. doi: 10.1152/ajpendo.90525.2008. [DOI] [PubMed] [Google Scholar]
- 25.Luciani DS, et al. Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death. Diabetes. 2009;58(2):422–432. doi: 10.2337/db07-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dror V, et al. Notch signalling suppresses apoptosis in adult human and mouse pancreatic islet cells. Diabetologia. 2007;50(12):2504–2515. doi: 10.1007/s00125-007-0835-5. [DOI] [PubMed] [Google Scholar]
- 27.Jahanshahi P, Wu R, Carter JD, Nunemaker CS. Evidence of diminished glucose stimulation and endoplasmic reticulum function in nonoscillatory pancreatic islets. Endocrinology. 2009;150(2):607–615. doi: 10.1210/en.2008-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cardozo AK, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes. 2005;54(2):452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- 29.Ravier MA, Sehlin J, Henquin JC. Disorganization of cytoplasmic Ca(2+) oscillations and pulsatile insulin secretion in islets from ob/ob mice. Diabetologia. 2002;45(8):1154–1163. doi: 10.1007/s00125-002-0883-9. [DOI] [PubMed] [Google Scholar]
- 30.Ravier MA, et al. Mechanisms of control of the free Ca2+ concentration in the endoplasmic reticulum of mouse pancreatic β-cells: interplay with cell metabolism and [Ca2+]c and role of SERCA2b and SERCA3. Diabetes. 2011;60(10):2533–2545. doi: 10.2337/db10-1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu HT, et al. Calcium signaling in chemorepellant Slit2-dependent regulation of neuronal migration. Proc Natl Acad Sci USA. 2004;101(12):4296–4301. doi: 10.1073/pnas.0303893101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guan CB, Xu HT, Jin M, Yuan XB, Poo MM. Long-range Ca2+ signaling from growth cone to soma mediates reversal of neuronal migration induced by slit-2. Cell. 2007;129(2):385–395. doi: 10.1016/j.cell.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 33.Huang ZH, et al. Slit-2 repels the migration of olfactory ensheathing cells by triggering Ca2+-dependent cofilin activation and RhoA inhibition. J Cell Sci. 2011;124(Pt 2):186–197. doi: 10.1242/jcs.071357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beauvois MC, et al. Glucose-induced mixed [Ca2+]c oscillations in mouse beta-cells are controlled by the membrane potential and the SERCA3 Ca2+-ATPase of the endoplasmic reticulum. Am J Physiol Cell Physiol. 2006;290(6):C1503–C1511. doi: 10.1152/ajpcell.00400.2005. [DOI] [PubMed] [Google Scholar]
- 35.Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA. 2004;101(50):17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thurmond DC, Gonelle-Gispert C, Furukawa M, Halban PA, Pessin JE. Glucose-stimulated insulin secretion is coupled to the interaction of actin with the t-SNARE (target membrane soluble N-ethylmaleimide-sensitive factor attachment protein receptor protein) complex. Mol Endocrinol. 2003;17(4):732–742. doi: 10.1210/me.2002-0333. [DOI] [PubMed] [Google Scholar]
- 37.Pigeau GM, et al. Insulin granule recruitment and exocytosis is dependent on p110gamma in insulinoma and human beta-cells. Diabetes. 2009;58(9):2084–2092. doi: 10.2337/db08-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nevins AK, Thurmond DC. Glucose regulates the cortical actin network through modulation of Cdc42 cycling to stimulate insulin secretion. Am J Physiol Cell Physiol. 2003;285(3):C698–C710. doi: 10.1152/ajpcell.00093.2003. [DOI] [PubMed] [Google Scholar]
- 39.Johnson JD, et al. Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc Natl Acad Sci USA. 2006;103(51):19575–19580. doi: 10.1073/pnas.0604208103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mehlen P, Delloye-Bourgeois C, Chédotal A. Novel roles for Slits and netrins: Axon guidance cues as anticancer targets? Nat Rev Cancer. 2011;11(3):188–197. doi: 10.1038/nrc3005. [DOI] [PubMed] [Google Scholar]
- 41.Latil A, et al. Quantification of expression of netrins, slits and their receptors in human prostate tumors. Int J Cancer. 2003;103(3):306–315. doi: 10.1002/ijc.10821. [DOI] [PubMed] [Google Scholar]
- 42.Forcet C, et al. The dependence receptor DCC (deleted in colorectal cancer) defines an alternative mechanism for caspase activation. Proc Natl Acad Sci USA. 2001;98(6):3416–3421. doi: 10.1073/pnas.051378298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Llambi F, Causeret F, Bloch-Gallego E, Mehlen P. Netrin-1 acts as a survival factor via its receptors UNC5H and DCC. EMBO J. 2001;20(11):2715–2722. doi: 10.1093/emboj/20.11.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stein E, Tessier-Lavigne M. Hierarchical organization of guidance receptors: silencing of netrin attraction by slit through a Robo/DCC receptor complex. Science. 2001;291(5510):1928–1938. doi: 10.1126/science.1058445. [DOI] [PubMed] [Google Scholar]
- 45.Bai G, et al. Presenilin-dependent receptor processing is required for axon guidance. Cell. 2011;144(1):106–118. doi: 10.1016/j.cell.2010.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang B, et al. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell. 2003;4(1):19–29. doi: 10.1016/s1535-6108(03)00164-8. [DOI] [PubMed] [Google Scholar]
- 47.Wong K, et al. Signal transduction in neuronal migration: Roles of GTPase activating proteins and the small GTPase Cdc42 in the Slit-Robo pathway. Cell. 2001;107(2):209–221. doi: 10.1016/s0092-8674(01)00530-x. [DOI] [PubMed] [Google Scholar]
- 48.Liu D, et al. Neuronal chemorepellent Slit2 inhibits vascular smooth muscle cell migration by suppressing small GTPase Rac1 activation. Circ Res. 2006;98(4):480–489. doi: 10.1161/01.RES.0000205764.85931.4b. [DOI] [PubMed] [Google Scholar]
- 49.Otani K, et al. Reduced beta-cell mass and altered glucose sensing impairs insulin secretory function in mice with pancreatic beta-cell knockout of the insulin receptor. Am J Physiol Endocrinol Metab. 2003;286(1):E41–E49. doi: 10.1152/ajpendo.00533.2001. [DOI] [PubMed] [Google Scholar]
- 50.Ueki K, et al. Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat Genet. 2006;38(5):583–588. doi: 10.1038/ng1787. [DOI] [PubMed] [Google Scholar]
- 51.Ueki K, Okada T, Ozcan U, Kulkarni R. Endogenous insulin-mediated signaling protects apoptosis induced by glucose in pancreatic beta-cells. Diabetes. 2003;52:A42–A42. [Google Scholar]
- 52.Xiao D, et al. Effect of actin cytoskeleton disruption on electric pulse-induced apoptosis and electroporation in tumour cells. Cell Biol Int. 2011;35(2):99–104. doi: 10.1042/CBI20100464. [DOI] [PubMed] [Google Scholar]
- 53.Posey SC, Bierer BE. Actin stabilization by jasplakinolide enhances apoptosis induced by cytokine deprivation. J Biol Chem. 1999;274(7):4259–4265. doi: 10.1074/jbc.274.7.4259. [DOI] [PubMed] [Google Scholar]
- 54.Gourlay CW, Ayscough KR. The actin cytoskeleton: A key regulator of apoptosis and ageing? Nat Rev Mol Cell Biol. 2005;6(7):583–589. doi: 10.1038/nrm1682. [DOI] [PubMed] [Google Scholar]
- 55.Yermen B, Tomas A, Halban PA. Pro-survival role of gelsolin in mouse beta-cells. Diabetes. 2007;56(1):80–87. doi: 10.2337/db06-0769. [DOI] [PubMed] [Google Scholar]
- 56.Alejandro EU, Johnson JD. Inhibition of Raf-1 alters multiple downstream pathways to induce pancreatic beta-cell apoptosis. J Biol Chem. 2008;283(4):2407–2417. doi: 10.1074/jbc.M703612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szabat M, Luciani DS, Piret JM, Johnson JD. Maturation of adult beta-cells revealed using a Pdx1/insulin dual-reporter lentivirus. Endocrinology. 2009;150(4):1627–1635. doi: 10.1210/en.2008-1224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.