

Significance
Treatment of HIV-1 infection in humans is achieved using combinations of highly effective antiretroviral therapy (ART) drugs to potently suppress viral replication and prevent the emergence of drug-resistant viruses. However, ART drugs must be taken indefinitely owing to rapid return of viremia upon termination of treatment. Highly potent broadly neutralizing antibodies (bNAbs) present a new potential therapeutic modality in the treatment of HIV-1 infection. Because of their comparatively longer half-lives relative to ART drugs and their ability to eliminate infected cells, bNAbs may alleviate some aspects of the lifelong treatment adherence burden of ART. Here we show that lowering the initial viral load with ART enables single bNAbs to effectively control an established HIV-1 infection in humanized mice.
Keywords: CD4bs, glycan, gp160
Abstract
Effective control of HIV-1 infection in humans is achieved using combinations of antiretroviral therapy (ART) drugs. In humanized mice (hu-mice), control of viremia can be achieved using either ART or by immunotherapy using combinations of broadly neutralizing antibodies (bNAbs). Here we show that treatment of HIV-1–infected hu-mice with a combination of three highly potent bNAbs not only resulted in complete viremic control but also led to a reduction in cell-associated HIV-1 DNA. Moreover, lowering the initial viral load by coadministration of ART and immunotherapy enabled prolonged viremic control by a single bNAb after ART was withdrawn. Similarly, a single injection of adeno-associated virus directing expression of one bNAb produced durable viremic control after ART was terminated. We conclude that immunotherapy reduces plasma viral load and cell-associated HIV-1 DNA and that decreasing the initial viral load enables single bNAbs to control viremia in hu-mice.
Antiretroviral therapy (ART) suppresses HIV-1 viremia to nearly undetectable levels within 12–48 wk in a majority of infected individuals (1, 2). This form of therapy combines at least three drugs, each of which can target a unique step in the viral life cycle including reverse transcription, integration, proteolytic processing, and viral entry (3). However, treatment must be continued indefinitely because ART interruption leads to rapid viremic rebound (4, 5), and attempts at treatment intensification by additional drugs have not improved viremic control (6, 7). Moreover, therapy is associated with a number of side effects and emergence of resistance, highlighting the need to explore new therapeutic modalities against HIV-1 (3).
Human monoclonal antibodies to HIV-1 can neutralize a broad range of viral isolates in vitro and can protect nonhuman primates against infection (8–10). In contrast, experiments in humanized mice (hu-mice) and humans initially suggested that antibodies were unable to control established HIV-1 infection (11–13). However, advances in single-cell antibody cloning (14–16) and structure-based design methods (17) uncovered far more potent antibodies (17–22) that were shown to suppress viremia in hu-mice (23, 24). Antibodies exhibiting this level of potency and breadth are rare among infected individuals (25) and typically arise only 2–3 y after initial infection (26, 27). These antibodies are extensively somatically mutated (14, 18, 20, 23, 28, 29), and all efforts to date to elicit them by immunization have failed (15, 30). In contrast to ART, which has no appreciable efficacy after termination of treatment, antibodies can control infection in hu-mice for up to 2 mo after cessation of therapy owing to their long half-life in vivo (23). However, the ability of highly potent broadly neutralizing antibodies (bNAbs) to sustain viremic control after termination of ART remains unknown.
Results
Building on our previous findings that mixtures of antibodies led to sustained viremic control (23, 24), we sought to characterize the extent of viral suppression using an optimized immunotherapy regimen. We selected three antibodies targeting three different epitopes on the gp120 portion of the viral spike: 3BNC117, a potent CD4 binding site (CD4bs) antibody with a longer half-life than NIH45-46G54W (45-46G54W) (21) (Fig. S1); PG16, which recognizes the V1/V2 loop region (22, 31); and 10-1074, an antibody that targets the base of the V3 stem (19). In two independent experiments, we treated HIV-1YU2 infected hu-mice 2–3 wk after infection (23, 32) for 6 wk with combination immunotherapy (1.0 mg per antibody, twice per week s.c.) (Fig. 1 and Fig. S2). Viral loads were monitored weekly using one of two different quantitative reverse-transcriptase PCR assays (Fig. 1 A and B and Fig. S2). All treated mice exhibited rapid and continuous plasma viral load suppression during the 6-wk treatment course, with a majority of mice dropping below the limit of viral load detection (Fig. 1 A–C and Fig. S2). We conclude that combined immunotherapy with 3BNC117, PG16, and 10-1074 is sufficient to control plasma HIV-1 viremia in hu-mice.
Fig. 1.

Combination immunotherapy with 3BNC117, PG16, and 10-1074. (A) Plasma viral loads for HIV-1YU2–infected hu-mice treated with combination immunotherapy were monitored at regular intervals by quantitative RT-PCR. Each line represents a single animal. Treatment began at day 0. Antibodies were administered s.c. at 1.0 mg per antibody, twice per week, for a total of 6 wk. Green line, average viral load for untreated mice (Fig. S4). (B) Fold change in viral load (shown in A) from the baseline measurement for each animal. Red line shows the geometric mean viral load change at each time point. Green line, as above. (C) Combination immunotherapy resulted in a significant drop in viral load in all animals (n = 6) after 6 wk of continuous treatment (*P < 0.05, Wilcoxon signed rank test, two-tailed). (D) Cell-associated HIV-1 DNA per 106 human peripheral blood mononuclear cells (PBMC) over time. Each line represents a single mouse from the same experiment as in A–C. (E) Fold change in cell-associated HIV-1 DNA (shown in D) from the baseline measurement for each animal. Red line shows the geometric mean change in cell-associated HIV-1 DNA levels at each time point. (F) Combination immunotherapy resulted in a significant drop in cell-associated HIV-1 DNA in all animals after 6 wk of continuous treatment (*P < 0.05, Wilcoxon signed rank test, two-tailed).
Cell-associated HIV-1 DNA is an additional measure of infection that may be more sensitive than circulating viral load (33, 34). To determine the effects of immunotherapy on cell-associated HIV-1, we measured total HIV-1 DNA in human lymphoid cells obtained from the blood by quantitative PCR (Fig. 1 D–F). The average starting HIV-1 DNA content in circulating lymphoid cells was 4.6 log10 DNA copies per 106 human cells (geometric average). Although cellular HIV-1 DNA content transiently increased in some mice during the treatment course, all mice had significantly reduced levels of cellular HIV-1 DNA after 6 wk of treatment, with an average reduction of 0.8 log10 DNA copies per 106 cells. Lymphoid tissues showed levels of viral DNA similar to those found in the blood after therapy (Fig. S3). Taken together, these data demonstrate that immunotherapy with 3BNC117, PG16, and 10-1074 successfully controls HIV-1 infection in hu-mice by reducing both plasma viral RNA and cell-associated viral DNA.
We next sought to test whether highly potent bNAbs can prolong suppression of HIV-1 infection in mice treated initially with ART. As expected from studies in humans (35, 36) and hu-mice (37), treatment of HIV-1–infected hu-mice with ART alone (tenofovir disproxil-fumarate, raltegravir, and emtricitabine, daily by oral gavage) lowered viremia by an average of 2.0 log10 copies per milliliter after 3 wk, but viral load rebounded to pretreatment levels shortly after ART was discontinued (Fig. S4). To test whether immunotherapy could prevent viral rebound after stopping ART, we treated mice with ART for 3 wk and added a bNAb mixture (45-46G54W, PG16, and 10-1074, 0.5 mg each, twice weekly s.c.) 5 d after starting ART (Fig. 2). Immunotherapy was continued for an additional 4 wk after ART was terminated. In contrast to mice treated with ART alone (Fig. S4), mice treated with a bNAb trimix did not exhibit viral rebound upon stopping ART (Fig. 2A). Although transient low-level viremia (<104 copies per milliliter) was observed in some mice (4/7), viremia in all cases returned below the limit of detection in the presence of antibody even after immunotherapy was stopped (Fig. 2 A and B).
Fig. 2.

Termination of ART in the presence of combined immunotherapy. (A) Plasma viral loads. HIV-1YU2–infected hu-mice were treated with ART beginning at day 0 for a total of 3 wk. Five days after starting ART, mice were additionally treated with combination immunotherapy using bNAbs 45-46G54W, 10-1074, and PG16 (0.5 mg each twice weekly s.c.). ART was terminated and combination immunotherapy was continued for an additional 4 wk, then withdrawn. Each line represents a single mouse; viral load measurements were taken at the indicated time points (symbols). Blue shading, ART only; purple shading, overlapping ART and immunotherapy; pink shading, immunotherapy only. Green line, as in Fig. 1A. (B) Individual mice from A after antibody therapy was stopped. Each plot shows a single animal. Blue lines/symbols, plasma viral load; red lines/symbols, gp120-binding human IgG in plasma. (C) Mutations in gp120 cloned from rebound viral RNA after antibody withdrawal. Each line represents a single clone; red and green marks indicate nonsynonymous and synonymous mutations relative to HIV-1YU2, respectively. (D) TZM-bl neutralization IC50 values for the indicated bNAbs against pseudovirus carrying representative mutations found in gp120 clones from C for each indicated animal.
Viral load was monitored for an additional 12 wk after stopping antibody therapy. In all cases, viral loads rebounded to pretreatment levels only when serum antibody titers were low or undetectable (Fig. 2B). To test whether viremic control after stopping ART was in fact due to antibody therapy, we obtained gp120 sequences from plasma virus that emerged after antibody titers had dropped. Although some viral clones carried mutations at sites known to confer resistance to one of the three antibodies in the trimix, no clones were identified bearing resistance to all three antibodies (Fig. 2C). Representative gp120 sequences from each mouse were tested for resistance to each of the antibodies in the trimix and to the trimix itself (Fig. 2D). In all cases, the viruses remained sensitive to neutralization by the trimix, indicating that antibody therapy was responsible for viremic control after ART was discontinued. We conclude that immunotherapy with 45-46G54W, PG16, and 10-1074 protects against viral rebound after discontinuation of ART.
Immunotherapy with a single bNAb alone is ineffective in suppressing viremia in hu-mice owing to rapid emergence of antibody-resistant viral mutants (ref. 23 and Fig. S1). Because immunotherapy with three antibodies was fully protective against viral rebound in ART-treated mice, we asked whether single bNAbs could also protect against viral rebound when viral load was initially suppressed with the help of ART. We administered ART and added one of four bNAbs (45-46G54W, 3BNC117, 10-1074, or PG16) for 3–4 wk then terminated ART and continued bNAb monotherapy alone (Fig. 3). In contrast to hu-mice receiving antibody monotherapy alone (ref. 23 and Fig. S1), infection remained controlled in 50–86% of the hu-mice receiving 45-46G54W (8/10), 3BNC117 (4/8), or 10-1074 (6/7) monotherapy after ART was terminated (Fig. 3). As expected, viremia rebounded quickly in mice that did not receive continued bNAb therapy after stopping ART (Fig. S5). The combination of PG16 plus ART differed from the other antibody monotherapy groups in that only 20% of the mice (1/5) remained controlled after ART termination (Fig. 3).
Fig. 3.
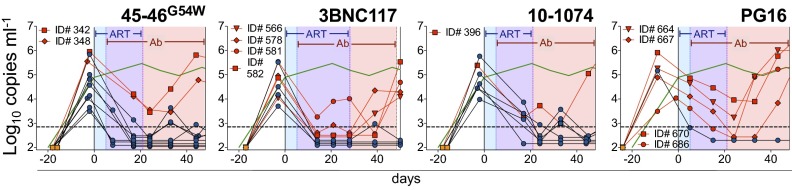
Combination of ART and single antibodies. Viral load measurements (symbols) from mice treated with ART and one of four monoclonal bNAbs are shown. Each line reflects a single mouse. Treatment was initiated at day 0 for all groups. Mice were treated for 5 d with ART alone and then with the combination of ART and the indicated antibody for 3 wk (45-46G54W, 10-1074, or PG16) or 4 wk (3BNC117). ART was then discontinued and bNAb monotherapy continued for an additional 3 wk (3BNC117) or 4 wk (45-46G54W, 10-1074, and PG16). Shading as in Fig. 2A; blue symbols/black lines, mice that remain controlled during immunotherapy after ART termination; red symbols/lines, mice that escape bNAb therapy; green line, as in Fig. 1A.
Plasma viremia was monitored for an additional 11–13 wk after termination of bNAb monotherapy, or until viral rebound was observed. Mice that were controlled by bNAb monotherapy exhibited viremic rebound (>3 × 103 copies per milliliter) after an average of 30 d for 45-46G54W, 43 d for 3BNC117, 45 d for 10-1074, and 35 d in the one mouse that was controlled by PG16 (Fig. 4). The time to viremic rebound for each treatment group correlated with antibody half-life, with all mice rebounding when serum antibody levels were low or undetectable (Fig. 4 and ref. 23). Three out of 17 mice failed to rebound after antibody levels became undetectable, indicating that prolonged viremic control was primarily due to the long half-life of the antibodies. We conclude that lowering the initial viral load with combined ART and immunotherapy facilitates viremic control by subsequent bNAb monotherapy alone in hu-mice.
Fig. 4.

Viremia in hu-mice after stopping antibody therapy. Mice controlled with antibody treatment (Fig. 3) were followed for up to 80 d after stopping antibody injection. Each graph represents a single mouse. Blue lines, viral load; maroon lines, plasma antibody concentration; red shading, end of antibody treatment period. Viral rebound occurred in nearly all cases when antibody concentrations were low or undetectable, with one exception (ID number 399, viral load shown in red line). Three of 17 mice failed to rebound (viral load >3 × 103 copies per milliliter).
To determine whether viral escape from antibody monotherapy was due to specific mutations in the HIV-1 envelope glycoprotein gene (env), we cloned and sequenced gp120 from HIV-1YU2–infected mice that rebounded during (Fig. 5A) or after (Fig. 5B) bNAb immunotherapy. Sequences cloned from mice that rebounded during antibody therapy always carried specific mutations in the antibody target sites, and these mutations rendered them antibody-resistant (Fig. 5A and Figs. S6 and S7). For example, escape from 45-46G54W and 10-1074 was associated with YU2A281T and YU2N332K, which are resistant to those respective antibodies (Fig. 5A and Figs. S6 and S7 and ref. 23). Similarly, mice that escaped 3BNC117 carried resistance mutations in the CD4bs at positions YU2(279–281) or YU2(458/459) (Fig. 5A and Figs. S6 and S7 and refs. 17 and 23), and PG16 escape viruses carried mutations at either YU2N160 or YU2T162, which remove the key N-linked glycosylation site targeted by this antibody (Fig. 5A and Figs. S6 and S7 and refs. 22 and 23). In contrast, viruses that emerged after immunotherapy was terminated did not contain antibody resistance mutations (with one exception, ID number 399) and remained sensitive to neutralization by the antibodies (Fig. 5B and Figs. S7 and S8). Thus, bNAb monotherapy alone can sustain viremic suppression in hu-mice when the viral load is initially lowered by combined ART and immunotherapy.
Fig. 5.

Viral gp120 sequences during and after immunotherapy. (A) Mutations in plasma virus gp120 sequences cloned from mice that escaped antibody therapy after ART was stopped (Fig. S6). Each line represents a single clone; red and green marks as in Fig. 2C. All escape variants suffered mutations at sites known to confer resistance to the respective antibody (highlighted in blue vertical shading; bold numbers refer to the HXBc2-aligned residue). (B) Mutations in gp120 sequences cloned from mice that rebounded after antibody levels became subtherapeutic did not contain mutations at sites known to confer resistance, with the exception of ID number 399 (Fig. S8).
Adeno-associated virus (AAV) vectors can direct long-term expression of anti–HIV-1 antibodies at sufficiently high levels to protect macaques from simian-HIV or hu-mice from HIV-1 infection (38, 39). To determine whether administration of antibody monotherapy by AAV can also achieve prolonged control of established HIV-1 infection, we treated mice with combined ART and AAVs directing the expression of either 3BNC117 or 10-1074. ART treatment interfered with AAV transduction (Fig. S9), so the viral load was initially lowered by treatment with ART and soluble biotin-labeled 10-1074 antibody (10-1074:bio, Fig. 6). Hu-mice with viral loads below the limit of detection 12 d after stopping ART were injected with 2.5 × 1011 genomic copies of a 10-1074–expressing AAV (AAV10-1074) and passive delivery of 10-1074:bio was stopped (Fig. 6A). Consistent with the half-life of 10-1074 (23), 10-1074:bio dropped to undetectable levels 4–7 wk after the last injection (Fig. 6B). However, AAV10-1074 sustained high levels of circulating 10-1074 (ca. 200 mg/mL) throughout the entire 67-d observation period (Fig. 6B). Of seven hu-mice treated with AAV10-1074, only one escaped antibody monotherapy (ID number 683), whereas the remaining six mice (86%) were controlled for the duration of the experiment (Fig. 6). Gp120 sequencing of virus from the mouse that escaped revealed mutations in the 10-1074 binding site that conferred resistance to that antibody (Fig. 6 C and D). Similar results were obtained for mice treated with AAV3BNC117 (Fig. S9). We conclude that a single injection of an AAV-encoded bNAb can durably control viremia in HIV-1–infected hu-mice.
Fig. 6.

AAV-directed immunotherapy. (A) HIV-1YU2–infected hu-mice were treated first with ART for 5 d and then with ART plus biotinylated 10-1074 antibody for 16 d (10-1074:bio, 0.5 mg twice per week s.c.). ART was then terminated and 10-1074:bio continued for an additional 12 d. Mice with viral loads below the limit of detection (solid blue or red symbols/lines) 12 d after stopping ART were injected with 2.5 × 1011 genomic copies of a 10-1074–expressing AAV (AAV10-1074) and passive 10-1074:bio was stopped. Viral loads were monitored for an additional 67 d. Only one of seven mice escaped AAV10-1074 therapy, ID number 683. Open symbols/dashed lines, mice above the limit of viral load detection 12 d after stopping ART; green line, as in Fig. 1A. (B) Individual mice treated with AAV10-1074. Each graph represents a single mouse. Blue or red lines/symbols, viral load; yellow lines/symbols, plasma 10-1074 concentration measured by gp120-specific anti-human IgG1 ELISA. Asterisks reflect the time point at which 10-1074:bio was no longer detectable on a gp120-specific anti-biotin ELISA. (C) Mutations in gp120 sequences cloned from the one mouse that escaped AAV10-1074. Each line represents a single clone; red and green marks as in Fig. 2C. Consensus mutations were found at two residues in the 10-1074 binding site (highlighted in blue vertical shading; bold numbers refer to the HXBc2-aligned residue). (D) Pseudovirus neutralization by 10-1074 of HIV-1YU2 control and mutant virus from mouse ID number 683 that escaped 10-1074 immunotherapy.
Whereas individual antiretroviral drugs interfere with the HIV-1 protease, reverse transcriptase, integrase, or viral entry, antibodies may affect infection by concurrently interfering with the virus in a number of different ways. For example, antibodies can block infection by free virions, but they can also direct the killing of infected cells that express cell surface gp160 or its fragments by antibody-dependent cell-mediated cytotoxicity/antibody-dependent cell-mediated viral inhibition (ADCC/ADCVI) (40, 41). In addition, antibodies may be able to block synapse formation and subsequent cell contact-mediated viral transmission (42–44). To determine whether the antibodies used in this study can interfere with cell contact-mediated transmission of HIV-1YU2, we compared their activity in established cell-free and cell contact-permitting transmission assays (45) in vitro (Fig. S10). All antibodies blocked cell-free virus transmission at the expected concentrations. In addition, the bNAbs 45-46G54W, 3BNC117, and 10-1074 blocked transmission of HIV-1YU2 under cell contact-permitting conditions at concentrations similar to those required for viremic suppression in vivo, each reaching IC50s ≤3 μg/mL and IC80s ≤15 μg/mL. PG16, which was the least effective of the antibodies in vivo, was unable to reach a consistent IC50 against HIV-1YU2 in this assay (Fig. S10 C and D). These data suggest that highly potent bNAbs can passively block cell contact-mediated viral transmission, a property that may be an important determinant of antibody therapeutic efficacy in hu-mice. Whether other bNAb-specific effects, such as ADCC or viral particle clearance, are additional mechanisms that contribute to HIV-1 therapy must be further explored.
Discussion
Monoclonal antibodies have become important components of the clinical armamentarium in treating malignancies and inflammation, but they are rarely used to treat infection.
Combinations of antibodies were tested in HIV-1–infected hu-mice and humans, but the results were discouraging, leading to the conclusion that antibodies could not be effective in treating this disease (11–13). Early experiments in hu-mice showed that treatment with a mixture of three antibodies had little apparent effect on viral load and led to rapid selection for resistant variants (11). In two human trials, antibody therapy following ART interruption showed only modest effects (12, 13). In one well-controlled study (12), eight chronically infected patients underwent scheduled ART interruption to determine the baseline kinetics and magnitude of viral rebound. The same patients were subsequently treated with a mixture of three bNAbs after ART was stopped. Only one out of eight showed prolonged viremic control during bNAb therapy (12).
An important caveat of both human trials is that only one of the three antibodies used (2G12) exerted any discernible pressure on the virus, and these studies were performed using antibodies that are far less potent than those now available. We have reinvestigated antibody therapy using recently discovered highly potent bNAbs in the context of ART interruption in hu-mice. In contrast to prior studies, our experiments in hu-mice indicate that immunotherapy can protect against viremic rebound after discontinuing ART. Surprisingly, even single antibodies were able to sustain viral suppression when the viral load was initially lowered with the aid of transient ART.
Experiments in macaques and in mice suggest that a single injection of a bNAb-encoding AAV can direct long-term expression of anti–HIV-1 antibodies and afford durable protection from HIV-1 challenge (38, 46). Whereas these studies highlight the utility of such vectors for passive vaccination, we show that bNAb-encoding AAVs can also be used in the context of HIV-1 therapy in hu-mice. Although it remains to be seen whether the AAV platform will be useful for long-term antibody therapy in humans, the idea that antibodies might comprise an inexpensive, single-shot durable therapeutic should be further explored.
Antibodies may control established HIV-1 infection by a variety of mechanisms that differ from most ART drugs. These include, among others, facilitating clearance of viral particles, decreasing the half-life of infected cells through effector signaling (ADCC/ACDVI), inhibiting infection by free virions, and blocking cell contact-mediated viral spread (as we show in vitro for 45-46G54W, 3BNC117, and 10-1074). That we observe declining levels of cell-associated HIV-1 DNA during immunotherapy is consistent with decreasing infected cell half-life and blocking viral spread, although additional experiments are required to dissect these mechanisms further. Because the antiviral mechanisms that underlie bNAb immunotherapy likely differ from those of ART drugs, the two modalities may prove complementary when used in combination, particularly in protocols that attempt viral eradication.
Hu-mice differ from humans in a number of important ways. For example, because of size differences the total viral load in hu-mice, and therefore the viral swarm diversity, is far smaller than in humans. Moreover, the virologic reservoirs in hu-mice have not been defined and are likely to differ from those found in humans. Unlike humans, hu-mice are not immunologically competent. They lack an effective innate immune compartment harboring functional effector cells, and they are unable to produce robust T- or B-cell immune responses. In immune competent hosts, each of these immunological components might amplify the effects of bNAb immunotherapy in suppressing HIV-1 infection.
In conclusion, immunotherapy using combinations of broadly neutralizing antibodies achieves viremic control by reducing both plasma viral load and cell-associated HIV-1 DNA. In mice treated initially with ART drugs, combination immunotherapy also protects against viral rebound after discontinuation of ART. Furthermore, single bNAbs given either as soluble protein or by AAV injection can durably sustain viremic control when the viral load is initially lowered by combining ART and immunotherapy. Whether these effects will also be seen in HIV-1–nfected individuals can only be determined in clinical trials.
Materials and Methods
Humanized Mice.
NOD Rag1−/−Il2rgnull (NOD.Cg-Rag1tm1Mom Il2rgtm1Wjl/SzJ) mice were purchased from The Jackson Laboratory and bred and maintained at the Comparative Bioscience Center of The Rockefeller University according to guidelines established by the Institutional Animal Committee. All experiments were performed with authorization from the Institutional Review Board and the Institutional Animal Care and Use Committee at The Rockefeller University. Hu-mice were produced as described (23). Briefly, neonatal mice (1–5 d of age) were sublethally irradiated and injected intrahepatically with purified CD34+ human hematopoietic stem cells derived from fetal liver tissue.
HIV-1 Infection.
Hu-mice were screened at 8 wk of age for reconstitution of human lymphocytes as earlier described (23). Mice with a measurable human graft were injected intraperitoneally with infectious HIV-1YU2 and screened for viremia at 2 to 3 wk postinfection by quantitative reverse-transcriptase PCR (23).
Antiretroviral Therapy.
Individual tablets of tenofovir disproxil-fumarate (TDF; Gilead Sciences), emtricitabine (FTC; Gilead Sciences), raltegravir (RAL; Merck), and efavirenz (EFV; Bristol-Myers Squibb) were crushed into fine powder form using a mortar and pestle and suspended in PBS. ART preparations were aliquotted into 200-μL doses in sterile Eppendorf tubes and stored at −20 °C until use. TDF, FTC, RAL, and EFV were administered by daily oral gavage at 2.46, 1.48, 1.23, and 2.46 mg per mouse, respectively, based on effective doses reported in the literature (47, 48) or human-to-animal dose translation studies (49).
Antibody Therapy.
Monoclonal preparations of bnAbs were isolated in Tris-glycine using Protein G Sepharose 4 Fast-Flow (GE Healthcare) from tissue-culture supernatant of HEK 293T (American Type Culture Collection) or HEK 293E cells transfected with plasmids encoding bnAb heavy- and light-chain Ig genes then buffer-exchanged with PBS and sterile-filtered using Ultrafree-CL centrifugal filters (0.22 μm; Millipore). Purified IgG (10.5 or 1.0 mg) was injected s.c. into hu-mice twice per week or as described.
Adeno-Associated Virus Vectors.
AAV vectors encoding the bnAbs 3BNC117 and 10-1074 were produced as described (50). Briefly, single-stranded AAV genomes carrying AAV serotype 2 inverted terminal repeats contained a human thyroglobulin promoter driving the expression of Ig heavy- and light-chain genes separated by a furin-2A cleavage motif. Vectors were packaged with the muscle- and liver-tropic AAV serotype 8 capsid protein and titered by quantitative PCR (50).
HIV-1 Plasma Viral Load.
Viral load was determined using one of two methods. Viral loads for Fig. S2 were measured using the Abbott HIV-RealTime assay on the ABBOTT M2000 platform. HIV was automatically extracted with the M2000SP using 100–400 µL plasma. In case of lower volumes, these were diluted with PBS and the calculated numbers were adjusted according to the dilution. Amplification and detection were performed with the ABBOTT M200RT. For all other experiments, viral load was measured using an in-house quantitative RT-PCR assay as described (23).
Cell-Associated HIV-1 DNA.
Mononuclear cells were isolated from mouse peripheral blood by Ficoll density gradient, or from peripheral lymph nodes and spleen by tissue homogenization and cell filtration. Homogenized spleen samples were briefly subjected to ACK lysis (Gibco) to remove erythrocytes. Total DNA was isolated using the QIAmp DNA blood mini kit (Qiagen) and eluted in a volume of 80 μL. Purified DNA was assayed by quantitative reverse-transcriptase PCR using the MX3005P QPCR system (Agilent Technologies). HIV-1–specific primers and the probe targeting the 5′-LTR were identical to those used for viral load quantitation. In some cases, a second set of HIV-1–specific primers targeting a highly conserved region in pol were used because they permitted an approximately fivefold higher sensitivity (forward primer 5′-TAATGGCAGCAATTTCACCA-3′, reverse primer 5′- GAATGCCAAATTCCTGCTTGA-3′, internal probe 5′-/5HEX/CCCACCAAC/ZEN/ARGCRGCCTTAACTG/3IABkFQ/-3′). To measure the number of cells in each sample, extracted samples were assayed in separate reactions for human CCR5 genomic DNA using the forward primer 5′-GTTGGACCAAGCTATGCAGGT-3′ and reverse primer 5′-AGAAGCGTTTGGCAATGTGC-3′ with the internal probe 5′-/5HEX/TTGGGATGA/ZEN/CGCACTGCTGCATCAACCCCA/3IABkFQ/-3′. All quantitative PCR (qPCR) reactions contained 25ul AmpliTaq Gold PCR master mix (Applied Biosystems), purified DNA extract, and nuclease-free water up to 50ul, with the following primer and probe concentrations: 450nM forward and reverse primers with 125nM probe (HIV-1 assays); 150nM forward and reverse primers with 41.5nM probe (CCR5 assay). When necessary, purified DNA extract was diluted fivefold in nuclease-free water before qPCR analysis. Reference samples contained an equal mixture of two plasmids, one encoding HIV-1YU2 and another encoding human CCR5, at 5 × 105 plasmid copies each. The lower limit of detection for both HIV-1 qPCR assays was found at 2.8 HIV-1 DNA copies per reaction, corresponding to 56 copies per sample for the LTR-specific primers and 12 copies per sample for the pol-specific primers. The lower limit of detection for the CCR5 qPCR assay was 5.8 copies per reaction, corresponding to 58 cells per sample (based on two copies per cell).
Cell-Associated HIV-1 RNA.
Total RNA was isolated from a sample volume equivalent to that used for extraction of cellular HIV DNA, using the QIAmp MinElute Virus Spin Kit (Qiagen) in combination with the Qiagen RNase-free DNase kit and eluted in a volume of 50 μL. RNA was quantified by quantitative RT-PCR, as was done for viral load. Cell-associated HIV-1 RNA was reported as RNA copies per 106 human cells based on the ratio of HIV-1 RNA copies per sample to CCR5 genomic DNA copies per equivalent sample measured in DNA extract.
Antibody Levels.
Total and gp120-specific human IgG1 levels were measured from mouse plasma by ELISA as described (23).
HIV-1 Genome Sequencing.
Viral sequencing for the HIV-1 env gene encoding gp120 was performed as described (23).
Pseudovirus Neutralization.
Antibody neutralization testing of pseudoviruses carrying the env sequences of HIV-1 isolates from hu-mice was performed by TZM-bl assay as described (23). Pseudovirus molecular clones were generated by insertion of env sequences cloned from HIV-1 infected hu-mice into the KpnI/MfeI restriction sites replacing the sequence for wild-type YU2 in the pSVIIIenv pseudovirus vector used previously (23).
Statistical Analysis.
Statistical analyses were performed using GraphPad Prism 5.0a for Mac OS X.
Supplementary Material
Acknowledgments
We thank Caroline Eden for protein production and immunoassays; Alexander Abadir, Han Gao, and Xiying Fan for protein production; and Reha-Baris Incesu for hu-mouse screening. We thank Marcus Dorner, Eva Billerbeck, Rachael N. Labitt, Chase Budell, Tamar Friling, Kevin Vega, and Brenna Flatley for assistance with hu-mouse production. F.K. was supported by the Stavros Niarchos Foundation. E.B., M.D., A.P., and C.M.R. were supported by the Starr Foundation. O.S. is supported by grants from the Agence Nationale de Recherche sur le Sida, Sidaction, AREVA Foundation, Vaccine Research Institute, the Labex Integrative Biology of Emerging Infectious Diseases program, the Seventh Framework Programme HIT Hidden HIV (Health-F3-2012-305762), and Institut Pasteur. This work was supported in part by the Bill and Melinda Gates Foundation with Comprehensive Antibody Vaccine Immune Monitoring Consortium Grant 1032144 (to M.S.S.) and Collaboration for AIDS Vaccine Discovery Grants 38660 (to P.J.B.) and 38619s (to M.C.N.). This work was also supported by the German Center for Infection Research (S.G. and H.B.), UL1 TR000043 Translational Science Award (Clinical and Translational Science Award) program, AI 100663-01 (to M.C.N.), Center for HIV/AIDS Vaccine Immunology and Immunogen Discovery, and AI 100148-01 (to P.J.B. and M.C.N.). P.J.B. and M.C.N. are Howard Hughes Medical Institute Investigators.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315295110/-/DCSupplemental.
References
- 1.Molina J-M, et al. CASTLE Study Team Once-daily atazanavir/ritonavir compared with twice-daily lopinavir/ritonavir, each in combination with tenofovir and emtricitabine, for management of antiretroviral-naive HIV-1-infected patients: 96-week efficacy and safety results of the CASTLE study. J Acquir Immune Defic Syndr. 2010;53(3):323–332. doi: 10.1097/QAI.0b013e3181c990bf. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett JA, DeMasi R, Quinn J, Moxham C, Rousseau F. Overview of the effectiveness of triple combination therapy in antiretroviral-naive HIV-1 infected adults. AIDS. 2001;15(11):1369–1377. doi: 10.1097/00002030-200107270-00006. [DOI] [PubMed] [Google Scholar]
- 3.Arts EJ, Hazuda DJ. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med. 2012;2(4):a007161. doi: 10.1101/cshperspect.a007161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamlyn E, et al. INSIGHT SMART and SPARTAC Investigators Plasma HIV viral rebound following protocol-indicated cessation of ART commenced in primary and chronic HIV infection. PLoS ONE. 2012;7(8):e43754. doi: 10.1371/journal.pone.0043754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sigal A, et al. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature. 2011;477(7362):95–98. doi: 10.1038/nature10347. [DOI] [PubMed] [Google Scholar]
- 6.Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012;37(3):377–388. doi: 10.1016/j.immuni.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deeks SG, et al. International AIDS Society Scientific Working Group on HIV Cure Towards an HIV cure: A global scientific strategy. Nat Rev Immunol. 2012;12(8):607–614. doi: 10.1038/nri3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mascola JR, et al. Protection of Macaques against pathogenic simian/human immunodeficiency virus 89.6PD by passive transfer of neutralizing antibodies. J Virol. 1999;73(5):4009–4018. doi: 10.1128/jvi.73.5.4009-4018.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shibata R, et al. Neutralizing antibody directed against the HIV-1 envelope glycoprotein can completely block HIV-1/SIV chimeric virus infections of macaque monkeys. Nat Med. 1999;5(2):204–210. doi: 10.1038/5568. [DOI] [PubMed] [Google Scholar]
- 10.Moldt B, et al. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc Natl Acad Sci USA. 2012;109(46):18921–18925. doi: 10.1073/pnas.1214785109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poignard P, et al. Neutralizing antibodies have limited effects on the control of established HIV-1 infection in vivo. Immunity. 1999;10(4):431–438. doi: 10.1016/s1074-7613(00)80043-6. [DOI] [PubMed] [Google Scholar]
- 12.Trkola A, et al. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med. 2005;11(6):615–622. doi: 10.1038/nm1244. [DOI] [PubMed] [Google Scholar]
- 13.Mehandru S, et al. Adjunctive passive immunotherapy in human immunodeficiency virus type 1-infected individuals treated with antiviral therapy during acute and early infection. J Virol. 2007;81(20):11016–11031. doi: 10.1128/JVI.01340-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scheid JF, et al. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature. 2009;458(7238):636–640. doi: 10.1038/nature07930. [DOI] [PubMed] [Google Scholar]
- 15.McCoy LE, Weiss RA. Neutralizing antibodies to HIV-1 induced by immunization. J Exp Med. 2013;210(2):209–223. doi: 10.1084/jem.20121827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moir S, Malaspina A, Fauci AS. Prospects for an HIV vaccine: Leading B cells down the right path. Nat Struct Mol Biol. 2011;18(12):1317–1321. doi: 10.1038/nsmb.2194. [DOI] [PubMed] [Google Scholar]
- 17.Diskin R, et al. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science. 2011;334(6060):1289–1293. doi: 10.1126/science.1213782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walker LM, et al. Protocol G Principal Investigators Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477(7365):466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mouquet H, et al. Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc Natl Acad Sci USA. 2012;109(47):E3268–E3277. doi: 10.1073/pnas.1217207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X, et al. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science. 2010;329(5993):856–861. doi: 10.1126/science.1187659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheid JF, et al. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science. 2011;333(6049):1633–1637. doi: 10.1126/science.1207227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker LM, et al. Protocol G Principal Investigators Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326(5950):285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein F, et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature. 2012;492(7427):118–122. doi: 10.1038/nature11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diskin R, et al. Restricting HIV-1 pathways for escape using rationally designed anti-HIV-1 antibodies. J Exp Med. 2013;210(6):1235–1249. doi: 10.1084/jem.20130221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simek MD, et al. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol. 2009;83(14):7337–7348. doi: 10.1128/JVI.00110-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gray ES, et al. CAPRISA002 Study Team The neutralization breadth of HIV-1 develops incrementally over four years and is associated with CD4+ T cell decline and high viral load during acute infection. J Virol. 2011;85(10):4828–4840. doi: 10.1128/JVI.00198-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liao HX, et al. NISC Comparative Sequencing Program Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013;496(7446):469–476. doi: 10.1038/nature12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu X, et al. NISC Comparative Sequencing Program Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333(6049):1593–1602. doi: 10.1126/science.1207532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mouquet H, et al. Memory B cell antibodies to HIV-1 gp140 cloned from individuals infected with clade A and B viruses. PLoS ONE. 2011;6(9):e24078. doi: 10.1371/journal.pone.0024078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burton DR, et al. A Blueprint for HIV Vaccine Discovery. Cell Host Microbe. 2012;12(4):396–407. doi: 10.1016/j.chom.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLellan JS, et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature. 2011;480(7377):336–343. doi: 10.1038/nature10696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baenziger S, et al. Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2-/-gamma c-/- mice. Proc Natl Acad Sci USA. 2006;103(43):15951–15956. doi: 10.1073/pnas.0604493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goujard C, et al. (2006) CD4 cell count and HIV DNA level are independent predictors of disease progression after primary HIV type 1 infection in untreated patients. Clin Infect Dis 42(5):709–715. [DOI] [PubMed]
- 34.Pasternak AO, Lukashov VV, Berkhout B. Cell-associated HIV RNA: A dynamic biomarker of viral persistence. Retrovirology. 2013;10:41. doi: 10.1186/1742-4690-10-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.García F, et al. Dynamics of viral load rebound and immunological changes after stopping effective antiretroviral therapy. AIDS. 1999;13(11):F79–F86. doi: 10.1097/00002030-199907300-00002. [DOI] [PubMed] [Google Scholar]
- 36.Harrigan PR, Whaley M, Montaner JS. Rate of HIV-1 RNA rebound upon stopping antiretroviral therapy. AIDS. 1999;13(8):F59–F62. doi: 10.1097/00002030-199905280-00001. [DOI] [PubMed] [Google Scholar]
- 37.Nischang M, et al. Humanized mice recapitulate key features of HIV-1 infection: A novel concept using long-acting anti-retroviral drugs for treating HIV-1. PLoS ONE. 2012;7(6):e38853. doi: 10.1371/journal.pone.0038853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Balazs AB, et al. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature. 2012;481(7379):81–84. doi: 10.1038/nature10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson PR, et al. Vector-mediated gene transfer engenders long-lived neutralizing activity and protection against SIV infection in monkeys. Nat Med. 2009;15(8):901–906. doi: 10.1038/nm.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mascola JR, Montefiori DC. The role of antibodies in HIV vaccines. Annu Rev Immunol. 2010;28:413–444. doi: 10.1146/annurev-immunol-030409-101256. [DOI] [PubMed] [Google Scholar]
- 41.Overbaugh J, Morris L. The antibody response against HIV-1. Cold Spring Harb Perspect Med. 2012;2(1):a007039. doi: 10.1101/cshperspect.a007039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Abela IA, et al. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog. 2012;8(4):e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang MY, et al. Potent and broad neutralizing activity of a single chain antibody fragment against cell-free and cell-associated HIV-1. MAbs. 2010;2(3):266–274. doi: 10.4161/mabs.2.3.11416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martin N, et al. Virological synapse-mediated spread of human immunodeficiency virus type 1 between T cells is sensitive to entry inhibition. J Virol. 2010;84(7):3516–3527. doi: 10.1128/JVI.02651-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sourisseau M, Sol-Foulon N, Porrot F, Blanchet F, Schwartz O. Inefficient human immunodeficiency virus replication in mobile lymphocytes. J Virol. 2007;81(2):1000–1012. doi: 10.1128/JVI.01629-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Girard MP, Picot V, Longuet C, Nabel GJ. Report of the Cent Gardes HIV Vaccine Conference: The B-cell response to HIV. Part 1: Broadly neutralizing antibodies. Fondation Mérieux Conference Center, Veyrier du Lac, France, 5–7 November 2012. Vaccine. 2013;31(29):2979–2983. doi: 10.1016/j.vaccine.2013.02.068. [DOI] [PubMed] [Google Scholar]
- 47.Denton PW, et al. Generation of HIV latency in humanized BLT mice. J Virol. 2012;86(1):630–634. doi: 10.1128/JVI.06120-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choudhary SK, et al. Suppression of human immunodeficiency virus type 1 (HIV-1) viremia with reverse transcriptase and integrase inhibitors, CD4+ T-cell recovery, and viral rebound upon interruption of therapy in a new model for HIV treatment in the humanized Rag2-/-gammac-/- mouse. J Virol. 2009;83(16):8254–8258. doi: 10.1128/JVI.00580-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22(3):659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 50.Wang L, et al. Systematic evaluation of AAV vectors for liver directed gene transfer in murine models. Mol Ther. 2010;18(1):118–125. doi: 10.1038/mt.2009.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.