

Significance
Influenza A viruses cause annual influenza epidemics and occasional severe pandemics. Their genome is segmented into eight RNA fragments, which offers evolutionary advantages but complicates genomic packaging. The existence of a selective mechanism ensuring specific packaging of one copy of each RNA into each virion is suspected, but its molecular details remain unknown. We identified a direct interaction between two viral genomic RNA segments of an influenza A virus and demonstrated that this interaction takes place in infected cells, is required for optimal viral replication, and favors copackaging of the interacting RNA segments. Collectively, our experiments indicate that viral RNA segments can promote selective packaging of the influenza A virus genome by forming a sequence-dependent supramolecular network of interactions.
Abstract
Influenza A viruses cause annual influenza epidemics and occasional severe pandemics. Their genome is segmented into eight fragments, which offers evolutionary advantages but complicates genomic packaging. The existence of a selective packaging mechanism, in which one copy of each viral RNA is specifically packaged into each virion, is suspected, but its molecular details remain unknown. Here, we identified a direct intermolecular interaction between two viral genomic RNA segments of an avian influenza A virus using in vitro experiments. Using silent trans-complementary mutants, we then demonstrated that this interaction takes place in infected cells and is required for optimal viral replication. Disruption of this interaction did not affect the HA titer of the mutant viruses, suggesting that the same amount of viral particles was produced. However, it nonspecifically decreased the amount of viral RNA in the viral particles, resulting in an eightfold increase in empty viral particles. Competition experiments indicated that this interaction favored copackaging of the interacting viral RNA segments. The interaction we identified involves regions not previously designated as packaging signals and is not widely conserved among influenza A virus. Combined with previous studies, our experiments indicate that viral RNA segments can promote the selective packaging of the influenza A virus genome by forming a sequence-dependent supramolecular network of interactions. The lack of conservation of these interactions might limit genetic reassortment between divergent influenza A viruses.
Influenza A viruses (IAVs) belong to the Orthomyxoviridae family and cause annual influenza epidemics and occasional pandemics that represent a major threat for human health (1). The IAV genome consists of eight single-stranded negative-sense RNA segments (vRNAs), ranging from 890 to 2,341 nucleotides (nts) and packaged as viral ribonucleoproteins (vRNPs) containing multiple copies of nucleoprotein (NP) and a RNA-dependent RNA polymerase complex (2–4). The central coding region (in antisense orientation) of the vRNAs is flanked by short, segment-specific untranslated regions and conserved, partially complementary, terminal sequences that constitute the viral polymerase promoter and impose a panhandle structure to the vRNPs (4–9). The segmented nature of the IAV genome favors viral evolution by genetic reassortment. This process, which takes place when a single cell is coinfected by different IAVs, can generate pandemic viruses that represent a major threat for human health (1). However, segmentation complicates packaging of the viral genome into progeny virions.
Although it had initially been proposed that the vRNAs are randomly packaged into budding viral particles, several lines of experiment suggest that IAVs specifically package one copy of each vRNA during viral assembly (7). First, electron microscopy and tomography revealed that the relative disposition of the eight vRNPs within viral particles is not random, even though some variability is tolerated, and they adopt a typical arrangement, with seven vRNPs surrounding a central one (10–12). Second, genetic and biochemical analysis revealed that the vast majority of IAV particles contain exactly one copy of each vRNA (7, 13, 14). Third, analysis of defective interfering RNAs (7, 15–17) and reverse genetic experiments (7, 18–25) identified specific bipartite packaging signals, most often located within the ends of the coding regions, in each segment. Of note, the terminal promoters are crucial for RNA packaging (8), but they cannot confer specificity to the packaging process (7).
A selective packaging mechanism requires the existence of direct RNA–RNA or indirect RNA–protein interactions between vRNAs (7). Because all vRNAs associate with the same viral proteins to form vRNPs and no cellular protein has been identified that would specifically recognize an IAV packaging signal, we (10) and others (7, 12, 19) hypothesized that direct interactions between vRNAs might ensure selective packaging. However, these interactions remain elusive. We recently showed that the eight vRNAs of both a human H3N2 IAV (10) and an avian H5N2 IAV (26) form specific networks of intermolecular interactions in vitro, but the functional relevance of these interactions was not demonstrated. Here, we used a biochemical approach to identify, at the nt level, an interaction between two in vitro transcribed vRNAs. Unexpectedly, this interaction occurs between regions not previously identified as packaging signals. We then demonstrated that this interaction is important for infectivity and packaging of the viral genome.
Results
Identification of the Interaction Between vRNA 2 and 8 at the nt Level, in Vitro.
Here, we analyzed the interaction between vRNA 2 [coding for the polymerase basic subunit 1 (PB1), the proapoptotic protein PB1-F2, and a N-terminally truncated form of PB1 named PB1-N40] and vRNA 8 [coding for non structural protein 1 (NS1) and non structural protein 2/ nuclear export protein (NS2/NEP)] of the exemplar avian H5N2 virus A/Finch/England/2051/91. We focused on this particular interaction because it was the strongest among all of the interactions that we previously detected in vitro between vRNAs from the A/Finch/England/2051/91 (26) or the A/Moscow/10/99 (10) viruses. We showed by native agarose gel electrophoresis that these two vRNAs form a complex when synthesized and coincubated in vitro (Fig. 1). Surprisingly, when we deleted the terminal regions of vRNA 8, which are known to contain segment-specific packaging signals (7, 15, 18, 21, 27), this complex was not disrupted (Fig. 1A and Fig. S1A). To identify the sequences involved in this interaction, we first analyzed a series of deletion mutants covering the entire vRNA 8 (Fig. 1 A and B and Fig. S1 A and B) or vRNA 2 (Fig. 1 C1 and C2 and Fig. S1C2), except the highly conserved terminal promoters. Remarkably, in both cases, only one deletion dramatically reduced formation of the complex. In the case of vRNA 8, which is shorter, the deletions were overlapping. As deletion of nts 235–456 strongly reduced the intermolecular vRNA interaction, whereas deletions encompassing nts 57–256 and 435–646 did not, we concluded that the region of vRNA 8 that interacted with vRNA 2 was located between nts 256 and 435 (Fig. 1B, lanes 11–13). The region of vRNA 2 interacting with vRNA 8 was located between nts 125 and 384 (Fig. 1C2).
Fig. 1.

Identification of the nts mediating interaction between vRNAs 2 and 8 in vitro. (A) Analysis by native agarose gel electrophoresis of the interaction between WT vRNAs and the effect of terminal deletions in vRNA 8. (B) Internal deletions in vRNA 8. (C1 and C2) Internal deletions in vRNA 2. (D) Mapping of vRNA 8 with oligos. (E, Upper) Complementarity between vRNAs 2 and 8; (Middle) predicted stem loop structure of the interacting sequences; and (Lower) point mutations introduced in vRNA 2Mut and 8Mut. (F) Oligos targeting vRNAs 2 and 8. (G) Point trans-complementary mutations in vRNAs 2 or/and 8. The intermolecular complex is marked by a large arrowhead. Conditions under which complex formation is reduced are indicated in gray above the gels.
Next, we used a series of oligodeoxyribonucleotides (oligos) spanning region 256–435, delineated by deletion analysis, to define precisely the region of vRNA 8 interacting with vRNA 2 (Fig. 1D and Figs. S1D and S2). Only one oligo, complementary to nts 257–285, dramatically reduced the interaction with vRNA 2, whereas the remaining oligos had no significant effect (Fig. 1D and Fig. S1D), even though they all hybridized to vRNA 8 (Fig. S2). Interestingly, when we looked for possible base pairing between the regions identified experimentally in vRNAs 8 and 2, using the GUUGle software (28), we found that nts 263–276 of vRNA 8 and 290–303 of vRNA 2 are strictly complementary (Fig. 1E). Interestingly, these two sequences might adopt local stem–loop structures that could potentially initiate intermolecular interactions by forming a kissing loop complex (29). In keeping with the proposed interaction, oligos complementary to nts 284–309 of vRNA 2 and to nts 257–285 of vRNA 8 both inhibited the interaction between the two vRNAs (Fig. 1F and Fig. S1F, lanes 4–5). Last, we introduced four trans-complementary point substitutions in the putative interacting sequences (Fig. 1E, Lower). We showed that these substitutions disrupted the interaction between mutant vRNAs and their wild-type (WT) partners (Fig. 1G and Fig. S1G, lanes 6–7). However, the two trans-complementary mutant vRNAs interacted with a similar efficiency to WT vRNAs (Fig. 1G and Fig. S1G, lane 8). Collectively these data establish the existence of an in vitro interaction between nts 290–303 of vRNA 2 and nts 263–276 of vRNA 8.
The Interaction Between vRNAs 2 and 8 Is Required for Optimal Viral Replication.
We next sought to assess the role of the interaction between vRNAs 2 and 8 in IAV replication. Importantly, the mutations introduced in vRNAs 2Mut and 8Mut did not change the amino acid sequence of PB1, PB1-N40, NS1, or NS2/NEP (Fig. S3) and did not significantly affect the synthesis of vRNAs 2 and 8 in infected cells (Table S1). We observed that viruses containing mutations in segment 2 or in segment 8 (named viruses 2Mut and 8Mut, respectively) exhibited replication defects in Madin–Darby canine kidney (MDCK) cells at both 24 and 48 h postinfection (Fig. 2A). On average, their tissue culture infectious dose 50 (TCID50) was reduced more than 10-fold (1.11 ± 0.33 log10) compared with the WT virus. In contrast, the virus containing mutations in both segments (named 2Mut8Mut) replicated at WT levels (Fig. 2A). Similarly, the amount of vRNA 7 (coding for M) was reduced by 1.18 ± 0.21 log10 in the viral supernatant of viruses 2Mut and 8Mut relative to the WT virus, whereas the supernatants of WT and 2Mut8Mut viruses contained similar amounts of vRNA 7 (Fig. 2B). Importantly, viral replication was restored to WT levels when the two mutant vRNAs were combined in the same virus, demonstrating that the interaction between vRNAs 2 and 8 takes place in infected cells (Fig. 2 A and B). These results might reflect either a decrease in viral particle production or reduced vRNA packaging by the 2Mut and 8Mut viruses.
Fig. 2.
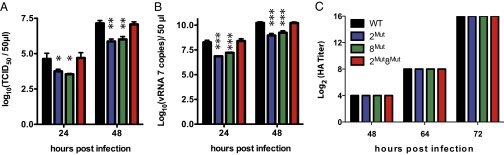
Replication of WT and mutant viruses. (A) Tissue culture infectious dose 50 (TCID50) of WT and mutant viruses. (B) Copy number of vRNA 7 in the supernatant of MDCK cells infected with WT or mutant viruses. Data in A and B were obtained from three independent experiments (biological replicates); each experiment was performed in duplicate (A) or in triplicate (B) (technical replicates). Results are reported as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001. (C) Hemagglutination assays performed on aliquots of viral supernatants. Assays performed in quadruplicate (technical replicates) on two independent series of samples (biological replicates) gave identical results.
Disruption of the Interaction Between vRNAs 2 and 8 Results in Global Packaging Defects.
To discriminate between these possibilities, we compared the HA titer of the WT and mutant viruses (Fig. 2C). Intriguingly, WT and mutant viruses had identical HA titers (Fig. 2C), suggesting that similar amounts of WT and mutant viral particles were produced. To probe the origin of the replication defect, we analyzed cross-sections of budding viruses from three independent experiments by electron microscopy, in which vRNPs appeared as dots inside the viral matrix (10–12) (Fig. 3 and Fig. S4). Only 4.88% (20/410) cross-sections of budding WT viruses from three independent infections appeared empty (Fig. 3A and Fig. S4A). These empty cross-sections might either correspond to viral particles that have not incorporated vRNPs or to viral particles sectioned below the longest vRNPs (10–12). In contrast, 41.40% (231/558) and 42.79% (270/631) of the cross-sections of viruses 2Mut and 8Mut appeared empty, respectively, reflecting a vRNP packaging defect (Fig. 3 B and C and Fig. S4 B and C). Empty viruses frequently appeared as clusters, and some budding sites seemed to produce exclusively empty viral particles (Fig. 3B, Right). Importantly, the vRNA packaging defect was partially restored in virus 2Mut8Mut, as only 18.81% (108/574) cross-sections were empty (Fig. 3D and Fig. S4D). The reason for the incomplete restoration of vRNP packaging in the 2Mut8Mut virus is unclear. We noticed that the point mutations introduced in vRNA 2 yield a truncated form (aa 1–57) of the PB1-F2 protein predicted to be expressed by the A/Finch/England/2051/91 virus. However, this protein has never been implicated in packaging (30).
Fig. 3.

Cross-sections of WT and mutant viruses. WT and mutant viruses were observed by electron microscopy. Cross-sections revealing a complete matrix layer without any dots corresponding to vRNPs inside are circled in red.
To further characterize the effect of the disruption of the interaction between vRNAs 2 and 8 on packaging, we quantified the relative amounts of vRNAs 2, 8, 6, and 7 in the supernatants containing the WT and mutant viruses. Viral RNA 7 codes for the matrix protein M1 and the ion channel M2 and interacts neither with vRNA 2 nor vRNA 8 in vitro (26), whereas vRNA 6 codes for NA and interacts weakly with vRNA 2 in vitro (26). Surprisingly, RT-quantitative (q)PCR revealed only minimal variations of the relative amounts of vRNAs 2, 6, 7, and 8 in the mutant viruses, which did not correlate with their loss in replicative efficiency (Fig. 4). Altogether, these data indicate that disruption of the interaction between vRNAs 2 and 8 has a general, rather than segment-specific, effect on packaging.
Fig. 4.
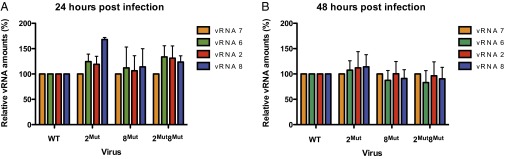
Relative amounts of vRNAs 2, 6, 7, and 8 in WT and mutant viral particles collected 24 and 48 h postinfection. For vRNA quantification, all viruses were normalized relative to the amount of vRNA 7 in the cell culture supernatant, and all vRNAs in the WT virus were set to 100%. Data were obtained from three independent experiments performed in triplicate (n = 3) and are presented as mean ± SEM.
The Interaction Between vRNAs 2 and 8 Favors Copackaging of These vRNAs.
To evaluate the importance of the interaction between vRNAs 2 and 8 on their selective incorporation into viral particles, we performed competition experiments by transfecting 293 T cells with nine reverse genetics plasmids (Table 1). WT and mutant vRNA 2 were allowed to compete for packaging in the presence of either WT or mutant vRNA 8, or vice versa, and the genotype of the progeny viruses was analyzed (Table 1). In these experiments, production of viral particles was strongly biased toward WT and double-mutant viruses, implying that vRNAs that were able to interact were preferentially copackaged. We further validated these conclusions by introducing competition between WT and mutant vRNAs 2 or 8 by first transfecting cells with a plasmid coding for WT or mutated segment 2 or 8, followed by infection with WT or mutant virus (Table S2). In line with previous competition experiments, we observed that WT and double-mutant viruses were highly overrepresented [206 (76.9%) of 268 viruses analyzed] among the viral progeny compared with single mutant viruses (Table S2). Altogether, these competition experiments indicate that interactions between vRNAs play a major role in vRNA packaging.
Table 1.
Competition between WT and mutant vRNAs 2 and 8 for packaging into progeny virions
| Experimental conditions and results | Competition no. |
|||
| 1 | 2 | 3 | 4 | |
| Genetic background | 8WT | 2WT | 8Mut | 2Mut |
| Competing vRNAs | 2WT + 2Mut | 8WT + 8Mut | 2WT + 2Mut | 8WT+ 2Mut |
| Most frequent genotype of the progeny viruses | WT | WT | 2Mut8Mut | 2Mut8Mut |
| Fraction of viruses with the most frequent genotype | 65/74 (88%) | 30/30 (100%) | 47/63 (75%) | 91/111 (82%) |
| P* | <0.0001 | <0.0001 | 0.0057 | <0.0001 |
Nine plasmids including WT segments 1 and 3–7 and the combination of WT and mutant segments 2 and 8 indicated in the first two rows of each column were used to transfect 293 T cells.
Probability, for each competition, that the distribution of the two competing vRNAs is random.
Discussion
The involvement of direct intermolecular interactions between vRNAs in their selective packaging into viral particles is a longstanding hypothesis (7, 19). We previously showed that vRNAs from an avian and a human IAV build up complex interaction networks in vitro (10, 26), but the biological relevance of these interactions was not demonstrated. Here, we combined the strengths of biochemical and virology approaches to demonstrate that one of these interactions is required for efficient vRNA incorporation into progeny virions. Thus, this study, together with previous ones (10, 26), provides direct supporting evidence of a mechanism in which one copy of each vRNA is selected and packaged as part of a supramolecular complex held together by direct base-pairing between vRNAs. The model is further supported by electron tomography studies, which revealed multiple contacts between vRNPs (10–12, 26). Small RNA hairpins, such as those in Fig. 1E, might protrude from the regular vRNP scaffold formed by NP to promote these contacts. The periodicity of the NP double helix would then constitute a key factor in the assembly of the vRNP supramolecular complex (2, 3). The interacting sequences are located at similar distances from the 3′ end of vRNAs 2 and 8 and form antiparallel base pairs. A parallel orientation of the vRNPs does not preclude a locally antiparallel orientation of the interacting strands (10); alternatively, some vRNPs might have an antiparallel orientation within virions.
In all IAVs, the eight vRNPs adopt a characteristic arrangement, with seven vRNPs surrounding a central one (10–12, 26). Thus, disrupting a vRNA/vRNA interaction might have different effects, depending on the position of the vRNAs involved and the number of interactions these vRNAs establish with other partners. Mutations in conserved codons that act as packaging signals frequently resulted in altered ratios of vRNAs in progeny virions (25, 31), whereas in other instances, equimolar amounts of vRNAs were packaged, but the overall vRNA content was reduced (32). Here, we detected no perturbation of the ratio of vRNAs 2, 6, 7, and 8 when the interaction between vRNAs 2 and 8 was disrupted, indicating a general packaging defect. This result suggests that assembly of one copy of each vRNP into a single supramolecular assembly might constitute a checkpoint for viral assembly. Indeed, different vRNAs species colocalize during Rab-11 (a small monomeric GTPase)–dependent trafficking in the cytoplasm (33), suggesting that the supramolecular vRNP assembly might form during this process. Disruption of a vRNA/vRNA interaction might destabilize this complex and affect vRNA trafficking.
The general consensus, derived from studies on human H1N1 IAVs (7, 18–25, 34), is that the segment-specific packaging signals reside within the 100 nts at the end of the vRNA coding regions. It is thus surprising that the sequences involved in the interaction between vRNAs 2 and 8 are located more than 250 nts from the vRNA ends. It is possible that previous studies overlooked the presence of packaging signals in more central vRNA regions because they relied on the presence of a central reporter gene (7, 18, 19, 22, 24, 27) or because they focused on the most conserved parts of the vRNAs, which are the terminal regions (21, 23, 25, 32). Indeed, recent tomography studies of IAV revealed multiple interactions between the vRNPs, all along the particles (12, 26). Alternatively, it is conceivable that packaging signals are distributed differently in human and avian IAVs, as the regions of avian and human vRNAs interacting in vitro are different (10, 26).
Although some highly conserved codons function as packaging signals (21, 25, 31, 32), the interacting sequences in vRNAs 2 and 8 of the IAV used in this study are only partially conserved in avian H5N2 IAVs (Fig. S5) and are not conserved in human H1N1 and H3N2 IAVs (Fig. S6). Given the role of this interaction in the copackaging of vRNAs 2 and 8, our study has important implications for genetic reassortment of IAVs, a process that can introduce pandemic viruses in the human population (1). Indeed, the lack of conservation of some of these interactions among IAVs might limit the number of vRNA combinations that are likely to be copackaged into virions, and thus restrict genetic reassortment. In support of this idea, genetically divergent viruses are known to reassort less frequently than genetically similar viruses, and packaging signals have recently been shown to play a crucial role in this process (35, 36). Furthermore, the precise identification of all intermolecular interactions in the supramolecular network formed by influenza A segments could pave the way for the eventual engineering of reassortant influenza viruses for the generation of vaccine seeds.
Materials and Methods
In Vitro Experiments.
Plasmids, cloning, site-directed mutagenesis, and RNA in vitro transcription and purification of vRNAs of the A/Finch/England/2051/91 (H5N2) virus were performed according to standard procedures described in detail elsewhere (26). Interaction between WT or mutant vRNAs 2 and 8 were determined as described in ref. 26, using 2 pmol of each RNA. After native agarose gel electrophoresis, the RNA weight fraction of each band in a lane was determined, and the fraction of intermolecular complex was determined by dividing the weight fraction of the corresponding band by the sum of the weight fractions of all bands in the lane. Oligo-mapping experiments were performed as described in ref. 26.
Cell Culture and Reverse Genetics.
MDCK and 293 T-cell cultures were performed as described (10). The eight vRNA segments from the A/Finch/England/2051/91 (H5N2) virus were cloned into the pHW2000 vector to generate viruses by reverse genetics, as described earlier (37, 38). Briefly, plasmids (1 µg each) were mixed in 100 µL Opti-MEM (Gibco-BRL), together with 2.5 µL/µg plasmid of TransIT-LT1 reagent (Mirus), according to the manufacturer's instructions, and added to 293 T cells in six-well tissue culture plates. Supernatants were harvested 48 h posttransfection, and viruses were amplified twice in 48-well plates by infecting MDCK cells in Eagle's minimal essential medium (EMEM) (Lonza) supplemented with 1 μg/mL TPCK-trypsin (trypsin treated with L-1-Tosylamide-2-phenylethyl chloromethyl ketone; Sigma). The virus stocks were produced by infecting MDCK cells in a T25 flask (4.3 × 106 cells per T25) with 5 mL EMEM supplemented with 1 μg/mL TPCK-trypsin.
Viral Growth Kinetics, End-Point Titration, HA Titers, and Quantification of vRNAs.
To compare the kinetics of replication of WT and mutant viruses, confluent MDCK cells in six-well plates (106 cells per well) were infected at a multiplicity of infection (m.o.i.) of 10−4. After a 1-h viral adsorption period, cells were overlaid with 5 mL EMEM supplemented with 1 μg/mL TPCK-trypsin and further incubated at 34 °C. After collecting samples (300 µL), the EMEM volume was completed to 5 mL. Harvested supernatants were centrifuged at 1,500 × g for 10 min and stored at −80 °C for analysis.
End-point titrations of the collected samples were performed on confluent layers of MDCK cells in 96-well plates, as described (38). Briefly, 50 μL of 10-fold serial dilutions of each virus were inoculated into four replicate wells. The 96-well microplates were incubated at 34 °C, and the presence of cytopathic effects was monitored 3 d later under the microscope. The tissue culture infectious dose 50 per 50 µL values were determined using the Reed and Muench statistical method.
The HA titer of the samples was determined using round-bottomed 96-well plates. Fifty microliters of buffer containing 3.75 mM NaOH, 5 mM KH2PO4, and 1.5 mM NaCl were added in each well, and twofold serial dilutions were performed by mixing 50 µL virus sample to the first well and transferring 50 µL to the next well. After addition of 50 µL of 0.5% hen erythrocytes in the same buffer to each well, the 96-well plates were incubated for 60 min at room temperature. The HA titer is the highest dilution factor that gives a positive reading.
The amounts of vRNAs 2, 6, 7, and 8 in the supernatants were determined by extracting total viral RNA from the samples using QIAamp viral RNA Mini Kits (Qiagen), according to the manufacturer's instructions, including treatment with RNase Free DNase. Quantification was performed by RT-qPCR on a Mastercycler ep realplex 2 (Eppendorf) machine, using the SuperScript III Platinum One-Step qRT-PCR system (Invitrogen) and segment-specific primers and probes, as described previously for vRNA 7 (39). For each sample, reactions contained 5 µL total viral RNA, 0.8 mM MgSO4, 0.27 µM probe, and 0.67 µM forward and reverse primers, in a final volume of 15 µL. Primers and probes for segments 2 and 8 were designed to allow detection of WT and mutant genes. Cycling conditions were 15 min at 45 °C and 3 min at 95 °C, followed by 50 cycles (95 °C for 15 s and 60 °C for 40 s).
To evaluate the effect of mutations in vRNAs 2 and 8 on their replication during a single viral cycle, confluent MDCK cells were infected at a m.o.i. of 5. After virus adsorption for 90 min at 4 °C, cells were washed twice with PBS and overlaid with 4 mL prewarmed EMEM supplemented with 1 μg/mL TPCK-trypsin and incubated for 3 h. After removal of the supernatant, cells were washed with PBS, and total cellular RNA was extracted from cells with RNeasy Mini Kits (Qiagen), as described earlier, including DNase treatment. Total RNA was quantified using a nanodrop ND-1000 (Thermo). RT-qPCR of segments 2, 6, 7, and 8 were performed as described earlier, with 500 ng total cellular RNA.
Competition Experiments.
For competition experiments using nine reverse genetics plasmids, 293 T cells were cotransfected as described earlier. Supernatants were harvested 48 h posttransfection and used to infect six-well plates of confluent MDCK cells, using 10-fold serial dilutions. After viruses adsorption for 90 min at 37 °C, cells were overlaid with 2 mL 1.1% (wt/wt) Difco Noble agar (Becton, Dickinson and Company) in minimum essential medium (MEM) (Gibco) supplemented with TPCK-trypsin (1 μg/mL) and incubated 3–5 d to visualize individual viral plaques. Viruses collected from individual plaques were then amplified on MDCK cells in 48-well plates and stored at −80 °C for molecular analysis. For tranfection/infection competition experiments, 108 293 T cells were transfected by mixing 0.5 µg plasmid and TransIT-LT1 reagent (Mirus), according to the manufacturer’s instructions. Six hours posttransfection, cells were infected with WT or mutant virus at a m.o.i. of 2. One hour later, cells were washed and incubated at 37 °C with EMEM supplemented with 1 μg/mL TPCK-trypsin. At 20 hours postinfection (h.p.i.), supernatants were harvested, and individual viruses were purified from plaques as described earlier and amplified on MDCK cells. Viral supernatants were centrifuged at 1,500 g for 10 min and stored at −80 °C for molecular analysis.
To determine the genotype of the viruses isolated from plaques, viral RNA was extracted using a RNeasy Mini kit (Qiagen), as described earlier. The Uni12 primer (5′-AGC AAA AGC AGG-3′; 200 ng) was annealed to the viral RNA (8.5 µL) for 5 min at 70 °C, and then cDNA synthesis was performed with 11 units M-MuLV RT (Euromedex) and 1 mM dNTP (Euromedex) for 1 h at 42 °C in a final volume of 20 µL. WT and mutant segments 2 and 8 were identified by PCR, using specific primer pairs. PCR reactions contained 5 µL cDNA, 200 ng each primer, 0.4 mM dNTP (Euromedex), and 1.25 unit GoTaq polymerase (Promega). PCR conditions were denaturation at 94 °C for 5 min, 30 cycles of amplification (1 min at 94 °C, 1 min at 62 °C, and 1 min at 72 °C), and a final elongation at 72 °C for 5 min. The amplification products were analyzed by 0.8% agarose gel electrophoresis.
Ultrathin Section Electron Microscopy.
Electron microscopy of 65-nm sections of infected cell culture samples embedded in Epon was performed on a Philips CM 120 transmission electron microscope at an acceleration voltage of 80 kV, as previously described (38).
Sequence Analysis.
The covariation and conservation of base-pair analysis used 308 full-length PB1 and NS sequences that were aligned and downloaded from the National Center for Biotechnology Information Influenza Virus Resource. Sequence analysis was performed with custom scripts written in BioPython (www.biopython.org). Phylogenetic trees were computed using BioNJ of the APE package (40) in the R environment (www.r-project.org). Pairwise distance matrices were calculated using the APE dist.dna function with default options.
Supplementary Material
Acknowledgments
We thank John McCauley (National Institute for Medical Research, London, United Kingdom), Eric Westhof, and Pascale Romby (Institut de Biologie Moléculaire et Cellulaire, Centre National de la Recherche Scientifique) for critical reading of our manuscript. This work was supported by the Centre National de la Recherche Scientifique (R.M.) and by VirNext (V.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1314419110/-/DCSupplemental.
References
- 1.Horimoto T, Kawaoka Y. Influenza: Lessons from past pandemics, warnings from current incidents. Nat Rev Microbiol. 2005;3(8):591–600. doi: 10.1038/nrmicro1208. [DOI] [PubMed] [Google Scholar]
- 2.Arranz R, et al. The structure of native influenza virion ribonucleoproteins. Science. 2012;338(6114):1634–1637. doi: 10.1126/science.1228172. [DOI] [PubMed] [Google Scholar]
- 3.Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. Organization of the influenza virus replication machinery. Science. 2012;338(6114):1631–1634. doi: 10.1126/science.1227270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palese P, Shaw M. In: Fields Virology. Knipe DM, Howley PM, editors. Philadelphia: Lippincott, Williams and Wilkins; 2006. pp. 1647–1689. [Google Scholar]
- 5.Coloma R, et al. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 2009;5(6):e1000491. doi: 10.1371/journal.ppat.1000491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu MT, Parvin JD, Gupta S, Krystal M, Palese P. Genomic RNAs of influenza viruses are held in a circular conformation in virions and in infected cells by a terminal panhandle. Proc Natl Acad Sci USA. 1987;84(22):8140–8144. doi: 10.1073/pnas.84.22.8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutchinson EC, von Kirchbach JC, Gog JR, Digard P. Genome packaging in influenza A virus. J Gen Virol. 2010;91(Pt 2):313–328. doi: 10.1099/vir.0.017608-0. [DOI] [PubMed] [Google Scholar]
- 8.Luytjes W, Krystal M, Enami M, Parvin JD, Palese P. Amplification, expression, and packaging of foreign gene by influenza virus. Cell. 1989;59(6):1107–1113. doi: 10.1016/0092-8674(89)90766-6. [DOI] [PubMed] [Google Scholar]
- 9.Neumann G, Hobom G. Mutational analysis of influenza virus promoter elements in vivo. J Gen Virol. 1995;76(Pt 7):1709–1717. doi: 10.1099/0022-1317-76-7-1709. [DOI] [PubMed] [Google Scholar]
- 10.Fournier E, et al. A supramolecular assembly formed by influenza A virus genomic RNA segments. Nucleic Acids Res. 2012;40(5):2197–2209. doi: 10.1093/nar/gkr985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noda T, et al. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature. 2006;439(7075):490–492. doi: 10.1038/nature04378. [DOI] [PubMed] [Google Scholar]
- 12. Noda T, et al. (2012) Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat Commun 3:639. [DOI] [PMC free article] [PubMed]
- 13.Chou YY, et al. One influenza virus particle packages eight unique viral RNAs as shown by FISH analysis. Proc Natl Acad Sci USA. 2012;109(23):9101–9106. doi: 10.1073/pnas.1206069109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laver WG, Downie JC. Influenza virus recombination. I. Matrix protein markers and segregation during mixed infections. Virology. 1976;70(1):105–117. doi: 10.1016/0042-6822(76)90240-3. [DOI] [PubMed] [Google Scholar]
- 15.Jennings PA, Finch JT, Winter G, Robertson JS. Does the higher order structure of the influenza virus ribonucleoprotein guide sequence rearrangements in influenza viral RNA? Cell. 1983;34(2):619–627. doi: 10.1016/0092-8674(83)90394-x. [DOI] [PubMed] [Google Scholar]
- 16.Nakajima K, Ueda M, Sugiura A. Origin of small RNA in von Magnus particles of influenza virus. J Virol. 1979;29(3):1142–1148. doi: 10.1128/jvi.29.3.1142-1148.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Odagiri T, Tashiro M. Segment-specific noncoding sequences of the influenza virus genome RNA are involved in the specific competition between defective interfering RNA and its progenitor RNA segment at the virion assembly step. J Virol. 1997;71(3):2138–2145. doi: 10.1128/jvi.71.3.2138-2145.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujii K, et al. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J Virol. 2005;79(6):3766–3774. doi: 10.1128/JVI.79.6.3766-3774.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujii Y, Goto H, Watanabe T, Yoshida T, Kawaoka Y. Selective incorporation of influenza virus RNA segments into virions. Proc Natl Acad Sci USA. 2003;100(4):2002–2007. doi: 10.1073/pnas.0437772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Q, et al. The influenza A virus PB2, PA, NP, and M segments play a pivotal role during genome packaging. J Virol. 2012;86(13):7043–7051. doi: 10.1128/JVI.00662-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gog JR, et al. Codon conservation in the influenza A virus genome defines RNA packaging signals. Nucleic Acids Res. 2007;35(6):1897–1907. doi: 10.1093/nar/gkm087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang Y, Hong Y, Parslow TG. cis-Acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J Virol. 2005;79(16):10348–10355. doi: 10.1128/JVI.79.16.10348-10355.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang Y, Huang T, Ly H, Parslow TG, Liang Y. Mutational analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. J Virol. 2008;82(1):229–236. doi: 10.1128/JVI.01541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marsh GA, Hatami R, Palese P. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J Virol. 2007;81(18):9727–9736. doi: 10.1128/JVI.01144-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marsh GA, Rabadán R, Levine AJ, Palese P. Highly conserved regions of influenza a virus polymerase gene segments are critical for efficient viral RNA packaging. J Virol. 2008;82(5):2295–2304. doi: 10.1128/JVI.02267-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gavazzi C, et al. An in vitro network of intermolecular interactions between viral RNA segments of an avian H5N2 influenza A virus: Comparison with a human H3N2 virus. Nucleic Acids Res. 2013;41(2):1241–1254. doi: 10.1093/nar/gks1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujii K, Ozawa M, Iwatsuki-Horimoto K, Horimoto T, Kawaoka Y. Incorporation of influenza A virus genome segments does not absolutely require wild-type sequences. J Gen Virol. 2009;90(Pt 7):1734–1740. doi: 10.1099/vir.0.010355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gerlach W, Giegerich R. GUUGle: A utility for fast exact matching under RNA complementary rules including G-U base pairing. Bioinformatics. 2006;22(6):762–764. doi: 10.1093/bioinformatics/btk041. [DOI] [PubMed] [Google Scholar]
- 29.Skripkin E, Paillart JC, Marquet R, Ehresmann B, Ehresmann C. Identification of the primary site of the human immunodeficiency virus type 1 RNA dimerization in vitro. Proc Natl Acad Sci USA. 1994;91(11):4945–4949. doi: 10.1073/pnas.91.11.4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen W, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001;7(12):1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 31.Hutchinson EC, Wise HM, Kudryavtseva K, Curran MD, Digard P. Characterisation of influenza A viruses with mutations in segment 5 packaging signals. Vaccine. 2009;27(45):6270–6275. doi: 10.1016/j.vaccine.2009.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hutchinson EC, Curran MD, Read EK, Gog JR, Digard P. Mutational analysis of cis-acting RNA signals in segment 7 of influenza A virus. J Virol. 2008;82(23):11869–11879. doi: 10.1128/JVI.01634-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou YY, et al. Colocalization of different influenza viral RNA segments in the cytoplasm before viral budding as shown by single-molecule sensitivity FISH analysis. PLoS Pathog. 2013;9(5):e1003358. doi: 10.1371/journal.ppat.1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duhaut SD, Dimmock NJ. Defective segment 1 RNAs that interfere with production of infectious influenza A virus require at least 150 nucleotides of 5′ sequence: Evidence from a plasmid-driven system. J Gen Virol. 2002;83(Pt 2):403–411. doi: 10.1099/0022-1317-83-2-403. [DOI] [PubMed] [Google Scholar]
- 35.Essere B, et al. Critical role of segment-specific packaging signals in genetic reassortment of influenza A viruses. Proc Natl Acad Sci USA. 2013 doi: 10.1073/pnas.1308649110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marshall N, Priyamvada L, Ende Z, Steel J, Lowen AC. Influenza virus reassortment occurs with high frequency in the absence of segment mismatch. PLoS Pathog. 2013;9(6):e1003421. doi: 10.1371/journal.ppat.1003421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci USA. 2000;97(11):6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moules V, et al. In vitro characterization of naturally occurring influenza H3NA- viruses lacking the NA gene segment: Toward a new mechanism of viral resistance? Virology. 2010;404(2):215–224. doi: 10.1016/j.virol.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 39. Duchamp MB, et al. (2010) Pandemic A(H1N1)2009 influenza virus detection by real time RT-PCR: Is viral quantification useful? Clin Microbiol Infect 16(4):317–321. [DOI] [PubMed]
- 40.Paradis E, Claude J, Strimmer K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics. 2004;20(2):289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.