

Significance
The electronic structure of oxyhemoglobin has been under intense scrutiny since the geometric structure of this bent, end-on bound Fe-O2 species was first determined over three decades ago, but a consensus description has not yet been reached. Here, solution and crystalline X-ray absorption spectroscopies have been combined with time-dependent density functional theory calculations to demonstrate the dominantly 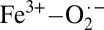 (ferric-superoxide) character of solution oxyhemoglobin. It is also shown that, in crystallo, a dominantly Fe2+-O2 (ferrous-dioxygen) character is favored, indicating multiconfigurational character, tunable by a change in state.
(ferric-superoxide) character of solution oxyhemoglobin. It is also shown that, in crystallo, a dominantly Fe2+-O2 (ferrous-dioxygen) character is favored, indicating multiconfigurational character, tunable by a change in state.
Abstract
Hemoglobin (Hb) is the heme-containing O2 transport protein essential for life in all vertebrates. The resting high-spin (S = 2) ferrous form, deoxy-Hb, combines with triplet O2, forming diamagnetic (S = 0) oxy-Hb. Understanding this electronic structure is the key first step in understanding transition metal–O2 interaction. However, despite intense spectroscopic and theoretical studies, the electronic structure description of oxy-Hb remains elusive, with at least three different descriptions proposed by Pauling, Weiss, and McClure-Goddard, based on theory, spectroscopy, and crystallography. Here, a combination of X-ray absorption spectroscopy and extended X-ray absorption fine structure, supported by density functional theory calculations, help resolve this debate. X-ray absorption spectroscopy data on solution and crystalline oxy-Hb indicate both geometric and electronic structure differences suggesting that two of the previous descriptions are correct for the Fe–O2 center in oxy-Hb. These results support the multiconfigurational nature of the ground state developed by theoretical results. Additionally, it is shown here that small differences in hydrogen bonding and solvation effects can tune the ground state, tipping it into one of the two probable configurations. These data underscore the importance of solution spectroscopy and show that the electronic structure in the crystalline form may not always reflect the true ground-state description in solution.
Hemoglobin (Hb) is the iron-containing heme protein essential for oxygen transport in all vertebrates. The protein is an assembly of four globular subunits, each containing a heme B (protoporphyrin IX group) (1). The first characterization of Hb was done in the mid-1800s (2, 3), with the magnetic properties first elucidated in early 1900s. Later, Pauling and Coryell (4) showed that whereas resting venous Hb is paramagnetic, the oxygenated arterial form (oxy-Hb) is diamagnetic in nature. The first structure determination (X-ray diffraction) was done by Perutz et al. (5) in 1960, who was awarded the Nobel prize for this work. In 1974, the geometry of O2 binding was shown to be end-on η1-Fe–O2, based on X-ray diffraction data on a biomimetic model complex (6), which was later confirmed by diffraction data on oxy-Hb. However, the debate over the electronic structure of O2 binding in Hb and the nature of the Fe–O2 bond continues today.
Although the diamagnetic nature of oxy-Hb has been described by several different models, three have persisted. Starting from deoxy-Hb (Fe2+, S = 5/2) and O2 (neutral, S = 1) and using a valence bond theory, Pauling proposed that the oxy-Hb is a Fe2+, S = 0 system that accepts one lone pair from the sp2 hybridized O2, forming a dative bond (Scheme 1) (7). The Weiss model involves one-electron reduction of O2 by the ferrous heme in deoxy-Hb (8), which leads to antiferromagnetic coupling between the Fe3+ (S = 1/2) and 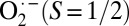 , resulting in a STotal = 0. The third model, proposed by McClure (9) and elaborated by Goddard and Olafson (10), involves an “Ozone model” and suggests a Fe2+(S = 1)–O2 (S = 1) electronic structure in which the four unpaired electrons (two each on Fe and O2) form a 3-center 4-electron bond (Scheme 1).
, resulting in a STotal = 0. The third model, proposed by McClure (9) and elaborated by Goddard and Olafson (10), involves an “Ozone model” and suggests a Fe2+(S = 1)–O2 (S = 1) electronic structure in which the four unpaired electrons (two each on Fe and O2) form a 3-center 4-electron bond (Scheme 1).
Scheme 1.
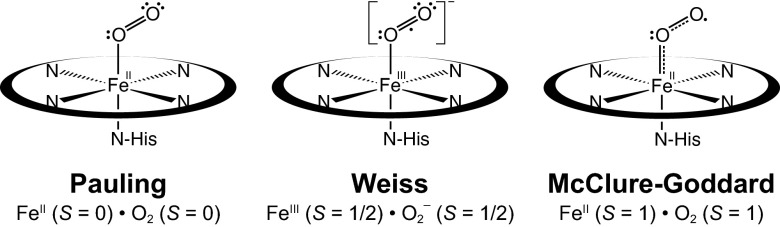
Schematic representation of the three proposed electronic structures of oxy-Hb.
Over the years, several theoretical and experimental methods have been used including simplified valence bond theory, semiempirical methods, density functional theory, and complete active space self-consistent field methods in support of the various models [see Shaik and coworkers (11) for a review]. Among the experimental techniques, Mössbauer data are interpreted in the context of the Pauling model (12), whereas resonance Raman (rR) data indicate a superoxide stretching frequency consistent with the Weiss model (13). X-ray crystallographic data indicate the presence of a very short O–O (∼1.23 Å) bond supporting either the Pauling or Ozone model (14). X-ray absorption spectroscopy (XAS) studies have focused on the position of the iron in the plane of the heme, with only qualitative assignments of oxidation state (15–18).
In this study, quantitative Fe K-edge XAS and density functional theory (DFT) calculations have been used to evaluate the electronic structure of oxy-Hb in conjunction with data on synthetic model complexes that have been used as references for the Pauling, Weiss, and Ozone descriptions. Additionally, Fe K pre-edge analysis has been performed using time-dependent DFT (TD-DFT) calculations, and data from oxy-Hb crystal structures have been evaluated and correlated to the literature rR data. Finally, XAS data on crystalline oxy-Hb have been analyzed to understand the electronic structure differences in solution and crystal forms.
Results and Discussion
A comparison of the Fe K rising-edge data for deoxy-, aquamet-, and oxy-Hb is given in Fig. 1A along with the corresponding UV-visible data (Fig. 1A, Inset) (19). Typically, the rising-edge energy positions of transition metal K-edges have been used as signatures of the oxidation state of the absorbing metal center (20). As the positive charge on the metal center increases (e.g., Fe2+ to Fe3+), the edge shifts to higher energy owing to an increase in Zeff. The energies (based on the first inflection points in Fig. 1B) occur at 7,117.5, 7,121.7, and 7,121.3 eV for deoxy-, aquamet-, and oxy-Hb, respectively (Table 1) and show that oxy-Hb is shifted ∼2 eV higher in energy relative to deoxy-Hb and is more aligned with ferric aquamet-Hb. Using this accepted method of oxidation state assignment, the data point to a Fe3+ Zeff in oxy-Hb. However, this method ignores the potential effects of spin-state on the absorbing atom and the rising-edge energy position. Note that in most literature examples, the spin-state is frequently not considered when oxidation state assignments are made based on rising-edge positions (17, 21). This is important, because deoxy-Hb is a high-spin ferrous (Fe2+, S = 2) system and aquamet-Hb is a high-spin ferric (Fe3+, S = 5/2) system, neither of which correspond to the iron center in the three proposed electronic structures of oxy-Hb.
Fig. 1.

(A) Fe K-edge XAS data for deoxy, aquamet, and oxyhemoglobin. (Inset) The corresponding UV-visible spectrum verifying the purity of the samples (19). (B) The Fe K-edge first derivative spectra with labeled first inflection-point energies.
Table 1.
Fe K-edge energies and pre-edge integrated areas
| XAS feature | Deoxy-Hb | Aquamet-Hb | Oxy-Hb | Hmet Hb | [Fe(TPP)] | [Fe(TPP)(Py)2] | [Fe(TPP)(THF)2] | [Fe(TPP)(ImH)2] | [Fe(TPP)(ImH)2]Cl |
| Rising edge,† eV | 7,117.5 | 7,121.7 | 7,121.3 | 7,121.5 | 7,115.4 | 7,121.0 | 7,117.9 | 7,120.7 | 7,122.2 |
| Pre-edge,‡ eV | 7,112.4 | 7,113.4 | 7,113.0 | 7,113.0 | 7,111.5 | 7,112.2 | 7,112.4 | 7,112.1 | 7,112.8 |
| Integrated area§ | 12.2 | 9.1 | 10.6 | 9.0 | 4.8 | 5.1 | 4.1¶ | 4.0 | 6.5 |
Hmet, hydroxymet.
Measured at the first inflection-point energy.
Intensity-weighted average energy position (resolution ± 0.1 eV, precision > ± 0.05 eV).
Calculated as peak-height × half width and scaled by 102 (error ∼5%).
Error in integrated area is higher owing to poor data quality.
Spin-State Effects on the Iron K-Edge.
The effect of spin-state on the rising-edge energy position was tested with three porphyrin model complexes. Fe K-edge XAS data were measured on intermediate-spin [Fe(TPP)] (Fe2+, S = 1), high-spin [Fe(TPP)(THF)2] (Fe2+, S = 2), and low-spin [Fe(TPP)(Py)2] (Fe2+, S = 0) (TPP is meso-tetraphenylporphyrin). These data are presented in Fig. 2. The rising-edge energy position for [Fe(TPP)], [Fe(TPP)(THF)2], and [Fe(TPP)(Py)2] occur at 7,115.4, 7,117.9, and 7,121.0 eV, respectively. For the same oxidation state (Fe2+), these data show a ∼2-eV spread in energy position. This is an important observation because a ∼2-eV edge-shift has been commonly attributed to a change in Zeff (17, 21). For these model complexes, conventional data analysis would erroneously indicate that the Fe center in [Fe(TPP)(Py)2] is one-electron oxidized relative to [Fe(TPP)]. Fig. 2 also shows that the energy shift does not increase linearly with spin-state (observed trend: S = 1 < S = 0 < S = 2). This, too, is counterintuitive to conventional wisdom, which suggests that because the 6-coordinate low-spin [Fe(TPP)(Py)2] (SI Appendix, Scheme S1) has the lowest Zeff on Fe owing to charge donation from the strongly coordinating ligands, and because the rising-edge typically shifts to higher energy with increase in Zeff, [Fe(TPP)(Py)2] should have the lowest rising-edge energy position. In reality, [Fe(TPP)(Py)2] has the highest energy edge position.
Fig. 2.

(A) Fe K-edge XAS spectra. (Inset) The correlation between rising energy position and the normalized (by the number of ligands) average Fe-ligand distance. (B) The first derivative spectra with the first inflection and rising-edge maximum energies.
The solution to this inconsistency was obtained by applying Natoli’s rule (22) to these three reference complexes, which says that given comparable systems, the rising-edge energy position is related to the average bond distance by
where E – E0 represents the relative rising-edge energy position and R is the average bond distance. Renormalizing R for the number of ligands in [Fe(TPP)] (four compared with six in the other two systems), a near inverse-linear relation between energy and R2 (R2 = 0.98) is obtained (Fig. 2, Inset). These data indicate that a comparison of the rising-edge energies can only be made for systems in the same spin-state.
Two of the proposed models for oxy-Hb suggest a ferrous iron center (Scheme 1). The Ozone model is intermediate-spin (S = 1) whereas the Pauling model is low-spin (S = 0). Thus, [Fe(TPP)] (Fe2+, S = 1) and [Fe(TPP)(Py)2] (Fe2+, S = 0) are good references for these electronic structure descriptions, respectively, from both an oxidation and spin-state perspective (SI Appendix, Fig. S1). A comparison of the energy positions with the rising-edge of oxy-Hb shows that [Fe(TPP)(Py)2] is similar, whereas [Fe(TPP)] is over ∼4 eV lower in energy (SI Appendix, Fig. S1 and Table 1). This clearly shows that the Ozone model is inconsistent with the electronic structure of oxy-Hb, leaving the Pauling (Fe2+, S = 0) and Weiss (Fe3+, S = 1/2) models in contention. Note that aquamet-Hb (Fe3+, S = 5/2) has the incorrect spin-state for comparison with the Weiss model (Fe3+, S = 1/2). Thus, the high-pH form, hydroxymet-Hb (Fe3+, S = 1/2), was chosen for comparison with oxy-Hb. Note also that because the bond distances do not change significantly between hydroxymet- and aquamet-Hb, following Natoli’s rule, the edge energies are very comparable (∼0.2-eV difference, Table 1). Similar trends in edge energy positions have also been observed in myoglobin (23). Based on the similarity of the rising-edge energy positions of oxy-Hb, [Fe(TPP)(Py)2], and hydroxymet-Hb, Fe K rising-edge analysis cannot distinguish between the Pauling and Weiss models.
Fe K Pre-Edge Analysis.
In contrast to the dipole-allowed Fe K-edge, the pre-edge feature at the onset of the rising-edge is a weak electric dipole-forbidden but quadrupole-allowed 1s → 3d transition. The pre-edge intensity and energy positions are affected by ligand field strength, site symmetry, and oxidation state, all of which have been widely applied to interrogate these properties (20, 24). A comparison of the Fe K pre-edge for oxy-Hb with that of [Fe(TPP)(Py)2] and hydroxymet-Hb is shown in Fig. 3A. Data for [Fe(TPP)] are included for comparison. The intensity-weighted average pre-edge energy positions and total intensities are given in Table 1. The pre-edge energy of the two ferrous systems, [Fe(TPP)] and [Fe(TPP)(Py)2], occur at 7,111.5 and 7,112.2 eV, whereas those of oxy-Hb and hydroxymet-Hb are shifted to higher energy at 7,113.0 eV. For reference, the pre-edge spectra of two well-known low-spin heme complexes, [Fe(TPP)(ImH)2] (Fe2+, S = 0) and oxidized [Fe(TPP)(ImH)2](Cl) (Fe3+, S = 1/2), are shown in Fig. 3B. These data for an oxidized and reduced couple clearly show a 0.7-eV shift upon oxidation, similar to that observed between [Fe(TPP)(Py)2] and oxy-Hb.
Fig. 3.

(A) Fe K pre-edge XAS spectra of [Fe(TPP)], [Fe(TPP)(Py)2], and hydroxymet-Hb representing the McClure “Ozone,” Pauling, and Weiss models, respectively, vs. oxy-Hb. (B) Low-spin ferrous and ferric reference systems [Fe(TPP)(ImH)2] and [Fe(TPP)(ImH)2](Cl). (C) TD-DFT simulated spectra of oxy-Hb, Hydroxymet-Hb, and [Fe(TPP)Py2]. (D) Comparison of experimental and TD-DFT–calculated Fe K pre-edge data for oxy-Hb, showing the presence of a low-energy pre-edge feature. The pseudo-Voigt fit line-shapes are included for clarity.
In recent years, core-level TD-DFT calculations have been successfully used to simulate Fe K pre-edges (25). To further probe the electronic structure of oxy-Hb and understand the pre-edge shifts reported above, DFT and TD-DFT calculations were combined with the pre-edge analysis method developed by Westre et al. (20). DFT calculations were first spectroscopically calibrated using experimental Fe–O and O–O distances as evaluating metrical parameters. To this end, a high-k (k = 15 Å–1) extended X-ray absorption fine structure (EXAFS) analysis was performed on oxy-Hb using the full multiple scattering approach implemented in FEFF (26–28). The nonphase shift-corrected Fourier transform and the corresponding EXAFS data (Inset) are presented in SI Appendix, Fig. S2A, with fit parameters given in SI Appendix, Table S1. The best fit shows that the first shell contains five Fe–N at 2.00 Å and one Fe–O at 1.83 Å.
Using the correlation developed by Cramer et al. (inverse linear relationship between O–O vibrational stretching frequency and bond distance) (29, 30), the rR O–O stretching frequency of oxy-Hb can be used to accurately estimate the O–O distance. Literature rR data show that the O–O stretch for oxy adducts of globins and other heme proteins are between 1,120 and 1,155 cm–1 (13). From this, an O–O distance for oxy-Hb between 1.318 Å and 1.335 Å was obtained (SI Appendix, Fig. S2B). Thus, the spectroscopically characterized Fe–O and O–O distances in solution oxy-Hb are 1.83 Å and 1.327 Å (average).
DFT geometry optimizations were then performed on the oxy-Hb active site using a truncated heme center with axial imidazole ligands (SI Appendix, Fig. S3). Because rR data suggest that the first-sphere hydrogen bonding (H-bond) plays a key role in stabilizing the oxy-adduct electronic structure (13), a histidine group H-bonded to O2 was included in the distal pocket, and a water molecule mimicking the glutamine H-bond to the axial histidine was included in the proximal pocket (SI Appendix, Fig. S3C). Using the BP86 functional, CP(PPP) and TZVP basis sets on Fe and the other atoms, respectively, an Fe–O bond distance of 1.83 Å and an O–O distance of 1.32 Å were obtained, which are in excellent agreement with the experiment.
The spectroscopically calibrated level of DFT determined above was next used to quantitatively calculate the Fe K pre-edge region of oxy-Hb, hydroxymet-Hb, and [Fe(TPP)](Py)2. The excellent agreement between experiment and theory (Fig. 3 A and C and SI Appendix, Table S2) point to the accuracy of DFT calculations and show that theory can successfully predict the expected energy shift from Fe2+(S = 0) to Fe3+(S = 1/2). TD-DFT calculations also predicted a low-energy feature in both oxy-Hb and hydroxymet-Hb. For hydroxymet-Hb, this feature is close to the more intense higher-energy pre-edge feature and is not observed when the appropriate broadening is applied. In contrast, in oxy-Hb this feature is clearly observed (Fig. 3D).
The TD-DFT results were then correlated to experimental Fe K pre-edge data using the method developed by Westre et al. (20) (SI Appendix, Fig. S4 and Table S3) The pre-edge of solution oxy-Hb was fit using pseudo-Voigt line-shapes. As is clearly seen, the pre-edge for solution oxy-Hb requires two pseudo-Voigts to adequately fit the data. These occur at 7,111.3 eV and 7,113.7 eV, respectively, and are in good agreement with the theoretical TD-DFT results. TD-DFT also reveals that the low-energy feature is primarily due to transitions from Fe 1s to the spin-polarized α and β lowest unoccupied molecular orbitals (LUMO), which have dominantly Fe 3dyz + O2 (π*) character, or, in other words, into states made available by the presence of a fifth Fe-based hole (d5 electronic configuration). The presence of this pre-edge feature suggest that the Fe in oxy-Hb is ferric in nature and that oxy-Hb is consistent with the Weiss model and a 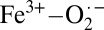 description.
description.
Pre-Edge Differences in Crystalline Oxyhemoglobin.
Fe K-edge XAS studies were also performed on isotropic crystalline oxy-Hb to investigate electronic structure differences in the crystalline phase. The Fe K rising-edge data for solution and crystalline oxy-Hb along with those of deoxy-Hb are presented in Fig. 4 (inset shows the expanded near-edge region). The data were closely monitored for photoreduction and successive scans were compared before inclusion in the Fe K-edge and pre-edge analysis presented herein (SI Appendix, Figs. S5 and S6, respectively). In the past, the near-edge region for heme-containing systems has been successfully interrogated using the multiple scattering code MXAN for structural information (18, 23). These studies have shown that the near-edge spectral differences can be attributed to ligand binding, and that the spectral shape is closely related to geometric structure. The near-edge data for solution and crystalline oxy-Hb were fit with MXAN and the results indicate an intact O2-bound heme site in both systems (SI Appendix, Fig. S7 and associated text).
Fig. 4.

(A) Normalized Fe K-edge XAS spectra of deoxy-Hb and solution and crystalline oxy-Hb. (Inset) The expanded near-edge region). (B) The expanded Fe K pre-edge region.
Fig. 4B shows the expanded Fe K pre-edge region presented in Fig. 4A. On going from solution to the crystalline form, the two features at 7,111.3 and 7,113.7 eV are replaced by a single feature at 7,112.3 eV, with a decrease in total intensity (SI Appendix, Table S3 and Fig. S4). This is surprising because the geometries, spin-states, and ligand-field strengths are expected to be very similar in the two phases. Thus, any difference in the pre-edge is expected to arise from change in Fe Zeff (effective nuclear charge), or, in other words, configurational makeup leaning toward the Pauling (Fe2+, S = 0) model. Because these two have identical ligand systems, their Fe K pre-edge data are compared with those of the isoleptic pair [Fe(TPP)(ImH)2]2+/3+, which represent the electronic structures of the Pauling (Fe2+, S = 0) and Weiss (Fe3+, S = 1/2) models, respectively (Fig. 3B) (a comparison with [Fe(TPP)(Py)2] and hydroxymet-Hb leads to quantitatively similar results). The spectral change between solution and crystalline oxy-Hb is similar to that between [Fe(TPP)(ImH)2]Cl and [Fe(TPP)(ImH)2] (SI Appendix, Fig. S8, respectively, show pseudo-Voigt fits) and supports an electronic structure rearrangement in crystalline oxy-Hb. Interestingly, the Fe K pre-edge of crystalline oxy-Hb (single pre-edge feature) is analogous to data from biomimetic model complexes (21). In a recent study (31), some of us also showed that these model complexes have a dominant Fe2+, S = 0 character, supporting our assignment here for the crystalline oxy-Hb data.
The electronic structure of crystalline oxy-Hb developed above is also supported by protein crystallography data. In metal-bound O2 systems, the O–O bond distance reflects the oxidation state of the O2 moiety with a ∼0.1-Å increase in the O–O bond distance corresponding to a one-electron reduction of O2 (i.e., the O–O distances in O2, 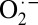 , and
, and 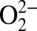 are ∼1.21–1.22, 1.30–1.35, and 1.39–1.45 Å, respectively). This has been both experimentally and theoretically demonstrated (29). Thus, given the
are ∼1.21–1.22, 1.30–1.35, and 1.39–1.45 Å, respectively). This has been both experimentally and theoretically demonstrated (29). Thus, given the 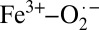 nature of solution oxy-Hb described above, the O–O distance should be in the range of 1.30–1.35 Å. SIAppendix, Fig. S9 presents a correlation of O–O and Fe–O bond distances in oxy-Hb obtained from crystal structures currently deposited in the Protein Data Bank (RCSB). This correlation reveals that although a wide variation in the Fe–O bond distance is observed (likely owing to partial/incomplete O2 binding) the O–O bond distances have a smaller variation. More importantly, the O–O distances are overwhelmingly in the range of 1.20–1.25 Å, with an average value of 1.23 Å. Because 1.23 Å is significantly smaller than the superoxide O–O distance and close to the dioxygen distance of 1.21 Å, the crystallographic data strongly support a Fe2+–O2 electronic structure for crystalline oxy-Hb.
nature of solution oxy-Hb described above, the O–O distance should be in the range of 1.30–1.35 Å. SIAppendix, Fig. S9 presents a correlation of O–O and Fe–O bond distances in oxy-Hb obtained from crystal structures currently deposited in the Protein Data Bank (RCSB). This correlation reveals that although a wide variation in the Fe–O bond distance is observed (likely owing to partial/incomplete O2 binding) the O–O bond distances have a smaller variation. More importantly, the O–O distances are overwhelmingly in the range of 1.20–1.25 Å, with an average value of 1.23 Å. Because 1.23 Å is significantly smaller than the superoxide O–O distance and close to the dioxygen distance of 1.21 Å, the crystallographic data strongly support a Fe2+–O2 electronic structure for crystalline oxy-Hb.
The Fe K-edge XAS and EXAFS results combined with the theoretical data presented here provide evidence that the electronic structure of crystalline oxy-Hb is Fe2+–O2, consistent with the Pauling model (Scheme 1), whereas that of solution oxy-Hb is 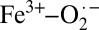 , as described by the Weiss model. This result bridges the gap between the X-ray diffraction (Fe2+–O2) and rR
, as described by the Weiss model. This result bridges the gap between the X-ray diffraction (Fe2+–O2) and rR 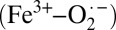 descriptions, shows that the protein exhibits substantial electronic structure differences between solution and crystalline forms, and unifies experiment with the recent quantum mechanics/molecular mechanics (QM/MM) and complete active space self-consistent field (CASSCF) theoretical study in which Shaik and coworkers (11) investigated the effect of the protein environment on the electronic structure of oxy-myoglobin (oxy-Mb). They showed that the ground state of oxy-Mb can be best described as a multiconfigurational wavefunction with both
descriptions, shows that the protein exhibits substantial electronic structure differences between solution and crystalline forms, and unifies experiment with the recent quantum mechanics/molecular mechanics (QM/MM) and complete active space self-consistent field (CASSCF) theoretical study in which Shaik and coworkers (11) investigated the effect of the protein environment on the electronic structure of oxy-myoglobin (oxy-Mb). They showed that the ground state of oxy-Mb can be best described as a multiconfigurational wavefunction with both 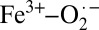 and Fe2+–O2 character, where inclusion of protein-related effects and a reasonably large MM component favors the Weiss model, whereas gas-phase calculations lead to the Pauling model (32). This result is consistent with the experimental result presented here on solution oxy-Hb, but different from that of crystalline oxy-Hb. Because QM/MM calculations reasonably mimic the protein environment, this raises an interesting issue. Is it possible that theory, even at the QM/MM level, cannot accurately describe such a multiconfigurational problem? Note that the 1.29-Å O–O distance obtained by QM/MM is still larger than the 1.23-Å X-ray diffraction distance. This points to electronic structure differences in the crystal compared with those obtained from theory (11). However, it is also possible that differences between oxy-Mb and oxy-Hb result in the differences observed in this study between crystalline oxy-Hb and the QM/MM study on oxy-Mb. Nevertheless, both point to an interesting idea, that the electronic structure of Hb–O2 lies in the multiconfigurational continuum between pure
and Fe2+–O2 character, where inclusion of protein-related effects and a reasonably large MM component favors the Weiss model, whereas gas-phase calculations lead to the Pauling model (32). This result is consistent with the experimental result presented here on solution oxy-Hb, but different from that of crystalline oxy-Hb. Because QM/MM calculations reasonably mimic the protein environment, this raises an interesting issue. Is it possible that theory, even at the QM/MM level, cannot accurately describe such a multiconfigurational problem? Note that the 1.29-Å O–O distance obtained by QM/MM is still larger than the 1.23-Å X-ray diffraction distance. This points to electronic structure differences in the crystal compared with those obtained from theory (11). However, it is also possible that differences between oxy-Mb and oxy-Hb result in the differences observed in this study between crystalline oxy-Hb and the QM/MM study on oxy-Mb. Nevertheless, both point to an interesting idea, that the electronic structure of Hb–O2 lies in the multiconfigurational continuum between pure 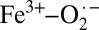 and Fe2+–O2 character, and that secondary changes in the protein environment, crystal packing, and so forth, can tip the electronic structure in either direction (Scheme 2).
and Fe2+–O2 character, and that secondary changes in the protein environment, crystal packing, and so forth, can tip the electronic structure in either direction (Scheme 2).
Scheme 2.
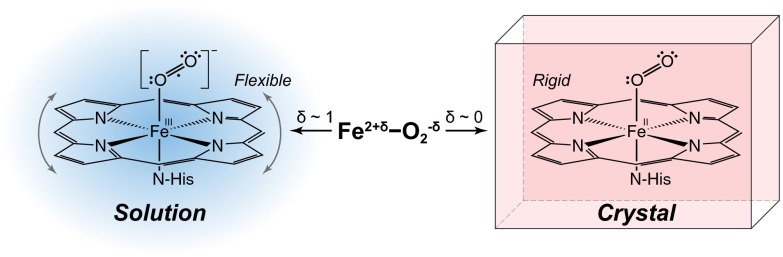
Schematic representation of the proposed electronic structure differences between solution and crystalline oxy-Hb. The δ-notation for charge is used to show the multiconfigurational nature of the ground state of oxy-Hb.
To assess this theory, a series of additional DFT calculations were performed to test the protein environment (H-bonding, first-shell ligands, and solvation effects; SI Appendix, Table S4).
These calculations resulted in a progressive shortening of the O–O bond distance as the solvation and H-bonding effects were removed (1.323 to 1.285 Å), approaching, but not quite reaching the crystallographic average of 1.23 Å. Thus, it is likely that in the crystalline form the lower dielectric constant and a weaker H-bonding interaction shift the electronic structure toward the Fe2+–O2 bonding description. Other effects, such as change in heme ring ruffling, could further modulate the electronic structure in the solution vs. the crystal. These calculations also show that the Fe–O bond changes from 1.83 to 1.76 Å (models 1 and 2, SI Appendix, Fig. S10 and Table S4), along with a 2.05- to 2.07-Å concomitant elongation of the Fe–N(His) bond when the H-bonding to the transaxial histidine is removed.
This allows for an interesting correlation with the biomimetic model complexes of oxy-Hb synthesized by Collman et al. (6) using modified porphyrin ligands (TpivPP) and transaxial methyl-imidazole (33). In their first crystal structure of an oxy-Hb model (6), they used 1-methyl imidazole (1-MeIm) as the transaxial ligand and obtained very short Fe–O and O–O distances of 1.75 and 1.21 Å, respectively. In the later study (33), they showed that changing 1-MeIm to 2-MeIm leads to an elongation of the Fe–O distance from 1.75 to 1.90 Å, whereas the O–O remains unchanged. Collman and coworkers attributed the change in Fe–O bond distance to steric interactions of the Me group and the porphyrin ring, which imparted a 0.116-Å difference in the position of iron in the heme plane between the two complexes (in 1-MeIm the iron was pulled toward the O2). DFT calculations show a similar difference in the position of the iron in the heme plane between the oxy-Hb model with (model 1) and without (model 2) the H-bonding to the proximal histidine ligand, albeit to a lesser extent (0.066 Å) (SI Appendix, Fig. S10). However, unlike the Collman complexes that demonstrate structural differences due to steric effects, the DFT calculations reveal an electronic difference between the two models. Here, the presence of the H-bond in model 1 weakens the (His)N–H interaction and results in an electron-rich N(His), leading to a stronger σ-donor interaction with the iron. In turn, this pulls the iron atom into the plane of the porphyrin and elongates the Fe–O bond from 1.76 Å to 1.83 Å. This also indicates that a large variation in the Fe–O distance is possible depending on secondary forces justifying the much larger spread in the Fe–O distance (relative to O–O) from crystallography (SI Appendix, Fig. S9). It is also interesting to note that the Fe K-edge XAS data for a series of “basket porphyrin” (b-p) complexes has been reported (21), including an oxy-Hb mimic. This ([(b-p)Fe–O2]) complex exhibits a pre-edge shift to lower energy by 0.6 eV relative to that of an Fe3+, S = 1/2 system ([(b-p)Fe3+–OH]), which is comparable to that observed between oxy-Hb and the Fe2+, S = 0 [Fe(TPP)(Py)2] system studied here. This indicates that the synthetic oxy-Hb mimics are dominantly Fe2+–O2 in nature and do not correctly describe the protein electronic structure because they lack the correct hydrogen bonding and protein effects.
Electronic Structure Estimation from Crystallography.
In recent years, high-intensity synchrotron sources have been used for X-ray diffraction measurements on biological macromolecular systems. This has revolutionized protein crystallography, resulting in an exponential increase in the number of deposited crystal structures in the Protein Data Bank (RCSB). However, the success of synchrotron sources comes with the disadvantage of X-ray beam-related damage to the biomolecule, resulting in changes in the diffraction pattern, and among other things the breaking of selected macromolecule bonds, caused by X-ray–generated radicals. This effect is even greater in metalloproteins that undergo beam-related photoreduction of the metal site, leading to geometry and oxidation state changes (34, 35). Hence, these measurements may not represent the true system in solution. Additionally, not much focus has been put on studying geometric and electronic structure differences in crystalline vs. solution phases. This study brings to light an important example in which the protein phase has a strong effect on tuning the electronic structure of the metal-active site in a metalloprotein and leads to inherent differences between solution and in crystallo spectroscopy. This emphasizes the importance of solution structure determination techniques, such as EXAFS, in understanding the geometry and electronic properties of the active site.
Conclusions
This study used solution Fe K-edge XAS to demonstrate the 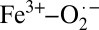 (S = 1/2 on Fe) character of oxy-Hb and shows experimental evidence for the validity of the Weiss model. In contrast, crystalline oxy-Hb is shown to have a dominantly Fe2+–O2 ground state (S = 0 on Fe). These results support the multiconfigurational description of oxy-Hb proposed by theory. DFT calculations show that small changes in H-bonding, along with other effects such as crystal-packing forces and heme ruffling, may also alter the configurational composition. Finally, this study reconciles the differences between crystallography and solution rR studies on the O–O bond distance in oxy-Hb and brings to light the important role of the protein phase in tuning the electronic structure of a multiconfigurational system.
(S = 1/2 on Fe) character of oxy-Hb and shows experimental evidence for the validity of the Weiss model. In contrast, crystalline oxy-Hb is shown to have a dominantly Fe2+–O2 ground state (S = 0 on Fe). These results support the multiconfigurational description of oxy-Hb proposed by theory. DFT calculations show that small changes in H-bonding, along with other effects such as crystal-packing forces and heme ruffling, may also alter the configurational composition. Finally, this study reconciles the differences between crystallography and solution rR studies on the O–O bond distance in oxy-Hb and brings to light the important role of the protein phase in tuning the electronic structure of a multiconfigurational system.
Materials and Methods
The procedure for model complex syntheses and the preparation of the different forms of hemoglobin with their UV-visible measurements can be found in SI Appendix along with details of the XAS measurements and DFT calculations.
XAS Measurements.
The Fe K-edge X-ray absorption spectra of the different forms of Hb and the model complexes presented here were obtained at the Stanford Synchrotron Radiation Lightsource (SSRL) on beamline 7-3 (SI Appendix gives details). Similar data collection and reduction protocol was followed for all datasets. Pyspline (36) was used for data workup and FEFF (28) and EXAFSPAK (37) were used for data fitting. The areas under the Fe K pre-edge region were quantified using the Edg_Fit program (37). A protocol similar to that published before was followed. Crystals of deoxy-Hb were grown in an O2-free glove box and anaerobically transferred to an O2-saturated chamber and exposed to O2 for ∼2–5 min. The crystals were immediately frozen thereafter. The Fe K-edge XAS data of crystalline oxy-Hb were measured on beamline 9-3 at SSRL. More details about data collection and analysis are given in SI Appendix.
DFT Calculations.
Gradient-corrected [generalized gradient approximation (GGA)], spin-unrestricted DFT and TD-DFT calculations for all protein systems and model complexes were performed using ORCA 2.8 (38) on a 12-cpu Linux cluster. Detailed information on functional and basis sets, solvation, and convergence criteria are given in SI Appendix.
Supplementary Material
Acknowledgments
R.S. thanks Dr. Patrick Frank with help in synthesis of the model complexes and preparation of hemoglobin samples. Stanford Synchrotron Radiation Lightsource (SSRL) operations are funded by the Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology program is supported by the National Institutes of Health (NIH); National Institute of General Medical Sciences; the National Center for Research Resources, Biomedical Technology Program; and the Department of Energy, Office of Biological and Environmental Research. This publication was made possible by NIH Grants P41GM103393 and P41RR001209.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1315734110/-/DCSupplemental.
References
- 1.Hsia CC. Respiratory function of hemoglobin. N Engl J Med. 1998;338(4):239–247. doi: 10.1056/NEJM199801223380407. [DOI] [PubMed] [Google Scholar]
- 2.Hoppe-Seylor F. Uber die oxydation in lebendemblute. Med-chem Untersuch Lab. 1866;1:133–140. [Google Scholar]
- 3. Funke O (1851) Über das milzvenenblut. Z Rat Med 1:172–218.
- 4.Pauling L, Coryell CD. The magnetic properties and structure of hemoglobin, oxyhemoglobin and carbonmonoxyhemoglobin. Proc Natl Acad Sci USA. 1936;22(4):210–216. doi: 10.1073/pnas.22.4.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perutz MF, et al. Structure of haemoglobin: A three-dimensional Fourier synthesis at 5.5-A. resolution, obtained by X-ray analysis. Nature. 1960;185(4711):416–422. doi: 10.1038/185416a0. [DOI] [PubMed] [Google Scholar]
- 6.Collman JP, Gagne RR, Reed CA, Robinson WT, Rodley GA. Structure of an iron(II) dioxygen complex; a model for oxygen carrying hemeproteins. Proc Natl Acad Sci USA. 1974;71(4):1326–1329. doi: 10.1073/pnas.71.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pauling L. Magnetic properties and structure of oxyhemoglobin. Proc Natl Acad Sci USA. 1977;74(7):2612–2613. doi: 10.1073/pnas.74.7.2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss JJ. Nature of the iron-oxygen bond in oxyhaemoglobin. Nature. 1964;202:83–84. doi: 10.1038/202083b0. [DOI] [PubMed] [Google Scholar]
- 9.McClure DS. Electronic structure of transition-metal complex ions. Radiat Res Suppl. 1960;2:218–224. [Google Scholar]
- 10.Goddard WA, 3rd, Olafson BD. Ozone model for bonding of an O2 to heme in oxyhemoglobin. Proc Natl Acad Sci USA. 1975;72(6):2335–2339. doi: 10.1073/pnas.72.6.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H, Ikeda-Saito M, Shaik S. Nature of the Fe-O2 bonding in oxy-myoglobin: Effect of the protein. J Am Chem Soc. 2008;130(44):14778–14790. doi: 10.1021/ja805434m. [DOI] [PubMed] [Google Scholar]
- 12.Oshtrakh MJ. Mössbauer spectroscopy of iron containing biomolecules and model compounds in biomedical research. J Mol Struct. 1999;480-481:109–120. [Google Scholar]
- 13.Das TK, Couture M, Ouellet Y, Guertin M, Rousseau DL. Simultaneous observation of the O---O and Fe---O2 stretching modes in oxyhemoglobins. Proc Natl Acad Sci USA. 2001;98(2):479–484. doi: 10.1073/pnas.98.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park SY, Yokoyama T, Shibayama N, Shiro Y, Tame JR. 1.25 A resolution crystal structures of human haemoglobin in the oxy, deoxy and carbonmonoxy forms. J Mol Biol. 2006;360(3):690–701. doi: 10.1016/j.jmb.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 15.Bianconi A, et al. Local Fe site structure in the tense-to-relaxed transition in carp deoxyhemoglobin: A XANES (x-ray absorption near edge structure) study. Proc Natl Acad Sci USA. 1986;83(20):7736–7740. doi: 10.1073/pnas.83.20.7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Durham P, et al. X-ray absorption near edge structure (XANES) for CO, CN and deoxyhaemoglobin: geometrical information. EMBO J. 1983;2(9):1441–1443. doi: 10.1002/j.1460-2075.1983.tb01605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenberger P, Shulman RG, Brown GS, Ogawa S. Structure-function relations in hemoglobin as determined by x-ray absorption spectroscopy. Proc Natl Acad Sci USA. 1976;73(2):491–495. doi: 10.1073/pnas.73.2.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Della Longa S, et al. Structure of the Fe-heme in the hemodimeric hemoglobin from Scapharca inaequivalvis and in the T721 mutant: An X-ray absorption spectroscopic study at low temperature. Eur Biophys J. 2001;29(8):559–568. doi: 10.1007/s002490000102. [DOI] [PubMed] [Google Scholar]
- 19.Zijlstra WG, Buursma A. Spectrophotometry of hemoglobin:absorption spectra of bovine oxyhemoglobin, deoxyhemoglobin, carboxyhemoglobin and methemoglobin. Comp Biochem Physiol A: Physiol. 1997;118B:743–749. [Google Scholar]
- 20.Westre TE, et al. A multiplet analysis of Fe K-edge 1s->3d pre-edge features of iron complexes. J Am Chem Soc. 1997;119(27):6297–6314. [Google Scholar]
- 21.Cartier C, et al. X-Ray absorption spectroscopy of Iron-(11) and -(111) basket-handle porphyrins. J Chem Soc, Dalton Trans. 1992;609:609–618. [Google Scholar]
- 22. Bianconi A, Dell'Ariccia M, Gargano A, Natoli CR (1983) Bond length determination using XANES. EXAFS and Near Edge Structure, Springer Series in Chemical Physics, eds Bianconi A, Incoccia A, Stipcich S (Springer, Berlin), Vol 27, pp 57–61.
- 23.Della Longa S, Pin S, Cortès R, Soldatov AV, Alpert B. Fe-heme conformations in ferric myoglobin. Biophys J. 1998;75(6):3154–3162. doi: 10.1016/S0006-3495(98)77757-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarangi R, et al. X-ray absorption edge spectroscopy and computational studies on LCuO2 species: Superoxide-Cu(II) versus peroxide-Cu(III) bonding. J Am Chem Soc. 2006;128(25):8286–8296. doi: 10.1021/ja0615223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeBeer George S, Petrenko T, Neese F. Prediction of iron K-edge absorption spectra using time-dependent density functional theory. J Phys Chem A. 2008;112(50):12936–12943. doi: 10.1021/jp803174m. [DOI] [PubMed] [Google Scholar]
- 26.Rehr JJ, Zabinsky SI, Albers RC, Albers RC. Mustre de Leon J Ab initio curved-wave x-ray-absorption fine structure. Phys Rev B Condens Matter. 1991;44(9):4146–4156. doi: 10.1103/physrevb.44.4146. [DOI] [PubMed] [Google Scholar]
- 27.Rehr JJ, Ankudinov AL. New developments in the theory and interpretation of X-ray spectra based on fast parallel calculations. J Synchrotron Radiat. 2003;10(Pt 1):43–45. doi: 10.1107/s0909049502017223. [DOI] [PubMed] [Google Scholar]
- 28.Rehr JJ, Mustre de Leon J, Zabinsky SI, Albers RC. Theoretical X-ray absorption fine structure standards. J Am Chem Soc. 1991;113(14):5135–5140. [Google Scholar]
- 29.Cramer CJ, Tolman WB, Theopold KH, Rheingold AL. Variable character of O-O and M-O bonding in side-on (eta(2)) 1:1 metal complexes of O2. Proc Natl Acad Sci USA. 2003;100(7):3635–3640. doi: 10.1073/pnas.0535926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho J, Sarangi R, Nam W. Mononuclear metal-o2 complexes: Crystallographic and spectroscopic characterization and reactivity. Acc Chem Res. 2012;45:1321–1330. doi: 10.1021/ar3000019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson SA, et al. Iron L-edge X-ray absorption spectroscopy of oxy-picket fence porphyrin: experimental insight into Fe-O2 bonding. J Am Chem Soc. 2013;135(3):1124–1136. doi: 10.1021/ja3103583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaik S, Chen H. Lessons on O2 and NO bonding to heme from ab initio multireference/multiconfiguration and DFT calculations. J Biol Inorg Chem. 2011;16:841–855. doi: 10.1007/s00775-011-0763-1. [DOI] [PubMed] [Google Scholar]
- 33.Jameson GB, et al. Models for the active site of oxygen-binding hemoproteins. Dioxygen binding properties and the structures of(2-Methylimidazole)-meso-tetra(α,α,α,α-o- pivalamidophenyl)porphyrinatoiron(II)-ethanol and Its dioxygen adduct. J Am Chem Soc. 1980;102:3224–3237. [Google Scholar]
- 34.Yano J, et al. X-ray damage to the Mn4Ca complex in single crystals of photosystem II: A case study for metalloprotein crystallography. Proc Natl Acad Sci USA. 2005;102(34):12047–12052. doi: 10.1073/pnas.0505207102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.George GN, et al. X-ray-induced photo-chemistry and X-ray absorption spectroscopy of biological samples. J Synchrotron Radiat. 2012;19(Pt 6):875–886. doi: 10.1107/S090904951203943X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tenderholt A. Pyspline and QMForge. Stanford, CA: Stanford Univ Press; 2007. [Google Scholar]
- 37.George GN. EXAFSPAK and EDG-FIT. Stanford, CA: Stanford Synchrotron Radiation Laboratory, Stanford Linear Accelerator Center; 2000. [Google Scholar]
- 38. Neese F (2008) ORCA: An ab initio, DFT and semiempirical electronic structure package. Version 2.6.35 (Univ of Bonn, Bonn)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.