


Abstract
Mutations in the gene for telomerase reverse transcriptase (hTERT) are associated with diseases including dyskeratosis congenita, aplastic anemia, pulmonary fibrosis and cancer. Understanding the molecular basis of these telomerase-associated diseases requires dependable quantitative measurements of telomerase enzyme activity. Furthermore, recent findings that the human POT1-TPP1 chromosome end-binding protein complex stimulates telomerase activity and processivity provide incentive for testing variant telomerases in the presence of these factors. In the present work, we compare multiple disease-associated hTERT variants reconstituted with the RNA subunit hTR in two systems (rabbit reticulocyte lysates and human cell lines) with respect to telomerase enzymatic activity, processivity and activation by telomere proteins. Surprisingly, many of the previously reported disease-associated hTERT alleles give near-normal telomerase enzyme activity. It is possible that a small deficit in telomerase activity is sufficient to cause telomere shortening over many years. Alternatively, mutations may perturb functions such as the recruitment of telomerase to telomeres, which are essential in vivo but not revealed by simple enzyme assays.
INTRODUCTION
Complete replication of chromosome ends in most human cells requires telomerase. Telomerase is a ribonucleoprotein (RNP) enzyme, consisting of a template-containing RNA subunit known as TR, TER or TERC (1), a reverse transcriptase subunit TERT (2,3) and multiple accessory proteins including dyskerin (4,5). In the absence of telomerase, chromosomes undergo progressive telomere shortening as seen in human somatic cells that do not express TERT and as documented in budding and fission yeast mutants and in mice with TR or TERT gene knockouts (6).
Continued proliferation of human cells requires maintenance of telomere length by telomerase or by alternative mechanisms. The causal relationship between telomere shortening and replicative senescence was established by ectopic expression of telomerase reverse transcriptase (hTERT) in human somatic cells (7). Insufficient telomerase activity can cause telomere shortening, decreased cell proliferation and tissue degeneration. The rare inherited disorder dyskeratosis congenita (characterized by defects in highly proliferative tissues such as skin and bone marrow) has been traced to insufficient telomerase activity due to mutations in the dyskerin protein subunit (8), the telomerase RNA (9) and TERT (10), or in telomere components such as TINF2 and RTEL1 (11–13). Recently, other diseases including idiopathic pulmonary fibrosis and aplastic anemia have been associated with telomerase deficiency (14). Telomerase deficiency also contributes to multiple forms of cancer, presumably because short telomeres lead to genome instability which contributes to oncogenesis (15–18).
Heterozygotes with one defective allele of hTERT or hTR are unable to maintain highly proliferative tissues because of haploinsufficiency (10,14,19). Interestingly, a related situation occurs in yeast, where telomerase RNA is haploinsufficient for telomere length maintenance (20). Non-synonymous single-nucleotide mutations in the hTERT gene occur at ∼5% frequency; 44 different non-synonymous mutations have been identified in the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu) as of January 2013, and this number will grow quickly as the cost of genome sequencing continues to plummet. Therefore, knowing which of these amino acid changes has phenotypic consequences will only grow in importance.
Most of the human genetic studies have assessed the activity of variant telomerases by reconstitution of hTERT and hTR in rabbit reticulocyte lysates (RRLs) or by transient transfection of genes encoding mutant telomerase components into tissue culture cells. Telomerase activity is then measured either by a PCR-based assay called the Telomeric Repeat Amplification Protocol (TRAP) assay or by direct analysis of extension of a telomeric DNA primer. These methods have been recently reviewed (4). Here, we use the direct enzyme assay because it provides excellent quantification and information about the step in the enzymatic cycle that is defective while avoiding potential PCR artifacts (21).
In past studies, the direct telomerase assay has been carried out with synthetic DNA oligonucleotides of telomeric sequence (TTAGGG repeats) as primers. However, genetic and biochemical evidence indicates that the single-stranded DNA at chromosome ends in vivo is bound by a protein heterodimer, POT1-TPP1. POT1-TPP1 stimulates telomerase processivity (22), and this higher processivity would appear to be required for one or two rounds of telomerase engagement to achieve the in vivo lengthening of 50 nt per cell cycle (23). One hTERT mutant, G100V, has been found to be insensitive to POT1-TPP1 stimulation (24); therefore, it seems important to assess whether any of the disease-associated alleles might also have such a defect.
Here, we perform a comparative analysis of 24 disease-associated hTERT variants using the direct telomerase enzyme assay. These non-synonymous mutations span most of the 1132-amino-acid TERT protein, including the TEN domain that anchors the DNA primer, the TR RNA-binding domain, the RT catalytic domain that resembles that of retroviral and retrotransposon RTs, and the C-terminal extension (Figure 1a). Our goals are to provide quantitative information about the effects of single amino acid substitutions on telomerase activity and processivity, to compare telomerase assembled in vitro with the enzyme assembled in human cells and to test the mutants for their ability to respond to POT1-TPP1 activation. Unanticipated findings suggest a reinterpretation of some disease-associated hTERT variants described previously.
Figure 1.

Disease-associated mutations in hTERT have wide-ranging effects on telomerase activity in the RRL assay. (a) Map of the hTERT protein showing location of disease-associated alleles studied here. Directly below, amino acid sequence motifs; DAT, mutations in these regions dissociate the in vivo and in vitro activities of telomerase. TEN, RBD, RT and CTE, protein structural domains. (b) Expression of variant hTERT proteins in the RRL system (Top panel, SDS–PAGE and autoradiography) and direct telomerase assays (Bottom panel, PAGE and autoradiography). (c) Activity of mutant telomerases relative to WT. (d) Processivity calculated by the linear regression method.
MATERIALS AND METHODS
Mutagenesis
Mutations in hTERT were made using the QuickChange II XL site-directed mutagenesis kit (Stratagene) with primers designed as described (25), and all mutated genes were completely sequenced. All primers were purchased from IDT.
RRL reconstitution of telomerase
C-terminal HA-tagged human TERT was expressed from phTERT-HA2. N-terminal 3×FLAG-tagged human TERT was expressed from phTERT-3×FLAG, and hTR was expressed from phTR using the TnT quick-coupled transcription/translation system (Promega) as previously described (24). Each 500 μl reaction contained 400 μl of TnT quick mix, 8 μl of 35S-methionine (Perkin-Elmer), 10 μl of PCR enhancer (Promega), 10 μl of 1 mM methionine, 10 μg of supercoiled hTERT plasmid and 10 μg of purified hTR RNA (transcribed from Fok I-cut phTR plasmid DNA), and water to a total of 500 μl. After incubation for 2 h at 30°C, the reconstituted telomerase complex was affinity-purified for 2 h at 4°C on anti-HA F7 agarose beads (Santa Cruz Biotechnologies) or anti-FLAG agarose beads (Sigma). The beads were washed with 1× human telomerase buffer with 30% glycerol four times and then resuspended in the same solution. The quantity of 35S-hTERT was determined by liquid scintillation counting and used to normalize the telomerase activities. Unused beads were quick-frozen in liquid nitrogen and stored at −80°C.
Reconstitution of telomerase in human cells
HEK293T or VA13 cells (both from ATCC) were transiently transfected with plasmids expressing N-terminal 3×FLAG-tagged human TERT (WT or mutants) and human telomerase RNA (hTR). Parent plasmids were a gift of J. Lingner (EPFL, Lausanne) (26), and we added sequences encoding epitope tags and mutations. Cells were grown for 2 days, and extracts were made using CHAPS lysis buffer (26). Telomerase was purified using anti-FLAG agarose beads and the beads used in telomerase activity assays. Unused beads were quick-frozen in liquid nitrogen and stored at −80°C and retained substantial activity for at least 1 year.
Telomerase activity assays
Human telomerase complexes reconstituted in RRLs or human cell lines were immunopurified and analyzed by direct telomerase assays (24). Each reaction mixture (20 μl) contained 1× human telomerase assay buffer (50 mM Tris–HCl at pH 8.0, 50 mM KCl, 1 mM MgCl2, 5 mM 2-mercaptoethanol, 1 mM spermidine), 1.0 μM telomeric DNA primer a5 (TTAGGGTTAGGGTTAGCGTTA, where the underlined mutation serves to position POT1-TPP1 on the 5′ end of the primer), 0.5 mM dATP, 0.5 mM dTTP, 2.92 μM dGTP and 0.33 μM 32P-dGTP (3000 Ci/mmol, 1 Ci = 37GBq), with 6 μl of immunopurified telomerase complex. Reactions containing 0.5 μM POT1 or 0.5 μM TPP1 or both contained 0.05 μM primer instead of 1.0 μM primer a5. Reactions were incubated for 1 h at 30°C, then stopped by adding 100 μl of 3.6 M NH4OAc containing 20 μg of glycogen. Samples were precipitated by addition of 500 μl of ethanol for 1 h at −80°C, then centrifuged for 15 min at 4°C. Pellets were washed with 70% ethanol and resuspended in 10 μl of water followed by 10 μl of 2× loading buffer (94% formamide, 0.1× TBE, 0.1% bromophenol blue, 0.1% xylene cyanol). Samples were denatured by boiling and then loaded onto a 10% polyacrylamide/7 M urea/1× TBE denaturing gel for electrophoresis. Gels were dried and quantified by using a PhosphorImager (GE Healthcare).
Western blots
Proteins were separated by SDS–PAGE on 4–12% polyacrylamide Bis Tris Glycine gels (Novex, NP0321). The anti-FLAG antibody (Sigma A8592) is horseradish peroxidase (HRP) conjugated; therefore, it required no secondary antibody. In some cases, protein levels were confirmed by western blots using the anti-TERT antibody Abcam Ab32020, a rabbit monoclonal antibody raised against the 20 C-terminal amino acids of hTERT (Supplementary Figure S1). We found Ab32020 to have good specificity for hTERT; the signal depended on protein overexpression, and the presence of one amino acid substitution in the F1127L mutant eliminated the signal (Supplementary Figure S1).
Protein expression and purification
Proteins hPOT1 and hTPP1-N (amino acids 89–334) were expressed and purified as reported previously (22,27). Concentrations were determined by the Bio-Rad Protein Assay, based on the Bradford method; hPOT1 was typically 40–50% active, as determined by quantitative DNA-binding titrations.
Processivity quantification
The number of counts in each lane of a telomerase activity gel was determined using PhosphorImager scanning and ImageQuant software (28). The graph of ln F versus repeat number n was fit with a linear regression, and processivity = −0.693/slope. The first repeat was not included in the fit because telomerase synthesizes one or two repeats before synthesis is linear. Alternatively, in the ‘> 15 repeats method’, relative processivity was estimated as the counts from repeat 15 and above divided by the total number of counts in the lane (29).
RESULTS
The two systems most commonly used for expression and assembly of recombinant telomerase are RRLs and cultured human cells. Whether these systems are more-or-less interchangeable has been tested in only a few cases (30–32). The two systems are likely to differ in the pathway of telomerase assembly, in the type and quantity of molecular chaperones that facilitate RNP formation and in the availability of accessory proteins such as dyskerin and Nop10. These differences could have differential effects on different hTERT mutants, with the human cell system presumably more likely to approximate the in vivo situation. One goal of the present work is to determine the degree of concordance between telomerase assays performed in these two systems.
Assay of telomerase core enzymes assembled in RRLs
From the practical standpoint of the telomerase researcher, the RRL system has the advantage that 35S-methionine can be incorporated into the hTERT during translation. This provides a straightforward means of normalizing activity measurements to the amount of hTERT present in the assay. (Normalizing instead to the amount of bound hTR would obscure the effect of RNA-binding deficiencies; a mutant with 1% RNA-binding activity, when normalized to hTR, would show 100% activity.) The quality of the protein expressed can be readily determined by SDS–PAGE; as seen in the top panel of Figure 1b, most hTERT alleles were expressed with similar efficiency and gave homogeneous full-length proteins (the exception was Y772C, which always showed a shorter product as well as the full-length protein; not shown).
Representative telomerase assays are shown in Figure 1b, bottom panel. A telomeric oligonucleotide primer is extended in the presence of 32P-dGTP; the telomerase enzyme stalls or dissociates when it reaches the end of the TR RNA template, producing the 6 nt repeats characteristic of telomerase. The reproducibility of the assay was assessed by performing triplicate reactions (Samples 1, 2 and 3) on each of two biological replicates (RRL Translation 1 and 2). Reactions performed independently by two different researchers using two different epitope tags on the TERT gave similar results (Supplementary Figure S2). Although we sometimes add a radiolabeled oligonucleotide before DNA precipitation as a loading control, this controls for only one of many steps that impact the precision of the assays. Therefore, in this study, we instead performed replicate assays to assess reproducibility. Occasional loss of a DNA pellet (12th lane in Figure 1b) is apparent even without a loading control.
‘Activity’ was measured as the total amount of radiolabeled nucleotide incorporated into the primer in a 1 h reaction (Figure 1c). ‘Processivity’, the average number of DNA repeats synthesized after a single primer-binding event, was determined by analyzing the distribution of products as described in ‘Materials and Methods’ section (Figure 1d); qualitatively, enzymes with wild-type or higher processivity produced a full ladder of extension products, whereas others [K570N and R865H; see also (34)] were defective in processivity, synthesizing mostly one repeat (Figure 1b). Enzymes with extremely low activity (such as G682D) could not be assessed for processivity. The quantification is summarized in Table 1.
Table 1.
Activity and processivity measurements for telomerases containing hTERT mutations
| Allele | Frequency | Disease | RRL activity | HEK activity | VA-13 activity | Published activity | Reference | RRL Processivity | HEK processivity |
|---|---|---|---|---|---|---|---|---|---|
| P33S | 0 | IPF | 0.72 ± 0.06 | 0.80 ± 0.2; (TRAP); ∼1 (direct) | (33;34) | 1.0 ± 0.1 | |||
| L55Q | 0 | IPF | 0.28 ± 0.006 | 0.37 ± 0.07 (direct); ∼0.5 (direct) | (35;34) | 1.1 ± 0.1 | |||
| P65A | 0 | AML | 0.61 ± 0.06 | 0 (TRAP) | (18) | 1.1 ÷ 0.1 | |||
| S191T | 0.0008 | None | 0.97 ± 0.29, 1.02 ± 0.10 | 1.14 ± 0.11 | 1.04 ± 0.05 | ND | 1.1 ± 0.2, 1.02 ± 0.16 | 0.99 ± 0.05 | |
| A202T | 0.0002 | AA | 0.88 ± 0.20 | 0.83 ± 0.19 | 0.53 ± 0.03 | <0.01 (TRAP) | (19) | 1.1 ± 0.1 | 1.1 ± 0.08 |
| A279T | 0.022 | None | 0.79 ± 0.20 | 0.81 ± 0.40 (TRAP); 0.95 ± 0.08 (direct) | (36;30) | 1.1 ± 0.1 | |||
| V299M | 0 | AML | 0.74 ± 0.20 | 1.10 (TRAP) | (18) | 1.1 ± 0.1 | |||
| H412Y | 0.0032 | a | 1.07 ± 0.20 | 1.20 ± 0.20 | 0.76 ± 0.03 | 0.36 − 0.50 (TRAP); 0.85 − 1.0 (direct) | (19,36;30) | 1.2 ± 0.1 | 1.05 ± 0.07 |
| 441del | 0 | AML | 0.82 ± 0.07 | 0.42 ± 0.08 (TRAP) | (18) | 1 ± 0.1 | |||
| R522K | 0 | AML | 1.05 ± 0.15 | 0.62 ± 0.03 (TRAP) | (18) | 1.1 ± 0.1 | |||
| K570N | 0 | AA | 0.19 ± 0.02, 0.22 ± 0.02 | 0.42 ± 0.03 | <0.01 (TRAP) | (37) | 0.09 ± 0.01 | 0.00 ± 0.0008 | |
| G682D | 0 | AA | 0.02 ± 0.006 | <0.01 (TRAP) | (37) | ND | |||
| V694M | 0 | AA | 0.42 ± 0.03, 0.41 ± 0.03 | 0.10 ± 0.02 | 0.16 ± 0.01 | <0.01 (TRAP) | (19) | 0.7 ± 0.1, 1.59 ± 0.15 | 0.77 ± 0.08 |
| P704S | 0 | DC, AA | 0.01 ± 0.006 | 0.13 ± 0.10 (TRAP) | (38) | ND | |||
| P721R | 0 | DC | 0.65 ± 0.11 | 0.2 − 1.0 (TRAP); ∼1 (direct) | (37;34) | 1.1 ± 0.1 | |||
| T726M | 0.0003 | AA | 0.79 ± 0.25 | 0.2 − 1.0 (TRAP) | (37) | 1 ± 0.1 | |||
| Y772C | 0 | AA | 0.12 ± 0.030 | <0.01 (TRAP) | (19) | 0.6 ± 0.1 | |||
| R865H | 0.0001 | IPF | 0.19 ± 0.08, 0.18 ± 0.02 | 0.17 ± 0.03 | 0.20 ± 0.01 | 0.28 (TRAP) | (33) | 0.2 ± 0.0, 0.09 ± 0.007 | 0.004 ± 0.001 |
| K902N | 0 | DC, AA | 0.15 ± 0.07, 0.02 ± 0.003 | 0.01 ± 0.001 | 0 (direct) | (10) | ND | ND | |
| S948R | 0 | None | 1.25 ± 0.61, 0.92 ± 0.07 | 0.58 ± 0.07 | ND | 1.4 ± 0.1, 1.55 ± 0.09 | 2.38 ± 0.40 | ||
| R979W | 0 | DC, AA | 0.38 ± 0.08 | 0.2 − 1.0 (TRAP) | (37) | 0.5 ± 0.1 | |||
| A1062T | 0.016 | a | 1.31 ± 0.40, 1.09 ± 0.08 | 1.08 ± 0.20 | 0.82 ± 0.14 | 0.62 ± 0.03 (TRAP) | (18) | 0.8 ± 0.1, 0.94 ± 0.09 | 1.01 ± 0.01 |
| V1090M | 0.0002 | AA | 0.73 ± 0.04 | <0.01 (TRAP) | (19) | 1 ± 0.1 | |||
| F1127L | 0 | DC | 0.71 ± 0.06 | 0.72 ± 0.09 | 0.2 − 1.0 (TRAP); ∼1 (direct) | (37;34) | 0.6 ± 0.1 | 0.442 ± 0.05 | |
| DN | 0 | None | ND | ∼0 | 0 | (39) | ND | ND |
Frequency: Allele frequency based on 12 000 genes, NHBLI Exome Sequencing Project. Frequency = 0, allele found in a family described in the literature but not in the Exome Sequencing Project.
RRL activity, HEK activity and VA13 activity, direct assay results from this study.
Published activity, references given in the same order as the activity values.
RRL processivity determined by the linear regression method, and HEK processivity determined by the >15 repeats method, all from this study.
Disease: IPF, idiopathic pulmonary fibrosis; AML, acute myloid leukemia; AA, aplastic anemia; DC, dyskeratosis congenita.
aThese alleles were originally reported as disease-associated, but the disease association is now thought to be modest [discussed in (14)].
All hTERT genes were subjected to complete DNA sequence analysis by the Sanger method to confirm the presence of the mutation and the absence of any other change. Nevertheless, in cases where the activity of an allele seemed to differ from that reported in the literature, we sought additional evidence that our site-specific mutagenesis did not produce a second mutation. This was accomplished by using oligonucleotide-directed mutagenesis to revert the mutant allele back to wild-type, a procedure that would retain any hypothetical mutations away from the targeted site (Supplementary Figure S3). The reversion to wild-type activity provided additional confidence that loss of activity was due only to the disease-associated mutation.
Assay of telomerase assembled in cultured human cells
This approach starts with overexpression of both hTR and hTERT in human embryonic kidney (HEK) 293T cells (26,40). An N-terminal 3×FLAG tag on hTERT gave a strong and unobstructed signal on western blots (Figure 2a), allowing the amount of hTERT protein in the extract to be measured and adjusted such that each assay contained an equal amount of hTERT. The 3×FLAG tag also allowed immunopurification of telomerase on antibody-coated beads, which were then used in a direct assay for telomerase activity with a telomeric oligonucleotide primer and 32P-dGTP (Figure 2b). Activity and processivity for telomerases containing disease-associated hTERT mutations were quantified, and the results are summarized in Table 1.
Figure 2.

Disease-associated variants of hTERT expressed in HEK293T cells with hTR and analyzed by the direct assay. (a) Western blot with an anti-hTERT antibody showing uniform expression of the alleles in triplicate. It is not known whether the minor breakdown of S948R hTERT contributes to its reduced activity or enhanced processivity. (b) Direct telomerase assays.
A number of control experiments provided confidence that the observed differences in activity were due to the hTERT mutations rather than variability in the assay. First, the activity obtained in independent transfections of the HEK293T cells was as reproducible as that seen with replicate assays of the same extract (Figure 2b). Second, the amounts of hTERT and hTR plasmids transfected into cells were optimized, and small fluctuations in the amount of DNA transfected would have little effect on the measured telomerase activity (Supplementary Figure S4). Third, the measured activity was a linear function of the amount of telomerase used (Supplementary Figure S5), a fundamental requirement for studies of enzyme activity. Finally, the 1-h reaction time was approximately in the linear range (Supplementary Figure S6a and b); the fall-off in incorporation at longer times was due to slow inactivation of the enzyme, as determined by enzyme pre-incubation studies (Supplementary Figure S6c and d). Based on these data, we would recommend a 20 or 30 min standard time for future studies.
The reproducibility of the data, as determined by SEM, was similar for the HEK cell assays and the RRL assays. The key question, then, is whether these two systems give equivalent values of telomerase activity. A direct comparison is shown in Figure 3. For most alleles, the two systems gave the same activity measurement, but in four cases, the differences were statistically significant (P < 0.0001). These results serve to emphasize that the information one really wants to know is the telomerase activity in the relevant human tissue, and measurements of telomerase assembled in human cells or in RRLs are imperfect surrogates for this information.
Figure 3.
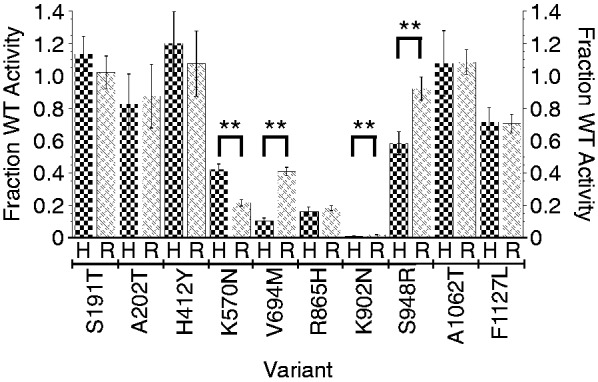
Comparison of relative activities of telomerases expressed in HEK cells (H) and RRLs (R). Error bars = SEM, n = 4–18. **Statistically significant differences (P < 0.0001).
To begin to assess whether the choice of human cell line might affect telomerase activity, we performed a more limited analysis transfecting hTR and various hTERT alleles into WI38 VA13/2RA cells, a telomerase-negative cell line that maintains telomeres by alternative lengthening of telomeres (41) [for a typical example of prior work, see (36)]. Telomerase activities of the hTERT alleles in the VA13 cells were consistent with those obtained in the HEK cells (Supplementary Figure S7). There were, however, some quantitative differences; for example, V694M had a relative activity of 0.16 ± 0.01 in VA13 cells compared with 0.10 ± 0.02 in HEK cells. We conclude that if it were sufficient to group hTERT alleles into three categories—essentially wild-type activity, substantially reduced activity (10–50%) and loss of function (<10%)—then HEK cells, VA13 cells and RRLs give equivalent results. However, at the present time, we do not know whether these broad categories are sufficient or whether instead a 10 or 20% reduction in telomerase activity might be important over a human lifespan.
hTERT mutants respond to POT1-TPP1 activation
The human telomere DNA-binding protein heterodimer POT1-TPP1 stimulates telomerase processivity (22) and recruitment to telomeres (29,42). Because the telomerase at chromosome ends is likely to be associated with these proteins in vivo, we tested the alleles in the presence of saturating amounts of POT1-TPP1. Representative telomerase enzyme assays are shown in Figure 4. Quantification revealed that all of the disease-associated alleles that retained some activity were activated by POT1-TPP1 (e.g. S948R, A1062T, F1127L and WT all showed >7-fold processivity enhancement relative to no POT1-TPP1 when the data in Figure 4 were analyzed by the >15 repeats method). The R865H processivity mutant clearly had processivity restored in the presence of POT1-TPP1, but the low level made quantification difficult. The extremely low activity of mutant K902N was slightly increased but not rescued by POT1-TPP1. We questioned whether the high-molecular-weight ‘smear’ near the top of the gel for active alleles really represented telomerase products; however, extending the electrophoresis time to resolve these products revealed that they represented a continuation of the 6 nt repeat ladder (Supplementary Figure S8); therefore, they were included in the quantification. In conclusion, although the previously described G100V mutant of hTERT shows that a POT1-TPP1-insensitive mutant is possible, none of the disease-associated mutants tested here had this property.
Figure 4.

Telomerases assembled from WT and mutant hTERT assayed for activity in the presence and absence of telomere proteins. Telomerase was reconstituted in RRLs and activity measured by the standard assay (−) or in the presence of 500 nM POT1 (P), 500 nM TPP1 (T) and 500 nM of each protein (P/T), concentrations high enough to saturate the oligonucleotide primer (22). +2 and +4, markers for primer extended by two and four nt, respectively.
DISCUSSION
Insufficient telomerase activity in highly proliferative tissues causes telomere shortening, which can lead to decreased cell proliferation and tissue degeneration or to genome instability and cancer. Because of haploinsufficiency, even a single codon substitution in one of the two alleles of hTERT can contribute to disease. Thus, the question of which hTERT sequence variants are hypomorphic is of great interest. Previous studies each compared a small number of telomerases reconstituted with hTERT proteins containing disease-associated amino acid substitutions. Here, we present a side-by-side analysis of a much larger set of disease-associated variants. Our quantitative analysis of the activities of 24 mutant telomerases gives new insights into the precision and accuracy of such measurements, and it reveals that many reported disease-associated mutations have unexpectedly little effect on telomerase enzyme activity per se.
Many disease-associated hTERT variants do not show much decrease in telomerase enzyme activity
Many published articles have shown that an hTERT variant is associated with dyskeratosis congenita or another disease of stem cell deficiency and that the subjects harboring the mutant gene have short telomeres (14,43). The reasonable expectation is then that this hTERT mutant will be defective in telomerase activity, which has been reported in many cases (14,43). We were therefore surprised to find that 10 of the 19 disease-associated alleles tested did not exhibit substantial loss of function of reconstituted telomerase (i.e. they had 61–100% of wild-type activity; as described later in the text, an allele with 60% activity is expected to give 80% overall activity when ‘averaged’ with the wild-type allele in a heterozygous individual). Robart and Collins (34) and Zhong et al. (42) also noted a lack of correlation between disease association and decreased telomerase activity for a limited set of alleles.
The lack of the expected correlation could have a number of explanations. First, a modest deficit in telomerase activity (10–20% decrease) could be sufficient to cause telomere shortening for a period of many years. This might be especially apparent in individuals who inherited short telomeres. Second, the activities of hTERT alleles in vivo may not be well represented by the simple in vitro enzyme assays. For example, an hTERT mutation could inhibit telomerase recruitment to telomeres in vivo but have no effect on enzyme activity per se (42,44). Third, an individual’s genetic background would be expected to enhance (or suppress) the effect of hTERT mutations. Candidate genes include those for transcription factors such as c-Myc and Ets2, which drive transcription of the hTERT gene (45), and those for telomere proteins including TPP1, which contributes to telomerase recruitment to telomeres and telomere elongation (22,29,42). Finally, some hTERT variants reported as disease-associated may not be causally related to the disease.
Comparison of methods for measuring activity
Two different methods of reconstituting the telomerase—transcription/translation in RRLs and transfection of plasmids into human cells—generally gave the same results. However, in the cases of V694M and S948R (and also the non-processive K570N), the SEM was small enough that we conclude that the two systems give different average values. There are many potential differences between the two systems—the assembly pathway, the availability of molecular chaperones, the concentration of telomerase-accessory proteins such as dyskerin and Nop10 and post-transcriptional and post-translational modifications—and it is not surprising that these could differentially affect the assembly of mutant and wild-type telomerase RNP complexes. If it were sufficient to know whether a telomerase mutation has little or no effect (>50% activity), is hypomorphic (10–50% active) or is essentially inactivating (<10% activity), then it appears that one can use either RRLs or human cells and achieve the same result, in agreement with the more limited comparisons done previously (30–32). As described earlier in the text, however, it is currently unknown whether these broad categories are sufficient to predict telomerase haploinsufficiency.
In stark contrast to the general agreement between telomerase reconstituted in vitro and in human cells, only about half of our direct assay measurements agreed even qualitatively with published TRAP assay values. Differences in cell lines are unlikely to explain the discrepancies because in our hands, multiple alleles gave similar activity in HEK cells as in the VA13 cells used in many previous reports. The discordance was mostly in the direction of the direct measurements showing greater activity. This was especially apparent with the P65A, A202T and V1090M alleles, which gave 61–88% of wild-type activity in our direct assays and <1% by TRAP. Our primer extension assays use a low dGTP concentration relative to that used in TRAP, which could result in a high TRAP/direct assay ratio for a mutant defective in nucleotide binding. Such a situation is opposite to what we observe; therefore, it cannot explain the low TRAP assay values. We have no basis for concluding whether false-negative TRAP measurements are revealing general challenges with using this assay (4) or are specific to a particular study.
The direct assay revealed that the K570N mutation destroys telomerase processivity. We considered that the discrepancy between our activity measurements (in the 20–40% range) and the TRAP assay (<1%) could be explained by short products not being effectively amplified in the TRAP assay. However, the R865H mutant also has low processivity (34), almost as low as K570N (see Table 1), and in that case, our direct assay of activity matched the published TRAP values. In any case, these comparisons do serve to point out one obvious advantage of the direct assay: it gives information about both activity and processivity, whereas the ‘ladder’ of products observed by TRAP is produced by PCR and is not related to enzyme processivity in any simple way (4,46).
Activity in heterozygotes
Because most disease-associated alleles of hTERT or of hTR occur in heterozygotes, a number of researchers have co-assembled mutant and wild-type alleles to test for dominant negative inhibition. In most cases, they observed activity that was intermediate between that of the individual alleles, indicating that the disease phenotype is due to haploinsufficiency rather than dominant negative inhibition (10,18,19,33,37,38,47,48). The one exception is the report by Sauerwald et al. (49), in which the catalytically inactive DN allele was found to poison the activity of a co-assembled wild-type subunit. We coexpressed the K570N, R865H, K902N and S948R alleles with wild-type hTERT in HEK293T cells and found the activity to be the average of the wild-type and mutant alleles (A.J.Z. and T.R.C., unpublished data). Taken together, these data support the conclusion that the activity of a variant allele plus a wild-type allele in a heterozygote can in most cases be calculated from the measured activity of the variant allele when present in pure form (i.e. a mutant with 10% activity would be predicted to give (10 + 100)/2 = 55% activity in the context of a heterozygote).
Activity in presence of telomere proteins
The findings that the human telomere end-binding protein complex, POT1-TPP1, stimulates telomerase activity and processivity (22) and that the interaction between TPP1 and telomerase is essential for activity in vivo (29,42) would appear to mandate assaying all mutant hTERTs in the presence of POT1-TPP1. However, all active telomerases tested here were stimulated by POT1-TPP1. The G100V mutant of hTERT (an engineered mutant, not found to be disease associated) is not stimulated by POT1-TPP1 (24), and some disease-associated alleles not studied here may be defective in this interaction (42).
Prediction of mutant hTERT activity in silico
Given the slow and tedious process of testing hTERT variants for activity by the direct telomerase enzyme assay, the field would benefit greatly from methods for predicting activity computationally. For some of the mutated sites, such as R865 and K902, inspection of the Tribolium TERT crystal structure (Supplementary Figure S9) shows close proximity to the active site and suggests that amino acid substitutions should be deleterious, and this is the case (activity levels of 18 and 2%, respectively). However, only the experimental approach could reveal the processivity defect in R865H, and in any case, the lack of structural information about the human telomerase RNP prevents this approach from being generally useful. The PolyPhen-2 software tool predicts the probability that an amino acid substitution will be damaging to the structure and function of a human protein using physical and evolutionary (comparative sequence) criteria (50). For about half of the variants, the PolyPhen-2 prediction agrees qualitatively with our activity measurements (Supplementary Figure S10). For most of the other variants, PolyPhen-2 is too pessimistic (i.e. it overestimates the effect of the mutation); alternatively, for one case (G682D) PolyPhen-2 predicts that the mutation should be benign when it is deleterious. Thus, although it should eventually be possible to predict the effects of single amino acid substitutions in silico, experimental measurements are likely to be required for the foreseeable future.
Concluding remarks
There is a general sense in the human telomerase field that the most common sequence variants are polymorphisms without phenotypic consequence, whereas the disease-causing variants are de novo mutations that are rare in the population. The four non-synonymous single-nucleotide variants found with highest frequency in the NHBLI Exome Sequencing Project (S191T, H412Y, A1062T and A279T; 10–275 occurrences/12 000 genes) all have high enzymatic activity in our study, and all of the deleterious variants are rare in the population (0 or 1 occurrence in the Exome Sequencing Project). Thus, although more data on allele frequencies are needed before making a firm conclusion, our work supports the prevailing generalization that most disease-causing hTERT mutations will be rare in the population.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online, including [51].
ACKNOWLEDGEMENTS
The authors thank David McKay (CU-Boulder) for molecular modeling and Mary Armanios (Johns Hopkins), Monica Bessler (Children’s Hospital of Philadelphia), Neal Young (NIH) and Dan Roden (Vanderbilt) for stimulating discussions. They thank Joachim Lingner (EPFL, Lausanne) for plasmids and discussions.
FUNDING
A sabbatical grant from DePauw University (to S.M.C.); EXROP student of the Howard Hughes Medical Institute (to K.C.). A.J.Z. is a senior scientist and T.R.C. is an investigator of the HHMI. NIH grant [R01 GM099705 to T.R.C. (in part)]. Funding for open access charge: Howard Hughes Medical Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1.Greider CW, Blackburn EH. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature. 1989;337:331–337. doi: 10.1038/337331a0. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura TM, Morin GB, Chapman KB, Weinrich SL, Andrews WH, Lingner J, Harley CB, Cech TR. Telomerase catalytic subunit homologs from fission yeast and human. Science. 1997;277:955–959. doi: 10.1126/science.277.5328.955. [DOI] [PubMed] [Google Scholar]
- 3.Meyerson M, Counter CM, Eaton EN, Ellisen LW, Steiner P, Caddle SD, Ziaugra L, Beijersbergen RL, Davidoff MJ, Liu Q, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90:785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 4.Podlevsky JD, Chen JJ. It all comes together at the ends: telomerase structure, function, and biogenesis. Mutat. Res. 2012;730:3–11. doi: 10.1016/j.mrfmmm.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nandakumar J, Cech TR. Finding the end: recruitment of telomerase to telomeres. Nat. Rev. Mol. Cell. Biol. 2013;14:69–82. doi: 10.1038/nrm3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strong MA, Vidal-Cardenas SL, Karim B, Yu H, Guo N, Greider CW. Phenotypes in mTERT(+)/(−) and mTERT(−)/(−) mice are due to short telomeres, not telomere-independent functions of telomerase reverse transcriptase. Mol. Cell. Biol. 2011;31:2369–2379. doi: 10.1128/MCB.05312-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 8.Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- 9.Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- 10.Armanios M, Chen JL, Chang YP, Brodsky RA, Hawkins A, Griffin CA, Eshleman JR, Cohen AR, Chakravarti A, Hamosh A, et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc. Natl Acad. Sci. USA. 2005;102:15960–15964. doi: 10.1073/pnas.0508124102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet. 2008;82:501–509. doi: 10.1016/j.ajhg.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ballew BJ, Yeager M, Jacobs K, Giri N, Boland J, Burdett L, Alter BP, Savage SA. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum. Genet. 2013;132:473–480. doi: 10.1007/s00439-013-1265-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am. J. Hum. Genet. 2013;92:448–453. doi: 10.1016/j.ajhg.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trudeau MA, Wong JM. Genetic Variations in Telomere Maintenance, with Implications on Tissue Renewal Capacity and Chronic Disease Pathologies. Curr. Pharmacogenomics Person. Med. 2010;8:7–24. doi: 10.2174/1875692111008010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 16.De Lange T. Telomere-related genome instability in cancer. Cold Spring Harb. Symp. Quant. Biol. 2005;70:197–204. doi: 10.1101/sqb.2005.70.032. [DOI] [PubMed] [Google Scholar]
- 17.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009;113:6549–6557. doi: 10.1182/blood-2008-12-192880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calado RT, Regal JA, Hills M, Yewdell WT, Dalmazzo LF, Zago MA, Lansdorp PM, Hogge D, Chanock SJ, Estey EH, et al. Constitutional hypomorphic telomerase mutations in patients with acute myeloid leukemia. Proc. Natl Acad. Sci. USA. 2009;106:1187–1192. doi: 10.1073/pnas.0807057106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaguchi H, Calado RT, Ly H, Kajigaya S, Baerlocher GM, Chanock SJ, Lansdorp PM, Young NS. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med. 2005;352:1413–1424. doi: 10.1056/NEJMoa042980. [DOI] [PubMed] [Google Scholar]
- 20.Mozdy AD, Cech TR. Low abundance of telomerase in yeast: implications for telomerase haploinsufficiency. RNA. 2006;12:1721–1737. doi: 10.1261/rna.134706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Cian A, Cristofari G, Reichenbach P, De Lemos E, Monchaud D, Teulade-Fichou MP, Shin-Ya K, Lacroix L, Lingner J, Mergny JL. Reevaluation of telomerase inhibition by quadruplex ligands and their mechanisms of action. Proc. Natl Acad. Sci. USA. 2007;104:17347–17352. doi: 10.1073/pnas.0707365104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang F, Podell ER, Zaug AJ, Yang Y, Baciu P, Cech TR, Lei M. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature. 2007;445:506–510. doi: 10.1038/nature05454. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Y, Sfeir AJ, Zou Y, Buseman CM, Chow TT, Shay JW, Wright WE. Telomere extension occurs at most chromosome ends and is uncoupled from fill-in in human cancer cells. Cell. 2009;138:463–475. doi: 10.1016/j.cell.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaug AJ, Podell ER, Nandakumar J, Cech TR. Functional interaction between telomere protein TPP1 and telomerase. Genes Dev. 2010;24:613–622. doi: 10.1101/gad.1881810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cristofari G, Lingner J. Telomere length homeostasis requires that telomerase levels are limiting. EMBO J. 2006;25:565–574. doi: 10.1038/sj.emboj.7600952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lei M, Podell ER, Cech TR. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat. Struct. Mol. Biol. 2004;11:1223–1229. doi: 10.1038/nsmb867. [DOI] [PubMed] [Google Scholar]
- 28.Latrick CM, Cech TR. POT1-TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J. 2010;29:924–933. doi: 10.1038/emboj.2009.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nandakumar J, Bell CF, Weidenfeld I, Zaug AJ, Leinwand LA, Cech TR. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature. 2012;492:285–289. doi: 10.1038/nature11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alder JK, Chen JJ, Lancaster L, Danoff S, Su SC, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl Acad. Sci. USA. 2008;105:13051–13056. doi: 10.1073/pnas.0804280105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie M, Podlevsky JD, Qi X, Bley CJ, Chen JJ. A novel motif in telomerase reverse transcriptase regulates telomere repeat addition rate and processivity. Nucleic Acids Res. 2010;38:1982–1996. doi: 10.1093/nar/gkp1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D'Souza Y, Chu TW, Autexier C. A translocation-defective telomerase with low levels of activity and processivity stabilizes short telomeres and confers immortalization. Mol. Biol. Cell. 2013;24:1469–1479. doi: 10.1091/mbc.E12-12-0889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC, Rosenblatt RL, Shay JW, Garcia CK. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl Acad. Sci. USA. 2007;104:7552–7557. doi: 10.1073/pnas.0701009104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robart AR, Collins K. Investigation of human telomerase holoenzyme assembly, activity, and processivity using disease-linked subunit variants. J. Biol. Chem. 2010;285:4375–4386. doi: 10.1074/jbc.M109.088575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA, 3rd, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007;356:1317–1326. doi: 10.1056/NEJMoa066157. [DOI] [PubMed] [Google Scholar]
- 36.Du HY, Pumbo E, Ivanovich J, An P, Maziarz RT, Reiss UM, Chirnomas D, Shimamura A, Vlachos A, Lipton JM, et al. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood. 2009;113:309–316. doi: 10.1182/blood-2008-07-166421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xin ZT, Beauchamp AD, Calado RT, Bradford JW, Regal JA, Shenoy A, Liang Y, Lansdorp PM, Young NS, Ly H. Functional characterization of natural telomerase mutations found in patients with hematologic disorders. Blood. 2007;109:524–532. doi: 10.1182/blood-2006-07-035089. [DOI] [PubMed] [Google Scholar]
- 38.Du HY, Pumbo E, Manley P, Field JJ, Bayliss SJ, Wilson DB, Mason PJ, Bessler M. Complex inheritance pattern of dyskeratosis congenita in two families with 2 different mutations in the telomerase reverse transcriptase gene. Blood. 2008;111:1128–1130. doi: 10.1182/blood-2007-10-120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, Beijersbergen RL, Knoll JH, Meyerson M, Weinberg RA. Inhibition of telomerase limits the growth of human cancer cells. Nat. Med. 1999;5:1164–1170. doi: 10.1038/13495. [DOI] [PubMed] [Google Scholar]
- 40.Fu D, Collins K. Distinct biogenesis pathways for human telomerase RNA and H/ACA small nucleolar RNAs. Mol. Cell. 2003;11:1361–1372. doi: 10.1016/s1097-2765(03)00196-5. [DOI] [PubMed] [Google Scholar]
- 41.Bryan TM, Marusic L, Bacchetti S, Namba M, Reddel RR. The telomere lengthening mechanism in telomerase-negative immortal human cells does not involve the telomerase RNA subunit. Hum. Mol. Genet. 1997;6:921–926. doi: 10.1093/hmg/6.6.921. [DOI] [PubMed] [Google Scholar]
- 42.Zhong FL, Batista LF, Freund A, Pech MF, Venteicher AS, Artandi SE. TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell. 2012;150:481–494. doi: 10.1016/j.cell.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armanios M, Blackburn EH. The telomere syndromes. Nat. Rev. Genet. 2012;13:693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banik SS, Guo C, Smith AC, Margolis SS, Richardson DA, Tirado CA, Counter CM. C-terminal regions of the human telomerase catalytic subunit essential for in vivo enzyme activity. Mol. Cell. Biol. 2002;22:6234–6246. doi: 10.1128/MCB.22.17.6234-6246.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu D, Dwyer J, Li H, Duan W, Liu JP. Ets2 maintains hTERT gene expression and breast cancer cell proliferation by interacting with c-Myc. J. Biol. Chem. 2008;283:23567–23580. doi: 10.1074/jbc.M800790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alder JK, Cogan JD, Brown AF, Anderson CJ, Lawson WE, Lansdorp PM, Phillips JA, III, Loyd JE, Chen JJ, Armanios M. Ancestral mutation in telomerase causes defects in repeat addition processivity and manifests as familial pulmonary fibrosis. PLoS Genet. 2011;7:e1001352. doi: 10.1371/journal.pgen.1001352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marrone A, Walne A, Tamary H, Masunari Y, Kirwan M, Beswick R, Vulliamy T, Dokal I. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood. 2007;110:4198–4205. doi: 10.1182/blood-2006-12-062851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Errington TM, Fu D, Wong JM, Collins K. Disease-associated human telomerase RNA variants show loss of function for telomere synthesis without dominant-negative interference. Mol. Cell. Biol. 2008;28:6510–6520. doi: 10.1128/MCB.00777-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sauerwald A, Sandin S, Cristofari G, Scheres SH, Lingner J, Rhodes D. Structure of active dimeric human telomerase. Nat. Struct. Mol. Biol. 2013;20:454–460. doi: 10.1038/nsmb.2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mitchell M, Gillis A, Futahashi M, Fujiwara H, Skordalakes E. Structural basis for telomerase catalytic subunit TERT binding to RNA template and telomeric DNA. Nat. Struct. Mol. Biol. 2010;17:513–518. doi: 10.1038/nsmb.1777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.