


Abstract
Localized hyper-mutability caused by accumulation of lesions in persistent single-stranded (ss) DNA has been recently found in several types of cancers. An increase in endogenous levels of reactive oxygen species (ROS) is considered to be one of the hallmarks of cancers. Employing a yeast model system, we addressed the role of oxidative stress as a potential source of hyper-mutability in ssDNA by modulation of the endogenous ROS levels and by exposing cells to oxidative DNA-damaging agents. We report here that under oxidative stress conditions the majority of base substitution mutations in ssDNA are caused by erroneous, DNA polymerase (Pol) zeta-independent bypass of cytosines, resulting in C to T transitions. For all other DNA bases Pol zeta is essential for ROS-induced mutagenesis. The density of ROS-induced mutations in ssDNA is lower, compared to that caused by UV and MMS, which suggests that ssDNA could be actively protected from oxidative damage. These findings have important implications for understanding mechanisms of oxidative mutagenesis, and could be applied to development of anticancer therapies and cancer prevention.
INTRODUCTION
Localized somatic hyper-mutability is a phenomenon of accumulating multiple mutations in a small section of the genome. It may lead to a rapid increase in fitness (1) or contribute to tumorigenesis (2,3). Several lines of evidence suggest that the persistence of long stretches of single-stranded DNA (ssDNA) either in the vicinity of a double-strand break (DSB) or at uncoupled replication forks can lead to accumulation of such clustered mutations (2–6). Both acute and chronic exposure to exogenous or endogenous mutagenic factors increase the incidence of mutations and amplify the probability of clustered, multiple genomic variations allowing for survival and further clonal expansion of malignant cells (7). The inability of the majority of DNA repair systems to process lesions in single-stranded substrates is likely to increase the contribution of ssDNA damage to hyper-mutability. Therefore mechanisms and origins of spontaneous and induced mutagenesis ssDNA are of special interest.
The levels of spontaneous mutations in ssDNA can be orders of magnitude higher than in dsDNA (4,6). Hyper-mutability may be caused by increased rate of errors during re-synthesis of the second DNA strand and/or by lesions inflicted by the products of cellular metabolism. Exposure to DNA-damaging agents (MMS, UV-irradiation and sulfites) further amplifies hyper-mutability of ssDNA (6,8,9). Oxidative stress defines the shape of multiple biological processes since normal aerobic metabolism generates a highly reactive, oxidizing environment for all macromolecules in living organisms. Increased levels of reactive oxygen species (ROS) are implicated in the etiology and progression of pathological conditions such as cancer (10), chronic inflammation (11) and neurodegenerative diseases (12). Oxidative stress is known to be a threat to genomic integrity (13–15).
ROS damage DNA and produce dozens of chemically distinct lesions that distort the structure and coding properties of DNA and may cause mutations and chromosomal aberrations. Despite numerous reports on the correlation between elevated cellular levels of ROS and increased mutations and chromosome aberrations rates (14,16), the precise mechanisms of ROS-induced genome destabilization remain poorly understood. Delineation of the pathways, contributing to ROS-induced genome instability is complicated by several factors including (i) redundancy and overlapping specificities of DNA damage repair and tolerance pathways (17); (ii) the ability of the replication fork to bypass oxidative damage with relatively high efficiency (18,19); and (iii) the absence of simple and reliable methods for detection and quantification of oxidative DNA damage in vivo as well as the low sensitivity of existing methods for determining endogenous ROS levels.
The major confounding factor in defining the potential to generate mutagenic lesions and the mutagenic signature of both endogenous and exogenous oxidative damage appears to be the redundancy of pathways for repairing oxidative DNA damage. The majority of enzymes involved in the DNA base excision repair (BER) pathway, which is the major pathway for repair of oxidative DNA damage, have a broad spectrum of substrates and can replace each other at the initial steps of repair. Furthermore, when the capacity of BER is severely compromised, the nucleotide excision repair (NER) pathway, normally involved in processing of bulky DNA lesions can function in the repair of a sub-set of oxidative DNA lesions (17). To make the picture even more complex, the post-replicative mismatch repair system can also recognize and remove 8-oxoG-A mis-pairs (20). Therefore, defining an unaltered mutagenic signature of oxidative damage requires simultaneous elimination of multiple systems of protection acquired during the evolution of aerobic organisms. In principle, this can be achieved through analysis of the mutagenesis in strains in which multiple DNA damage-handling genes are compromised. Although such approaches has been used by several groups (21,22) it is likely, that misinterpretation of the mutation signature data occurs due to the indirect effects of multiple mutations in the cellular background. For example, a complete elimination of both BER and NER pathways in yeast requires disruption of at least four DNA repair genes. Such quadruple mutants show significant changes in transcription patterns of different genes (23,24), as compared to defects in either BER or NER caused by a single gene disruption. Such pleiotropic deregulation could significantly affect the mutation spectra, making it difficult to recapitulate the spectrum of oxidative mutagenic lesions in cells corrupted for both BER and NER. An alternative to inactivation of BER and NER would to utilize an ssDNA substrate that cannot be processed by the major DNA repair pathways even if those pathways are completely functional in the rest of the genome.
In this study we employed budding yeast strains with an ssDNA mutation reporter, which is not influenced by indirect effects and is free from ambiguity in determining the damaged nucleotide giving rise to a base substitution. This allowed generation of a more accurate picture of the mutagenic properties of ROS. We also assessed the relative contributions of oxidative stress to hyper-mutability and gross chromosomal rearrangements (GCRs) by measuring mutation frequencies and defining the signature of oxidative DNA damage in a sensitive ssDNA reporter system. Utilization of this tool allowed identification of mutagenesis-prone targets of oxidative damage and made it possible to compare the mutagenic impact of ROS on ssDNA to that of other types of DNA-damaging agents such as UV-irradiation, methyl methanesulfonate (MMS) and sulfites. We also addressed the role of translesion synthesis (TLS) polymerase zeta in oxidative stress-induced mutagenesis. Our results challenge several established views on mutagenesis in higher organisms and could have important implications for development of the therapeutic strategies aiming at selective killing of the cancer cells via increase in their mutational load.
MATERIALS AND METHODS
Genetic analysis and media
Genetic procedures (transformation, tetrad analysis, etc.), cell growth and selection conditions were performed as previously described (25). Strains were grown at 30°C. cdc13-1ts mutants were grown at room temperature (23°C). In experiments which required telomere-uncapping cells were incubated at 37°C.
Strain construction
Yeast strains used in this study are isogenic to CG379 (26). The common genotype of haploid strains was MATα his7-2 leu2-3,112 trp1-289 can1Δ cdc13-1 ura3Δ. Strains also contained a CAN1-URA3 mutation reporter similar to the one used by Chan et al. (9) but missing an insert of the ADE2 open reading frame. CAN1-URA3 reporter was placed either in sub-telomeric or in mid-chromosome position as described in Results section. To avoid accumulation of mutations during propagation of strains defective in DNA repair and/or ROS scavenging, we constructed a series of homozygous diploid strains heterozygous for mutations affecting ROS accumulation and/or DNA repair. These strains were built by first initiating mating type switching after introducing YEp-HO plasmid and then by series of consecutive gene disruptions in the resulting diploid strains JFS1327 and JFC1328, containing CAN1-URA3 sub-telomeric reporter construct used to assess mutagenesis in ssDNA and in control diploid strains JFS1325 and JFS1326 with mid-chromosome location of CAN1-URA3 reporter. The genes of interest were disrupted by transformation with the PCR fragments (primers sequences are available upon request) containing genes for antibiotic resistance flanked by the upstream and downstream sequences of the corresponding genes (Supplementary Table S5). Deletions were confirmed by PCR.
Measurement of CAN1 mutation frequencies and frequencies of GCRs
Genetic defects leading to accumulation to ROS often lead to reduced growth rate increasing the probability of selecting a suppressor after long propagation. In order to reduce propagation before measuring mutation rates, diploids heterozygous for the gene of interest (Supplementary Table S5) were sporulated and dissected. Fresh spore colonies of the genotypes of interest were inoculated and grown for 72 h in rich liquid media. Following 1:10 dilution into rich media cells were incubated in a shaker at 37°C and/or 23°C for 3.5 h. Then cells were quickly washed with water, and exposed to oxidative-damaging agents (5 mM hydrogen peroxide or 150 µM paraquat in water) for 1 h at room temperature. Cells were washed in water and plated on synthetic complete medium containing canavanine and lacking arginine to measure the frequency of canavanine-resistant mutants. For sod1Δ cells canavanine-containing media was supplemented with L-methionine and L-lysine to final concentration of 200 mg/l and 300 mg/l, respectively. A diluted suspension was also plated on medium lacking canavanine to estimate number of cells in culture. At least four independent haploid spore isolates were examined for each genotype at any given experiment. The mutation frequencies were calculated as a ratio of cells resistant to canavanine to the total number of viable cells in culture. To estimate the frequencies of GCRs plates with CanR clones were replica plated onto media lacking uracil. CanR Ura− clones from independent cultures were streaked on YPD and single colony was used to inoculate the culture from which genomic DNA was prepared using Epicentre MasterPure Yeast DNA Purification Kit (Illumina, Madison, WI). The loss of left arm of chromosome V was confirmed by PCR with the primers complimentary to telomere-proximal URA3 sequence (oDG_97 5′-TGAAGAAGCTGCATTTGC-3′ and oDG_98 5′-TTGTTAGCGGTTTGAAGCAG-3′) and by CHEF as described in (16).
Statistical methods
Statistical evaluations were done with non-parametric Fisher exact, chi-square and Mann–Whitney tests.
Identification of the mutations in ssDNA reporter
Independent CanR Ura− or CanR clones from cultures originated from several haploid isolates were streaked on YPD plates. After confirmation of the phenotype by replica plating on corresponding media, single colonies of interest were used to set up the cultures for purification of the genomic DNA as described above. CAN1 or CAN1 and URA3 loci were PCR amplified and sequenced with primers listed in Supplementary Table S6. Complete list of mutations is presented in Supplementary Table S7. TaKaRa Ex Taq DNA polymerase (Takara Bio Inc., Japan) was used for PCR; manufacturer protocol was followed in choosing of the settings for the PCR reactions.
RESULTS
ROS causes mutations in ssDNA
Previous studies have indicated that the frequency of spontaneous mutagenesis in ssDNA can be two orders of magnitude higher, than in dsDNA (4,6). At least 90% of spontaneous mutations in ssDNA relied on error-prone TLS by DNA polymerase (Pol) zeta, which is essential for different kinds of damage-induced mutagenesis (reviewed in (6,27,28)). This suggests that endogenous DNA damage could be a predominant cause of spontaneous mutations in those experiments. We hypothesized that major contributors to spontaneous ssDNA hyper-mutability are ROS. To address this hypothesis we used a reporter system similar to those developed by Chan et al. (9), to measure mutation frequencies in ssDNA and to detect clusters of simultaneous mutations as changes in phenotypes controlled by two closely spaced ORFs. In these strains the wild-type CAN1 and URA3 genes on the left arm of chromosome V were deleted, the CAN1 and URA3 open reading frames were inserted into the LYS2 gene placed within ∼8.9 kb from an artificially constructed telomere at the left tip of chromosome V (Figure 1). The DNA between the telomere and the lys2::URA3-CAN1 in this construct does not contain any essential genes. The yeast strains carried a cdc13-1 temperature sensitive mutation, conferring a defect in telomere capping. At the non-permissive temperature 37°C, 5′→3′ resection occurs, generating long stretches of ssDNA adjacent to telomeric regions (29,30); after 3.5 h cells arrest at G2. At that time cells were exposed to oxidative DNA-damaging agents for 1 h at room temperature. They were subsequently plated onto canavanine-containing media for mutant selection and, after dilution, onto rich growth media to determine cell viability. Mutation frequencies were calculated as a ratio of cells resistant to canavanine to the total number of viable cells in culture.
Figure 1.
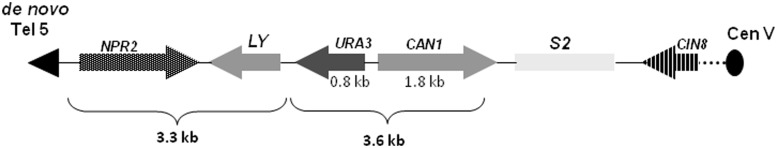
Schematic depiction of the reporter for ssDNA mutagenesis. CAN1 and URA3 open reading frames had been placed in the LYS2 locus on chromosome V. Arrows indicate the direction of transcription of the genes. Left arm of de novo constructed telomere is represented by a triangle; an oval depicts the centromere.
Since cells were not dividing when ssDNA was formed and throughout the time of exposure to endogenous and exogenous-damaging agents and restoration to dsDNA, these frequencies directly reflect the probability of mutant allele formation. Cells with an additional mutation in the URA3 reporter gene were identified by replica plating of canavanine-resistant colonies onto selective media lacking uracil. As a control, the URA3-CAN1 reporter was inserted into the LYS2 gene located within its unperturbed linkage group in the middle of the right arm of chromosome II (2) to address mutation in dsDNA. The 345 kbp segment of DNA between this mid-chromosome reporter and telomere contains 35 essential genes, which eliminates the opportunity for survival of GCR events in this region. The long distance between the telomere and the reporter construct precludes a stretch of ssDNA from encompassing reporter genes after telomere uncappping at 37°C. In order to reduce the chance of selecting growth enhancers in slow growing mutants with elevated levels of ROS we measured mutation rates in at least six independent meiotic haploid isolates after sporulating heterozygous diploids (see ‘Materials and Methods’ section).
To address the contribution of endogenous ROS to hyper-mutability of ssDNA regions, we first measured the frequencies of mutations in the CAN1 locus in the strains with decreased ROS-scavenging capacities. Superoxide dismutases and catalases play an important role in oxygen radical and hydrogen peroxide detoxification, respectively (reviewed in (31)). Inactivation of yeast Cta1 catalase or Sod1 superoxide dismutase leads to elevation in endogenous ROS levels (32) and to an increase in mutation rates in a dsDNA reporter (33). Surprisingly, disruption of SOD1 and/or CTA1 genes, did not result in a change in mutation frequencies in ssDNA (Figure 2). At the same time mutation frequencies for the mid-chromosome dsDNA reporter in sod1Δ mutants were significantly higher than in the wild-type strain (P = 0.002) at 37°C. We conclude that the major source of spontaneous mutagenesis in ssDNA is not endogenous ROS, even in mutants with decreased capacity of ROS scavenging. The lack of impact of the endogenous ROS level could be due to effectiveness and redundancy of intracellular ROS-scavenging systems and/or to ssDNA-specific mechanisms of protection against ROS within the cells. Alternatively, it is possible that ROS sub-species produced in ROS-scavenging defective strains, while mutagenic for dsDNA, has no effect on ssDNA. Also, the contribution of endogenous ROS to ssDNA mutagenesis could be over-shadowed by the high levels of alternative sources of spontaneous hyper-mutability in ssDNA, such as decreased fidelity of DNA synthesis during restoration to dsDNA state.
Figure 2.

Frequencies of spontaneous mutations in the CAN1 locus in strains with compromised ROS-scavenging activities. Mutation frequencies at the CAN1 locus were calculated as described in Materials and Methods section. Average mutation frequency measured for at least six independent cultures incubated at 37°C for 3.5 h to induce telomere un-capping and at 23°C in control experiments. Asterisk indicates statistically significant difference (P < 0.005) in frequencies of mutations as determined by Mann–Whitney test. NS indicates the absence of significant differences.
Even though it is unlikely that DNA glycosylases could efficiently remove oxidative lesions in ssDNA substrates (with the exception of uracil-DNA glycosylases (9,34)), and nucleotide incorporation across the abasic sites created by glycosylases would be often mutagenic, we sought to exclude the possibility that DNA glycosylases dealing with oxidative damage in dsDNA are also involved in protection of ssDNA against spontaneous mutagenesis. Spontaneous mutation frequencies in ssDNA reporter were determined in ogg1Δ, ntg1Δ, ntg2Δ and ntg1Δ ntg2Δ backgrounds. The ranges of mutation frequencies in wt strains (1.6 − 2.3 × 10−4) did not differ from the strains with glycosylase(s) knockouts (2.1 − 3.2 × 10−4). Thus, we confirmed that these DNA glycosylases are not involved in repair of endogenous lesions in ssDNA.
Acute exposure of wild-type cells to 5 mM hydrogen peroxide resulted in only a 2-fold increase in mutation frequency in the ssDNA reporter (Figure 3). However, such exposure caused an ∼300 × 106 absolute increase in mutation frequency superimposed on the already high frequency of spontaneous mutations in the ssDNA reporter within the wild-type strain. This is 12-fold greater than the mutation frequency added by hydrogen peroxide in the dsDNA mid-chromosome reporter. Although the relative increase in mutation frequencies by hydrogen peroxide in a sub-telomeric reporter of wild-type cells was modest, exposure to the same dose of hydrogen peroxide caused a detectable increase in mutation frequency in dsDNA (mid-chromosome reporter), indicating that the chosen dose was indeed mutagenic to chromosomal DNA.
Figure 3.

Exogenous oxidative damage induces mutagenesis in ssDNA. Mutation frequencies at the CAN1 locus were calculated as described in Materials and Methods section. Average mutation frequency measured for at least four independent cultures incubated at 37°C for 3.5 h induce telomere uncapping followed by exposure to 5 mM hydrogen peroxide or mock treatment are presented. Mid-chromosome reporter (described in Results section) was used as a control under the same experimental conditions. Numbers above the bars reflect the absolute frequency of mutation, numbers with ‘+’ indicate how many additional mutations were deposited at CAN1 locus following exposure to hydrogen peroxide. Standard deviations of average are presented in Supplementary Table S4.
Reducing the cellular capacity to scavenge hydrogen peroxide by deletion of CTA1 resulted in a statistically significant increase in hydrogen peroxide-induced mutation frequency in the ssDNA reporter, without influencing cell survival (average survival ∼92%). Thus, exogenous oxidative damage causes an increase in the ssDNA mutation frequencies, and cells with compromised ROS-scavenging capacity are sensitized to exogenous ROS-induced mutagenesis.
To eliminate the possibility that sensitivity of ssDNA to an oxidative DNA-damaging agent was a unique effect exerted by hydrogen peroxide, we tested the mutagenicity of another oxidative agent, paraquat. The precise mechanism of paraquat cytotoxicity is not completely understood, but is believed to involve an increase in cellular levels of superoxide anion (35). In agreement with the results for hydrogen peroxide-induced ssDNA mutagenesis, a significant increase in mutagenesis by paraquat was observed in sod1Δ strains, known to be highly sensitive to this chemical. However, survival and mutagenesis were essentially unchanged in a wild-type strain (Supplementary Figure S1).
Based on the outcome of the experiments involving two different oxidative DNA-damaging agents we conclude that even though oxidative stress can increase the frequency of mutations in ssDNA, the contribution of oxidative DNA damage to ssDNA mutagenesis is obscured by very high levels of spontaneous mutagenesis originating from other sources. These data and the lack of increase in mutation frequencies in ROS-scavenging mutants suggest that intracellular ROS are not the major source of spontaneous hyper-mutability of ssDNA.
TLS contributes to ROS-induced ssDNA mutagenesis
The CAN1-URA3 reporter for ssDNA mutagenesis measurements utilized in this study allows for a direct, forward selection of CanR Ura− cells. Such mutants could arise either as a consequence of independent mutations in both genes or due to the loss of the entire arm of chromosome V between the telomere and the CAN1 open reading frame. To distinguish between these two mutational outcomes we used a diagnostic PCR with primers specific for the telomere-proximal URA3 locus in the ssDNA reporter (Materials and Methods section). Our experiments with glycosylase knock-out mutants demonstrated that mutations in ssDNA (unlike in dsDNA) cannot be prevented by BER (see above) and therefore, the spectrum and frequencies of base substitutions and small indels in the ssDNA reporter allows unambiguous assessment of the site and the frequency of occurrence of pre-mutagenic lesions (at least for lesions which cannot be bypassed without an error during the re-synthesis of dsDNA). In contrast, assessment of potential GCR events is not as clear. GCR events could be avoided in ssDNA as well as in dsDNA by template switching to the available sister chromatid during re-synthesis of the resected sub-telomeric region. Also, the unknown fraction of cells in which TLS failed may not survive rather than generate GCR. Thus, the frequency of GCR would be defined by the efficiency of a TLS alternative pathway as well as by other confounding factors. We found that the overall frequency of spontaneous GCRs in rev3Δ strains did not increase significantly (Figure 4). Surprisingly, induction of oxidative damage by hydrogen peroxide in rev3Δ and rev3Δ cta1Δ double mutants substantially decreased the frequency of GCRs. Additional studies are required to address the mechanisms of such a decrease.
Figure 4.

Estimated frequencies of spontaneous and oxidative stress-induced GCRs. Frequencies of GCRs were calculated as the ratio of the number of CanR colonies unable to grow on the media lacking uracil to the total number of cells in the culture. Median frequency of Ura− CanR colonies per genotype per experimental condition for at least four independently selected cultures is presented. Asterisk indicates statistically significant differences (Mann–Whitney test, P < 0.03).
We noticed that the fraction of GCRs among spontaneous CanR clones in rev3Δ mutants was greater than in wild-type cells (Supplementary Figure S2), suggesting that the decrease in point mutation frequencies and not in GCRs is the major cause of overall lower CanR mutation rates in rev3Δ strains. Indeed, in contrast to the negligible contribution of TLS to GCR, elimination of TLS DNA Pol zeta significantly reduced frequencies of ROS-induced point mutations (measured as the frequency of single CanR mutants) in all backgrounds (Figure 3 and Supplementary Figure S1). These results can be explained by ROS causing mutagenic lesions in ssDNA. Alternatively, it is possible that in the context of ssDNA, Pol zeta as well as the replicative polymerases display a higher error propensity under the conditions of oxidative stress (see Discussion section)
Low mutation density in mutation clusters caused by ROS
The density of mutations in clusters induced by moderately toxic doses of UV, MMS and sulfites in ssDNA in the vicinity of DSBs or at uncapped telomeres is several orders of magnitude higher than the mutation density in dsDNA (6,8,9). Because such different types of DNA damage elicit closely spaced mutations, we sought to determine whether oxidative damage also induces clustered mutations in ssDNA. Since exogenous ROS significantly contribute to the increase in ssDNA mutagenesis in strains deficient for ROS-scavenging, we decided to analyse the sequences of CanR Ura− double mutants arising in cta1Δ and sod1Δ strains following hydrogen peroxide and paraquat exposure, respectively (Table 1). We reasoned that CanR Ura− clones with double or even multiple mutations within the two open reading frames most likely arise from cells in which 5′ to 3′ resection encompassed the entire sub-telomeric region, including the CAN1 and URA3 open reading frames, i.e. from cells where a long stretch of ssDNA had been exposed to the damaging agents. Importantly, when no oxidative agent was applied, PCR analysis of the CAN1-URA3 locus (Materials and Methods section) of the CanR Ura− clones revealed that they all arose as a consequence of GCR; none were caused by multiple point mutations.
Table 1.
Density of oxidative agent-induced mutations in CAN1-URA3 ssDNA reporter
| Number of mutations per CAN1-URA3 reporter |
|||||||
|---|---|---|---|---|---|---|---|
| Oxidative agent | Strain background | Number of mutants | Total number of mutations | D (mutations per kb) | |||
| 2 | 3 | 4 | |||||
| Peroxide | cta1Δ | 23 | 3 | 0 | 26 | 55 | 0.04 |
| Paraquat | sod1Δ | 20 | 0 | 0 | 20 | 40 | n.d |
Surprisingly, only 3 out of 26 of peroxide- and 0 of 20 paraquat-induced CanR Ura− clones in cta1Δ and sod1Δ backgrounds, respectively, contained more than a single mutation in each of the CAN1 and URA3 open reading frames. None of the CanR Ura− clones contained four or more mutations. Such low multiplicity of mutations differed greatly from the density of the mutations reported for UV-induced damage in the lys2 reporter system (1,6), which is comparable in size to the CAN1-URA3 reporter employed in our study. Up to six independent mutations were found in the LYS2 locus and more than 30% of the Lys- clones contained more than two lesions. The reported mutation density for the wild-type strain in those experiments was 0.31 mut/kb (1), whereas the mutation density of oxidative stress-induced mutations in the CAN1-URA3 reporter in sensitized cta1Δ mutants was as low as 0.04 mut/kb (density of unselected mutations calculated as in (1) and ref. therein). Incubation of sod1Δ mutants with 150 µM paraquat increased the mutation frequencies 30-fold and decreased cell survival to ∼4% (Supplementary Figure S1); nevertheless, none of the sequenced CanR Ura− clones contained more than two mutations. Importantly, treatment with paraquat resulted in mutation frequencies comparable to those caused by MMS in (8) and paraquat-induced cell killing was greater than the lethality caused by MMS. Additional biochemical and genetic studies are required to understand the difference in mutation density within clusters caused by ROS compared to those caused by alkylation (MMS), deamination (sulfites) or bulky (UV) lesions in DNA (see Discussion section)
TLS-independent mutations of cytosines are the major source of mutagenesis by hydrogen peroxide- and paraquat-induced oxidative stress in ssDNA
Determining the mutagenic targets of a specific DNA-damaging agent in vivo generally relies on analysis of the mutation spectrum. The use of traditional dsDNA reporter systems to assign mutagenic lesions to certain nucleotides is problematic, because any given mutational change originated in dsDNA could be caused by the damage of either nucleotide in a pair. The deduction of nucleotide lesion spectra based on mutation spectra in dsDNA reporters is further complicated by functional DNA repair systems, which may disproportionally repair lesions in different bases and sequence context. The ssDNA reporter system in this study enabled direct identification of the mutated nucleotides, without interference from BER and NER. A lesion can be unambiguously assigned to one of the two nucleotides within a base pair because the 5′ to 3′ direction of the DNA strand resection in the reporter is known. This defines which DNA strand is retained following resection and which nucleotide within a base pair is the target for damage or was erroneously copied by a DNA polymerase.
We calculated the frequencies of different types of mutation events (Figure 5) by multiplying the total mutation frequency by the fraction of the specific type of the mutation event within the mutation spectrum (Supplementary Table S1). Comparison of spontaneous mutation spectra in the CAN1 locus between rev3Δ and REV3 strains indicated that error-prone bypass of guanines is the greatest source of mutations in the REV3 strain (Figure 5, ‘no treatment’ panel). In a REV3 strain mutations at G occur ∼30- and ∼10-fold more frequently than in rev3Δ CTA1 and rev3Δ cta1Δ strains, respectively· Insertions of A across positions of G in ssDNA occurred most often, since G to T transversions constituted the majority of substitutions of guanine (Supplementary Table S2). DNA polymerase zeta was also responsible for 90–95% of spontaneous substitution frequencies of A and C (Figure 5). The contribution of substitutions of T to spontaneous mutagenesis was negligible in all genotypes. Presently, it is not possible to ascertain whether DNA polymerase zeta erroneously bypasses endogenously damaged nucleotides or whether the mutations are caused by mutagenic bypass of undamaged nucleotides due to a proposed defective replisome-induced mutagenesis mechanism (36).
Figure 5.

Frequencies of endogenous and oxidative damage-induced substitutions, complex mutations and insertions/deletions calculated by multiplication of the fraction of each type of mutation (Supplementary Table S1) to the total frequency of spontaneous mutations or following exposure to 5 mM hydrogen peroxide in the strains of different genetic backgrounds.
In the analyses of mutations induced by oxidative agents (hydrogen peroxide in wild type and in cta1Δ and paraquat in sod1Δ strains, respectively), we included only mutations from isolates with simultaneous inactivation of CAN1 and URA3, because those were likely to have originated from lesions in ssDNA, as discussed above. In ROS-scavenging proficient strains, the majority of CanR Ura− clones arose from GCR events whereas ssDNA double mutants were observed at low frequencies. We reasoned that since in all backgrounds there was ≥10-fold increase in mutation frequencies in the sub-telomeric ssDNA reporter as compared to mid-chromosome (dsDNA) reporter, all the CanR mutants, including CanR Ura+ have originated from independent mutagenesis events in ssDNA of non-dividing cells. In striking contrast to the spontaneous mutation spectra, base substitutions at cytosines in the putative ssDNA strand comprised the largest part of mutations in all the strains subjected to oxidative stress (Supplementary Table S1).
To test the possibility that the burden of 8oxoG accumulated in ssDNA following oxidative stress could be obscured by error-free bypass of 8oxoG by TLS DNA polymerase eta, we measured the frequencies of CanR mutations in rad30Δ strains (Supplementary Table S3). Frequencies of the oxidative stress-induced mutations in cta1Δ and cta1Δ rad30Δ strains, as well as in wt and rad30Δ were not significantly different (Mann–Whitney test P = 0.44 and 0.65, respectively). Thus, since Ogg1 does not remove 8oxoG from ssDNA (see above) it is unlikely that hydrogen peroxide induce significant number of lesions at G in ssDNA.
Incorporation of oxidized nucleotides during DNA replication causes mutations in prokaryotic and eukaryotic organisms (37,38). An increase in A–C and G–T transversions would be indicative of incorporation of 8oxodG across A and 2-OH-dA across G, respectively, in ssDNA under oxidative stress conditions. Since mutations at A are less frequent than at C and G (Figure 5) and G–T transversions is not the major class of oxidative stress-induced mutations at G (Supplementary Table S2), it is unlikely that that insertion of oxidized nucleotides during the re-synthesis of the second strand of DNA contributes significantly to oxidative stress-induced mutagenesis in ssDNA.
In summary, oxidative damage-induced mutations occurred with the highest frequency at cytosines. This differed from different from spontaneous mutations, where the most frequent events were indels or mutations at guanines. The proportion of mutations at C among all mutations under oxidative stress conditions was significantly higher compared to that for spontaneous mutagenesis (P < 0.0001; Fisher’s exact test).
We then explored the role of Pol zeta TLS in ssDNA mutagenesis caused by oxidative stress. In agreement with other reports (36,39) the Rev3 deletion caused a strong decrease in frequencies of hydrogen peroxide-induced complex mutations (Figure 5). We also noticed that the effect of Rev3 deletion on oxidative mutagenesis in ssDNA was moderate (2.4-fold decrease in the mutation frequencies in rev3Δ strain) as compared to the effects on mutagenesis caused by UV, MMS and sulfites (6,8,9). The rev3Δ mutants retained ∼25% of ssDNA oxidative stress-induced mutagenesis in cta1Δ or in sod1Δ and even 42% in a wild-type background (Figure 3 and Supplementary Figure S1). In contrast to other types of DNA lesions, the frequency of base substitutions at cytosines did not decrease in the absence of Rev3 (Figure 5), suggesting that the mutator effect of damage to cytosines or their erroneous bypass caused by oxidative stress conditions is Rev3 independent. The reasonable candidate for such a lesion would be deaminated cytosine creating uracil (U) in ssDNA. If U remains in ssDNA until second-strand DNA synthesis, it would lead to C to T mutations, which are prevalent in all three mutation spectra (hydrogen peroxide-induced in wild type and cta1Δ and paraquat-induced in sod1Δ). Since uracil in DNA is as good a template for A pairing as a T, such mutations would be TLS independent. However, it was previously established that U, which was created in ssDNA in vivo through enzymatic deamination by endogenously expressed APOBEC3G appears to be a good substrate for uracil-DNA glycosylase (Ung1). The resulting mutations were approximately equal for C–T (45%) and C–G (51%) with the small fraction of C–A (3%) (9). In our study distribution of C changes caused by oxidative agents in wild type, cta1Δ and sod1Δ (C–T—56%, C–G—22%, C–A—22%) clearly differed from the distribution obtained with APOBEC3G enzymatic cytosine deamination (P = 0.005, chi-square test). If the major mutagenic lesions at C arise as a consequence of oxidative damage-induced C–U deamination, deletion of Ung1 should have resulted in an increase of C–T transitions. Importantly, we did not observe such increase in ung1Δ strains (Supplementary Table S2). The distribution of hydrogen peroxide-induced changes at C in the ung1Δ strain (C–T—50%, C–G—20%, C–A—30%) did not differ from that of wild-type strain (P = 0.8, chi-square test). These results indicate that C deamination is not the major mutagenic event in ssDNA, and that different type(s) of ROS-induced DNA lesions may contribute significantly to mutagenesis at C (see ‘Discussion’ section).
DISCUSSION
Oxidative stress and mutability of ssDNA
Recently, hyper-mutability of ssDNA and its potential relevance to cancer was established (2,3). Analysis of genome-wide mutation datasets from four types of human cancers identified strand-coordinated clusters of mutations, often occurring in the vicinity of the genome rearrangement breakpoints. Even though the precise mechanism of generation of such clusters is largely unknown, clusters as well as single mutation events were significantly enriched for the mutation motifs characteristic for the APOBEC family of cytosine deaminases, mutator enzymes highly preferring ssDNA to dsDNA, which indicated that there is a sufficient amount of ssDNA to account for a sizable fraction of mutagenesis in cancers. It appeared plausible that physiologically relevant, persistent increases in ROS levels caused by chronic inflammation and/or defective energy metabolism in cancer cells or a transient redox imbalance due to signal transduction processes could also contribute to mutagenesis of ssDNA.
Utilizing yeast as model system we found that, in contrast to our expectations, an increase in endogenous levels of ROS caused by defined genetic defects did not mutagenize ssDNA. Importantly, mutants caused by ROS-generating treatments had a much lower density of mutations as compared to MMS-, UV- and sulfite-induced mutants, which often contained clusters of multiple mutations. These findings suggest the existence of either an unknown pathway of repairing BER substrates in ssDNA or specialized mechanisms for ssDNA protection against oxidative damage. Also, it is possible that ssDNA at uncapped telomeres is protected from oxidative, but not from UV- and MMS-induced damage by telomere-specific ssDNA binding proteins. Although exposure to exogenous oxidative agents increased the mutability of ssDNA it is clear that high spontaneous levels of ssDNA mutagenesis are caused by sources different from endogenous ROS. Nevertheless, even though the consequences of exposure to oxidative DNA-damaging agents were less relevant to localized hyper-mutability compared to the effects of MMS-induced alkylating damage and UV-induced damage, compromising the endogenous cellular antioxidant defenses significantly increased the frequency of ROS-induced mutations in ssDNA.
The above findings could have important translational implications. Several lines of evidence suggest that altered metabolism of cancer cells places them under continuous ROS stress due to the Warburg effect (40). Exposure of cancer cells to chronic, endogenous oxidative stress could exploit the higher vulnerability of cancer cells to DNA damage-inducing therapeutic agents [reviewed in (41)]. It was shown recently that a small molecule targeting ROS response can cause selective killing of cancer cells independently of their p53 status (42). Our results suggest that therapies aimed at diminishing activities of superoxide dismutase or catalase, could selectively increase the mutational load in ssDNA of actively replicating cancer cells, overloaded with ROS. Such increased mutational pressure could lead to cell death due to multiple mutations in essential genes or via apoptosis caused by activation of replicative checkpoints. In contrast, increasing the endogenous levels of ROS in normal tissues by suppressing antioxidant defenses should not cause deleterious effects.
Cytosines are the major source of ROS-induced mutagenesis in ssDNA
8-Oxoguanine is thought to be the most frequently occurring DNA lesion caused by oxidation (summarized in (43)). However, it remains unclear whether this lesion is most mutagenic since analyses of mutation spectra indicate that C–T transitions are prevalent among spontaneous base substitutions in vivo (44,45). In order to address the mutational potential of different nucleotides under oxidative stress conditions we utilized a reporter in which precise identity of the pre-mutagenic lesions is not obscured by action of DNA excision repair systems. It also allowed discrimination between two bases within a mutated base pair, because the strand that is being resected during the telomere-uncapping step is known. These features of the ssDNA reporter system revealed that cytosines are the major source of oxidative damage-induced mutations in ssDNA and implicate cytosine as the major source of the mutations in a GC pair. Our findings raise several important questions. Are cytosine modifications the most common oxidative DNA lesions, and are they the most mutagenic or both? It is possible that oxidative deamination is the major source of mutagenesis at C under oxidative stress conditions, even though the fraction of C–T substitutions among all C substitutions is much higher compared to conditions when, in the presence of Ung1, APOBEC3G-induced deamination produces almost equal numbers of C–T and C–G substitutions. Accordingly, if C deamination is not the only source of mutations at C, what types of oxidative cytosine modification prompt mis-pairing with dATP by the replicative DNA polymerases? It has been shown that products of cytosine oxidation and deamination, namely, 5-hydroxyuracil and uracil glycol, efficiently cause C–T substitutions in vivo when single-stranded oligos, containing such modified bases are replicated in Escherichia coli (46). 5-Hydroxycytosine, a product of direct oxidation of cytosine, exhibits weaker mutagenic properties. Its miscoding properties are dictated by instability in the replicative polymerase active site, which allows dATP misincorporation in a process similar to that described for incorporation opposite to abasic sites (47). In addition, chemical interaction of dCTP with oxidants generates 5-hydroxycytidine, which can be incorporated into DNA and is highly mutagenic when bypassed by E. coli Pol I (48). Taken together, these results suggest that the products of oxidation and/or deamination of cytosine are important genome-destabilizing products of oxidative damage in vivo. Development of analytical methods for rapid detection of such modified nucleotides should provide precise and meaningful measurements of endogenous oxidative stress.
Among the four common DNA bases, cytosines were found to be mutated most frequently following exposure to exogenous oxidative damage. Nevertheless, the majority of spontaneous mutations were caused by error-prone, DNA polymerase zeta-mediated bypass by of guanines. These observations allow us to speculate that endogenously—produced ROS may not be the major contributors to spontaneous mutagenesis or that exogenous oxidative agents are metabolized in the cells to ROS sub-types that are different from those produced endogenously and cause a different spectrum of mutagenic base modifications. It is important to note that it is possible that the high mutagenic potential of C under oxidative stress results from a greater propensity for replicative DNA polymerases to incorporate erroneous nucleotides across non-damaged C in ssDNA in a highly oxidative intracellular environment.
In summary, our study of ROS-induced ssDNA mutagenesis suggests that ROS are not the major source of spontaneous ssDNA mutagenesis and that TLS polymerase zeta contributes significantly to both ROS-induced and spontaneous ssDNA mutagenesis. Under oxidative stress, C is the most mutagenic nucleotide (Figure 6). Its mutagenic potential is TLS independent, whereas spontaneous base substitutions in ssDNA occur mainly due to DNA polymerase zeta-dependent mutagenic bypass of guanines.
Figure 6.

Mutagenic potential of the nucleotides and role of TLS Pol zeta in ROS-induced mutagenesis. Propensity of the pre-mutagenic lesions to cause spontaneous and oxidative stress-induced mutations and the role of TLS in the mutagenesis is schematically depicted. Fraction of the mutations occurred at each nucleotide calculated from Supplementary Table S1 for each experimental condition. The height of corresponding letter is proportional to the contribution of each nucleotide to ssDNA mutagenesis. Bar graph reflects the relative frequencies of mutations at CAN1 locus in ssDNA reporter for each experimental condition in wt (Rev3+) and rev3Δ (Rev3−) strains.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: National Institute of Environmental Health Sciences [ES011163 to P.W.D.]; Intramural Research Program of the National Institute of Environmental Health Sciences [ES065073 to M.A.R.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Drs L.P.Morris, K.Chan and E. Werner for critical reading of this manuscript and helpful discussions.
REFERENCES
- 1.Burch LH, Yang Y, Sterling JF, Roberts SA, Chao FG, Xu H, Zhang L, Walsh J, Resnick MA, Mieczkowski PA, et al. Damage-induced localized hypermutability. Cell Cycle. 2011;10:1073–1085. doi: 10.4161/cc.10.7.15319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts SA, Sterling J, Thompson C, Harris S, Mav D, Shah R, Klimczak LJ, Kryukov GV, Malc E, Mieczkowski PA, et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell. 2012;46:424–435. doi: 10.1016/j.molcel.2012.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strathern JN, Shafer BK, McGill CB. DNA synthesis errors associated with double-strand-break repair. Genetics. 1995;140:965–972. doi: 10.1093/genetics/140.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hicks WM, Kim M, Haber JE. Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science. 2010;329:82–85. doi: 10.1126/science.1191125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y, Sterling J, Storici F, Resnick MA, Gordenin DA. Hypermutability of damaged single-strand DNA formed at double-strand breaks and uncapped telomeres in yeast Saccharomyces cerevisiae. PLoS Genet. 2008;4:e1000264. doi: 10.1371/journal.pgen.1000264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306–313. doi: 10.1038/nature10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang Y, Gordenin DA, Resnick MA. A single-strand specific lesion drives MMS-induced hyper-mutability at a double-strand break in yeast. DNA Repair (Amst) 2010;9:914–921. doi: 10.1016/j.dnarep.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan K, Sterling JF, Roberts SA, Bhagwat AS, Resnick MA, Gordenin DA. Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLoS Genet. 2012;8:e1003149. doi: 10.1371/journal.pgen.1003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat. Rev. Cancer. 2003;3:276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 11.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 13.Vafa O, Wade M, Kern S, Beeche M, Pandita TK, Hampton GM, Wahl GM. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 14.Ragu S, Faye G, Iraqui I, Masurel-Heneman A, Kolodner RD, Huang ME. Oxygen metabolism and reactive oxygen species cause chromosomal rearrangements and cell death. Proc. Natl Acad. Sci. USA. 2007;104:9747–9752. doi: 10.1073/pnas.0703192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc. Natl Acad. Sci. USA. 2010;107:7461–7466. doi: 10.1073/pnas.1002459107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Degtyareva NP, Chen L, Mieczkowski P, Petes TD, Doetsch PW. Chronic oxidative DNA damage due to DNA repair defects causes chromosomal instability in Saccharomyces cerevisiae. Mol. Cell Biol. 2008;28:5432–5445. doi: 10.1128/MCB.00307-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Swanson RL, Morey NJ, Doetsch PW, Jinks-Robertson S. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol. Cell Biol. 1999;19:2929–2935. doi: 10.1128/mcb.19.4.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moriya M, Ou C, Bodepudi V, Johnson F, Takeshita M, Grollman AP. Site-specific mutagenesis using a gapped duplex vector: a study of translesion synthesis past 8-oxodeoxyguanosine in E. coli. Mutat. Res. 1991;254:281–288. doi: 10.1016/0921-8777(91)90067-y. [DOI] [PubMed] [Google Scholar]
- 19.Henderson PT, Delaney JC, Gu F, Tannenbaum SR, Essigmann JM. Oxidation of 7,8-dihydro-8-oxoguanine affords lesions that are potent sources of replication errors in vivo. Biochemistry. 2002;41:914–921. doi: 10.1021/bi0156355. [DOI] [PubMed] [Google Scholar]
- 20.Ni TT, Marsischky GT, Kolodner RD. MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol. Cell. 1999;4:439–444. doi: 10.1016/s1097-2765(00)80346-9. [DOI] [PubMed] [Google Scholar]
- 21.Collura A, Kemp PA, Boiteux S. Abasic sites linked to dUTP incorporation in DNA are a major cause of spontaneous mutations in absence of base excision repair and Rad17-Mec3-Ddc1 (9-1-1) DNA damage checkpoint clamp in Saccharomyces cerevisiae. DNA Repair (Amst) 2012;11:294–303. doi: 10.1016/j.dnarep.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Mudrak SV, Welz-Voegele C, Jinks-Robertson S. The polymerase eta translesion synthesis DNA polymerase acts independently of the mismatch repair system to limit mutagenesis caused by 7,8-dihydro-8-oxoguanine in yeast. Mol. Cell Biol. 2009;29:5316–5326. doi: 10.1128/MCB.00422-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evert BA, Salmon TB, Song B, Jingjing L, Siede W, Doetsch PW. Spontaneous DNA damage in Saccharomyces cerevisiae elicits phenotypic properties similar to cancer cells. J. Biol. Chem. 2004;279:22585–22594. doi: 10.1074/jbc.M400468200. [DOI] [PubMed] [Google Scholar]
- 24.Salmon TB, Evert BA, Song B, Doetsch PW. Biological consequences of oxidative stress-induced DNA damage in Saccharomyces cerevisiae. Nucleic Acids Res. 2004;32:3712–3723. doi: 10.1093/nar/gkh696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amberg DC, Burke D, Strathern JN Cold Spring Harbor Laboratory. Methods in Yeast Genetics : A Cold Spring Harbor Laboratory Course Manual. 2005 edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- 26.Morrison A, Bell JB, Kunkel TA, Sugino A. Eukaryotic DNA polymerase amino acid sequence required for 3′—-5′ exonuclease activity. Proc. Natl Acad. Sci. USA. 1991;88:9473–9477. doi: 10.1073/pnas.88.21.9473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gan GN, Wittschieben JP, Wittschieben BO, Wood RD. DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res. 2008;18:174–183. doi: 10.1038/cr.2007.117. [DOI] [PubMed] [Google Scholar]
- 28.Rattray AJ, Shafer BK, McGill CB, Strathern JN. The roles of REV3 and RAD57 in double-strand-break-repair-induced mutagenesis of Saccharomyces cerevisiae. Genetics. 2002;162:1063–1077. doi: 10.1093/genetics/162.3.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao H, Cervantes RB, Mandell EK, Otero JH, Lundblad V. RPA-like proteins mediate yeast telomere function. Nat. Struct. Mol. Biol. 2007;14:208–214. doi: 10.1038/nsmb1205. [DOI] [PubMed] [Google Scholar]
- 30.Booth C, Griffith E, Brady G, Lydall D. Quantitative amplification of single-stranded DNA (QAOS) demonstrates that cdc13-1 mutants generate ssDNA in a telomere to centromere direction. Nucleic Acids Res. 2001;29:4414–4422. doi: 10.1093/nar/29.21.4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jamieson DJ. Oxidative stress responses of the yeast Saccharomyces cerevisiae. Yeast. 1998;14:1511–1527. doi: 10.1002/(SICI)1097-0061(199812)14:16<1511::AID-YEA356>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 32.Mesquita A, Weinberger M, Silva A, Sampaio-Marques B, Almeida B, Leao C, Costa V, Rodrigues F, Burhans WC, Ludovico P. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc. Natl Acad. Sci. USA. 2010;107:15123–15128. doi: 10.1073/pnas.1004432107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang ME, Rio AG, Nicolas A, Kolodner RD. A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc. Natl Acad. Sci. USA. 2003;100:11529–11534. doi: 10.1073/pnas.2035018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brooks SC, Adhikary S, Rubinson EH, Eichman BF. Recent advances in the structural mechanisms of DNA glycosylases. Biochim. Biophys. Acta. 2013;1834:247–271. doi: 10.1016/j.bbapap.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cocheme HM, Murphy MP. Complex I is the major site of mitochondrial superoxide production by paraquat. J. Biol. Chem. 2008;283:1786–1798. doi: 10.1074/jbc.M708597200. [DOI] [PubMed] [Google Scholar]
- 36.Northam MR, Robinson HA, Kochenova OV, Shcherbakova PV. Participation of DNA polymerase zeta in replication of undamaged DNA in Saccharomyces cerevisiae. Genetics. 2010;184:27–42. doi: 10.1534/genetics.109.107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tajiri T, Maki H, Sekiguchi M. Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat. Res. 1995;336:257–267. doi: 10.1016/0921-8777(94)00062-b. [DOI] [PubMed] [Google Scholar]
- 38.Rai P, Onder TT, Young JJ, McFaline JL, Pang B, Dedon PC, Weinberg RA. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc. Natl Acad. Sci. USA. 2009;106:169–174. doi: 10.1073/pnas.0809834106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harfe BD, Jinks-Robertson S. DNA polymerase zeta introduces multiple mutations when bypassing spontaneous DNA damage in Saccharomyces cerevisiae. Mol. Cell. 2000;6:1491–1499. doi: 10.1016/s1097-2765(00)00145-3. [DOI] [PubMed] [Google Scholar]
- 40.Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51:794–798. [PubMed] [Google Scholar]
- 41.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Friedberg EC. DNA Repair and Mutagenesis. 2nd edn. Washington, DC: ASM Press; 2006. [Google Scholar]
- 44.Schaaper RM, Dunn RL. Spontaneous mutation in the Escherichia coli lacI gene. Genetics. 1991;129:317–326. doi: 10.1093/genetics/129.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wright JH, Modjeski KL, Bielas JH, Preston BD, Fausto N, Loeb LA, Campbell JS. A random mutation capture assay to detect genomic point mutations in mouse tissue. Nucleic Acids Res. 2011;39:e73. doi: 10.1093/nar/gkr142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kreutzer DA, Essigmann JM. Oxidized, deaminated cytosines are a source of C –> T transitions in vivo. Proc. Natl Acad. Sci. USA. 1998;95:3578–3582. doi: 10.1073/pnas.95.7.3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zahn KE, Averill A, Wallace SS, Doublie S. The miscoding potential of 5-hydroxycytosine arises due to template instability in the replicative polymerase active site. Biochemistry. 2011;50:10350–10358. doi: 10.1021/bi201219s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feig DI, Sowers LC, Loeb LA. Reverse chemical mutagenesis: identification of the mutagenic lesions resulting from reactive oxygen species-mediated damage to DNA. Proc. Natl Acad. Sci. USA. 1994;91:6609–6613. doi: 10.1073/pnas.91.14.6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.