


Abstract
Engineered nucleases, which incise the genome at predetermined sites, have a number of laboratory and clinical applications. There is, however, a need for better methods for controlled intracellular delivery of nucleases. Here, we demonstrate a method for ligand-mediated delivery of zinc finger nucleases (ZFN) proteins using transferrin receptor-mediated endocytosis. Uptake is rapid and efficient in established mammalian cell lines and in primary cells, including mouse and human hematopoietic stem-progenitor cell populations. In contrast to cDNA expression, ZFN protein levels decline rapidly following internalization, affording better temporal control of nuclease activity. We show that transferrin-mediated ZFN uptake leads to site-specific in situ cleavage of the target locus. Additionally, despite the much shorter duration of ZFN activity, the efficiency of gene correction approaches that seen with cDNA-mediated expression. The approach is flexible and general, with the potential for extension to other targeting ligands and nuclease architectures.
INTRODUCTION
Engineered nucleases, including zinc finger nucleases (ZFNs), TAL effector nucleases and RNA-guided endonucleases are useful for targeted genome modification [reviewed in (1–4)]. They incise the genome at predetermined sites to create DNA double-strand breaks. These breaks can be repaired by two general mechanisms. Non-homologous end joining rejoins broken ends directly, sometimes with deletions or insertions leading to loss of gene function. Homologous recombination uses a donor template to synthesize DNA across the break site. This leads to genome modification when the donor template differs from the original sequence, as when a wild-type sequence is used to replace its mutant counterpart, resulting in gene correction. Examples of laboratory and clinical applications for engineered nucleases include the creation of knockout animal models (5–7), generation of HIV-resistant hematopoietic stem cells (8) and correction of the X-linked severe combined immunodeficiency mutation in human cells (9).
One of the limiting factors in the use of ZFNs and other engineered nucleases, particularly for clinical applications, is the need to gain better control over the amount of nuclease delivered and the duration of nuclease activity. Vector-mediated cDNA expression, which is the standard delivery method, suffers from the limitation that it promotes prolonged and uncontrolled expression, potentially leading to off-target cutting and significant toxicity. Another limitation of cDNA expression, as evidenced by recent work with homing endonucleases (another type of rare-cutting nuclease), is variation in nuclease levels between cells in the target population, which has substantial effects on repair outcome (10). Transfection of nuclease-encoding mRNAs in place of cDNAs shortens the duration of ZFN expression and has shown promise in the generation of genetically modified animals (5–7), although it has not yet been fully evaluated for human somatic cells.
An entirely different approach is based on direct delivery of nucleases as proteins. Recent work provides proof of concept that ZFNs can be delivered in this manner, by taking advantage of their intrinsic cell-penetrating capability (11). The zinc finger domains of ZFNs fall into the category of the so-called ‘supercharged’ proteins, i.e. proteins with a very high net positive charge to mass ratio (12,13). As is the case with positively charged cell-penetrating peptides, supercharged proteins enter cells directly through multiple pathways, including macropinocytosis and clathrin-dependent endocytosis (14). Although successful for ZFNs, this method of uptake has some limitations, as it requires relatively high protein concentrations, is limited to highly charged proteins and likely occurs by a nonspecific mechanism.
We hypothesized that addition of a targeting ligand to ZFNs might help overcome these limitations. Ligand-mediated targeting significantly enhances delivery of a variety of drugs, proteins and nanoparticles [reviewed in (15,16)]. For intracellular protein delivery, the ligand is often attached via a scissile (‘self-immolative’) disulfide linker, which allows separation of ligand-cargo complex in the intracellular reducing environment (17,18). In the present study, we used transferrin as the ligand for ZFN delivery. We show that transferrin-ZFNs are taken up rapidly at 50–100 nM concentrations, under conditions where there was no detectable uptake of unmodified ZFNs. ZFNs delivered by the ligand-targeting method escape the endocytic compartment and enter the nucleus, where they mediate target gene cleavage and gene correction with an efficiency approaching that seen with vector-mediated cDNA expression. Ligand-mediated nuclease delivery has the potential for broad application, as it can be readily adapted for ligands other than transferrin and for other engineered nuclease architectures.
MATERIALS AND METHODS
Cells and culture conditions
HEK-293/A658 (19) and U2OS 2-6-3 (20) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMAX-1 (Invitrogen Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum, and penicillin/streptomycin. Primary ROSA26GFP*/* fibroblasts were obtained by standard methods using mouse ears as the tissue source (21).
Human CD34+ hematopoietic stem-progenitor cells (HSPCs) were purchased from StemCell Technologies (Vancouver, BC, Canada) and expanded for 48 h in serum-free IMDM (Invitrogen Life Technologies) supplemented with BIT 9500 (StemCell Technologies), 100 ng/ml human SCF, 100 ng/ml human Flt3-L and 20 ng/ml human thrombopoietin (StemCell Technologies). Mouse HSPCs [lineage marker negative, sca-1 positive, c-kit positive (LSK)] were obtained from mouse bone marrow and expanded as described (22). After 9 days, cells were stained with biotinylated lineage marker cocktail antibodies (anti-TER119, anti-Gr1, anti-Mac1, anti-B220, anti-CD8, anti-CD4), streptavidin-APC-Cy7, anti-Sca-1-APC and anti-c-Kit-PE (all from BD Pharmingen, San Diego, CA). LSK cells were isolated by flow cytometry.
Preparation of ZFNs and transferrin-ZFN conjugates
GFP-ZFN1 and GFP-ZFN2 (23) were cloned into the Not/BamHI sites of pHIS-parallel (24) and transformed into Escherichia coli Rosetta 2 (EMD Millipore, Gibbstown, NJ, USA). Expressed proteins contained the following elements: hexahistidine tag, Tev protease site, FLAG epitope tag, nuclear localization sequence, triple tandem zinc finger domain and FokI dimerization and catalytic domain. Escherichia coli cultures were grown in an NBS 110 fermentor to OD600 ≈ 2, cooled to 18°C and incubation was continued to OD600 ≈ 4. Cultures were adjusted to 0.2 mM isopropyl thiogalactoside, 100 μM ZnCl2 and incubation was continued 16–18 h to OD600 ≈ 14. Cells from 3 l of culture were resuspended in 200 ml of lysis buffer [20 mM Tris–HCl (pH 8.0), 300 mM NaCl, 10 mM β-mercaptoethanol, 0.1% Tween-20, 100 μM ZnCl2], sonicated and centrifuged at 10 000g for 25 min at 4°C. Cleared lysates were subjected to chromatography using a 5 ml of immobilized HIS-Select nickel affinity column (Sigma-Aldrich, St. Louis, MO, USA), which was equilibrated in lysis buffer containing 25 mM imidazole and eluted in lysis buffer adjusted to pH 7 and containing 150 mM imidazole. Non-conjugated ZFNs were further purified by Superdex S-75 chromatography (GE Healthcare Life Sciences, Piscataway NJ) in phosphate buffered saline (PBS) supplemented with 0.25 M NaCl. ZFN-containing fractions were adjusted to 10% glycerol, aliquoted and stored at −80°C.
For conjugation, human holo-transferrin (10 mg in 1 ml of PBS) (Akron Biotechnology, Boca Raton, FL) was reacted with 1 mM sulfosuccinimidyl 6-[3′(2-pyridyldithio)-propionamido] hexanoate (SPDP; Pierce Biotechnology, Rockford, IL, USA) for 1 h at room temperature. After desalting (PD10 column, GE Life Sciences, Piscataway, NJ), 1.4 mg of the activated transferrin was incubated overnight at 4°C with a 2-fold excess of nickel affinity-purified ZFN. Conjugates were isolated by Superdex S-75 chromatography and stored in the same way as non-conjugated ZFNs.
In vitro ZFN cleavage assays
Linear substrate DNA was prepared by AlwNI digestion of pPC264 plasmid DNA (21). Reactions contained 20 mM Tris–HCl (pH 8.0), 150 mM NaCl, 2.5 mM MgCl2, 10 mM β-mercaptoethanol, 100 μM ZnCl2, 0.1 mg/ml bovine serum albumin, 2.6 nM gel-purified DNA and ZFNs or transferrin-ZFN conjugates, in a volume of 20 μl. Reactions were incubated for 1 h at 30°C and analyzed by agarose gel electrophoresis. Quantification was performed by scanning SYBR Green I-stained gels (Invitrogen, Carlsbad, CA) using a Typhoon Trio imager (GE Healthcare Life Sciences).
Measurement of uptake in immunofluorescence assays
Adherent cells were seeded at a density of 5 × 104 on cover slips in 24-well plates. After 24 h, 1 µg of ZFN1/2 expression plasmid (21) was delivered using Lipofectamine 2000 (Invitrogen Life Technologies). Alternatively, transferrin-ZFNs were delivered by washing cells twice with serum-free medium and incubating with 100 nM transferrin-ZFN in 200 µl of serum-free medium for 1 h at 37°C. Suspension cells (HSPCs) were washed with IMDM containing 1% bovine serum albumin, then incubated with transferrin-ZFNs in IMDM supplemented with 100 μM ZnCl2.
For immunofluorescence, cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 20 min at room temperature, washed, permeabilized with 0.1% Triton X-100 for 5 min on ice and blocked with 2% bovine serum albumin for 1 h at room temperature. Cells were stained with 1:200 anti-FLAG monoclonal antibody (#GTX80656; GeneTex Inc. Irvine, CA) at 4°C overnight and 1:500 Texas Red goat anti-mouse IgG2b (sc-2981; Santa Cruz Biotechnologies, Santa Cruz, CA) at room temperature for 1 h. After washing, cover slips were mounted using UltraCruz Mounting Medium DAPI (Santa Cruz Biotechnologies). Image stacks (0.2 µm) were collected using a Deltavision microscope (Applied Precision Inc., Issaquah, WA, USA) and deconvolved. Signal was quantified from projections of depth-coded Z-stacks.
Measurement of uptake in immunoblotting assays
Cells (1.75 × 105 per well in poly-l-lysine-coated plates) were transfected or treated with 200 nM transferrin-ZFN for 1 h at 37°C, washed twice with DMEM and incubation was continued in DMEM. At intervals, lysates were prepared in RIPA buffer containing protease and phosphatase inhibitors (Santa Cruz Biotechnology). Equal protein amounts (∼45 µg) of each sample were resolved by SDS–PAGE using 4–20% precast gels and transferred to Immun-Blot polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules CA). These were blocked with 5% bovine serum albumin in Tris-buffered saline with 0.1% Tween 20 and probed with anti-FLAG primary antibody (#F1804; Sigma-Aldrich, St. Louis, MO) at 4°C overnight and horseradish peroxidase-conjugated secondary antibody (#626820; Invitrogen) at room temperature for 1 h. Membranes were stripped and re-probed with anti-α-actin (Santa Cruz Biotechnologies). Immune complexes were visualized with the ECL system (Bio-Rad Laboratories). Scanned images were quantified using Image J (http://rsbweb.nih.gov/ij).
In situ cleavage assay
For experiments shown in Figure 5, U2OS 2-6-3 cells (20) were transfected with 2 μg/well pSV2-lacI-ECFP (created by swapping enhanced cyan fluorescent protein (ECFP) into pSV2-lacI-EYFP). After 24 h, cells were incubated with 120 nM each of transferrin-ZFN1 and transferrin-ZFN2 at 37°C for 2 h. Cells were fixed as described previously and stained with 1:200 rabbit anti-53BP1 (#4937, Cell Signaling Technology, Danvers, MA, USA) at 4°C overnight and with 1:500 Alexa Fluor 594-goat anti-rabbit IgG (#A11012, Invitrogen Life Technologies) for 1 h at room temperature. Two independent observers scored cells blindly for co-localization of LacI-ECFP and 53BP1 foci. Experiments in Supplementary Figure S4 were performed similarly except that cells were transfected with cDNA vectors expressing ZFN1, ZFN2 or ZFN1/2 rather than being treated with transferrin-ZFN. Additionally, cells were transfected with mCherry-53BP1 (rather than stained for endogenous 53BP1) and fixation and imaging was as described (25).
Figure 5.

In situ cleavage of genomic target site by transferrin-ZFN1/transferrin-ZFN2 pair. (A) Schematic diagram of the U2OS 2-6-3 transgene array, modified from (20). Approximately 200 copies of this array are integrated at a single genomic site. Each repeat in the tandem array contains 256 copies of the lac operator recognition site and a single ZFN-cleavable CFP cDNA sequence. The transgene is not induced in these experiments, and several other elements not used here, and relevant only to gene expression are depicted in gray: 96 copies of a tetracycline response element, a minimal CMV promoter, a peroxisomal targeting signal (SKL), 24 copies of the MS2 translational operator, a rabbit beta-globin intron/exon module and a polyadenylation signal (20). (B) Merged images of fluorescent lac repressor (LacI-ECFP) and the DSB marker 53BP1 in untreated control cells (top) or in two representative fields of transferrin-ZFN treated cells (two lower rows). Insets show colocalization of Laci-ECFP and anti-53BP1 staining in treated cells. (C) Tabulation of co-localization in the indicated numbers of untreated and transferrin-ZFN treated U2OS 2-6-3 cells.
Gene correction
HEK-293/A658 cells (1.75 × 105) were seeded on coverslips in a 24-well plate and transfected with 0.1 μg/well ZFN1/2 expression plasmid, 0.9 µg of donor template plasmid or both. After 24 h, some groups were incubated with 100 nM each of transferrin-ZFN1 and transferrin-ZFN2 for 2 h at 37°C, washed twice and incubation was continued in growth medium for 3 days to allow green fluorescent protein (GFP) expression. Live cells were observed by fluorescence microscopy, and fixed cells were analyzed by flow cytometry.
Viability assay
Cells were incubated with 100 nM each of transferrin-ZFN1 and transferrin-ZFN2 for 90 min at 37°C, washed twice and culture was continued in growth medium for the indicated times. Cell viability was determined with the [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)] assay according to the vendor’s instructions (Biotium, Hayward, CA) and absorbance measured on an enzyme-linked immunosorbent assay plate reader with a test and reference wavelength of 570 and 630 nm, respectively.
RESULTS
Principle of the method
A schematic representation of transferrin-ZFN conjugation is shown in Figure 1A. Human holo-transferrin was activated by controlled reaction with SPDP, a crosslinker for amine-to-sulfhydryl conjugation. Activated transferrin was reacted with ZFNs to form a cleavable disulfide bond with a single non-liganded cysteine residue present near the N-terminus of the ZFN.
Figure 1.
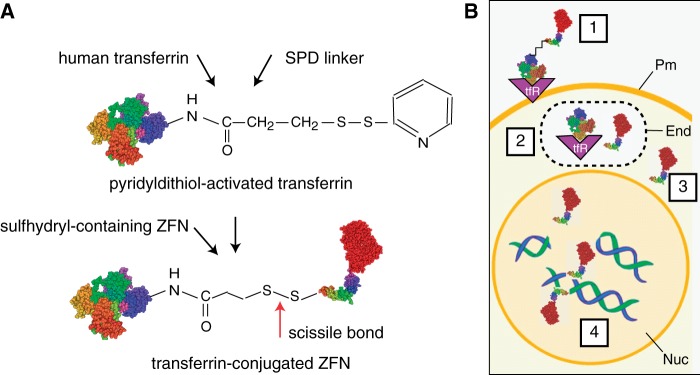
Receptor mediated delivery concept. (A) Conjugation scheme. Human holo-transferrin was activated by incubation with SPDP, and conjugates were isolated and incubated with purified ZFN as described in ‘Materials and Methods’ section. A scissile disulfide bond joins the transferrin and the ZFN. (B) Cellular delivery. 1. The transferrin-nuclease complex binds to its cognate receptor, which induces uptake of the cargo to the cell interior in the early recycling endosome. 2. Under the reducing conditions of the endosomal milieu, the ZFN protein is released from the ligand-receptor complex by ‘self-immolation’ of the disulfide bond. 3. ZFN protein escapes the endosome. 4. ZFN protein translocates to the cell nucleus where the two ZFN subunits bind to opposing DNA strands and cleave the target sequence. Pm, plasma membrane; End, endosome; Nuc, nucleus.
Figure 1B shows the pathway for transferrin receptor-mediated uptake and delivery of ZFNs to the cell nucleus. Transferrin-ZFNs bind the ubiquitous plasma membrane transferrin receptor (step 1). They are internalized to the endosomal compartment, where the reducing environment promotes cleavage of the scissile disulfide bond (step 2). ZFNs then exit the endosomal compartment (step 3), enter the nucleus and bind to and cleave their target sequence within a GFP transgene (step 4).
Production and characterization of transferrin-ZFNs
To prepare transferrin-ZFNs, recombinant ZFN proteins were expressed in E. coli and purified by immobilized nickel affinity chromatography (Supplementary Figure S1). ZFNs were incubated with activated transferrin, and conjugates were isolated by gel filtration chromatography, where they eluted ahead of unreacted transferrin and ZFN (Figure 2A). The column fractions were analyzed by SDS–PAGE (Figure 2B). Under non-reducing conditions, conjugates migrated near the top of SDS–PAGE gels, whereas under reducing conditions, the disulfide linker was cleaved to regenerate more rapidly migrating free transferrin and ZFNs. Comparison of the amount of ZFN that eluted in the transferrin-ZFN peak versus the amount that eluted later (Figure 2B, lower panel, fractions B4–B7 versus C2–C6) suggests that one-quarter to one-third of the input ZFN was recovered as conjugate.
Figure 2.

Production and cleavage activity of transferrin-ZFN conjugates. (A) Purification. S-75 gel filtration chromatography of transferrin and ZFN containing reaction mix. Elution positions of conjugates and reactants are indicated. (B) SDS–PAGE analysis of fractions from panel (A) as indicated. Sample buffer was either non-reducing (top) or reducing (bottom). Reduction of transferrin-ZFN2 (tf-ZFN2) yields free ZFN2, free transferrin and an additional band migrating just ahead of free transferrin, the identity of which is not known. (C) ZFN and tf-ZFN DNA cleavage activity. Figure shows titration of ZFN2 or tf-ZFN2 with ZFN1 held constant. Titrations of ZFN1 and tf-ZFN1 were similar (Supplementary Figure S2). Position of substrate and products are indicated. (D) Quantification of data from panel (C) showing substrate and products as a percentage of total DNA in each lane. (E) Cleavage activity of tf-ZFN1 and tf-ZFN2 in combination.
We investigated the effect of conjugation on ZFN activity and specificity. Efficient ZFN-mediated cleavage requires heterodimerization of ZFN1 (which recognizes a DNA sequence on one side of the cleavage site) and ZFN2 (which recognizes a DNA sequence on the other). In the example shown, varying amounts of transferrin-ZFN2 or non-conjugated ZFN2 were mixed with a constant amount of ZFN1 and tested for the ability to cleave a linear target DNA. About twice as much transferrin-ZFN2 was required to give the same cleavage, indicating a modest loss of activity and no loss of specificity (Figure 2C and D). Similar results were obtained when varying amounts of transferrin-ZFN1 or non-conjugated ZFN1 were mixed with a constant amount of ZFN2 and tested in the same way (Supplementary Figure S2). Finally, incubation of substrate DNA with an equimolar mixture of transferrin-ZFN1 and transferrin-ZFN2 also gave complete and specific cleavage (Figure 2E). We noted that there was also some site-specific cleavage by transferrin-ZFN2 alone at this concentration, perhaps reflecting homodimerization in solution, followed by transient interaction of transferrin-ZFN2 with its half site. We have seen the same phenomenon with non-conjugated ZFNs (LJ and WSD, unpublished).
Receptor-ligand-mediated delivery of transferrin-ZFNs to cells
We evaluated functional characteristics of transferrin-ZFNs in cells. For this purpose, we assembled a collection of different cell types, including HEK293/A658, a human embryonic kidney cell line modified to create a gene correction model (19); U2OS 2-6-3, a human osteosarcoma line bearing a multicopy ZFN target array, (20) and ROSA26GFP*/*, a primary fibroblast strain derived from a mouse gene correction model (21). We also tested human and mouse HSPCs (human CD34+ cells and mouse LSK cells), obtained as described in ‘Materials and Methods’ section. In all cases, cells were exposed to transferrin-ZFN for 1 h, fixed and stained. For established cell lines and strains, we performed conventional vector-mediated cDNA expression as a positive control.
Figure 3 presents a gallery of immunofluorescence images. Each panel represents a single optical section obtained by deconvolution microscopy. Transferrin-ZFN uptake was detected in all cells lines tested. Uptake occurred in virtually 100% of cells in each population, as seen in the micrographs (Figure 3), and confirmed in some instances by flow cytometry (data not shown). ZFN uptake was seen in both the nucleus and cytoplasm, in a ratio that varied between cell types. Where tested, the amount and distribution of intracellular ZFNs was similar in transferrin-ZFN treated and cDNA-transfected populations. Control experiments in the HEK-293 T cells showed that transferrin-ZFN uptake was competed by free transferrin, that there was no detectable uptake of non-conjugated control ZFNs when tested at the same concentration. As expected, there was no signal in mock-treated control cells. Together, results indicate that delivery is efficient, that the method is applicable to a wide range of cell types and that the uptake is mediated via the transferrin receptor.
Figure 3.

Cellular delivery of transferrin-conjugated ZFN. (A) ZFN protein levels in HEK293/A658 cells visualized by indirect immunofluorescence using ant-FLAG primary antibody. Cells were transfected with ZFN1/2 cDNA. Alternatively, they were incubated with 100 nM transferrin-ZFN1, or 100 nM non-conjugated ZFN1, as indicated, for 1 h at 37°C. Competition experiments were performed with a 10-fold molar excess of free holo-transferrin. Z-stacks were collected with a Deltavision microscope and deconvolved. Each panel corresponds to a single Z-section. (B) Quantification of nuclear uptake of tf-ZFN1 protein based on anti-FLAG fluorescence intensity. HEK293/A658 cells were incubated for 60 min with various concentration of tf-ZFN1 as indicated, then fixed and stained. Measurements were based on one representative Z-section from each of 25 cells total (from five different fields) for each experimental group. Graph shows mean and standard deviation. (C) Quantification of nuclear uptake of tf-ZFN1 as in panel (B), except that tf-ZFN1 concentration was fixed at 100 nM, and continuous incubation was performed for the indicated times. (D) ZFN protein levels in U2OS 2-6-3 human osteosarcoma cells. Cells were transfected with ZFN1/2 cDNA expression plasmid or incubated with tf-ZFN1 (100 nM, 60 min) as indicated. (E) Same as panel (D) but with murine adult fibroblasts. (F) Same as panel (D) but with primary human HSPCs (CD34+). (G) Same as panel (D) but with primary mouse HSPCs (LSK). Panels compare ZFN distribution with labeled free transferrin, note difference in localization. Scale bars, 10 μm for panels (A–E), 5 μm for panels (F) and (G).
In HEK293/A658 cells, ZFN concentrations of 50–100 nM were sufficient for maximum uptake, with no further increase at 200 nM (Figure 3B). Intranuclear ZFN levels reached a peak after 1 h of incubation, with longer incubations leading to a steady decline (Figure 3C).
Uptake in HSPCs (Figure 3F and G) is significant because these are the relevant cell type for many potential applications of engineered nuclease technology. HSPCs, particularly in the mouse, are refractory to DNA transfection, suggesting that receptor-mediated delivery may have a particular advantage over conventional methods.
The transferrin-ZFN delivery approach is based on the idea that transferrin (the carrier) and ZFN (the cargo) dissociate following cleavage of the disulfide linker in the early endosome. We tested this idea directly in the mouse HSPCs by comparing the fate of transferrin (detected using an AlexaFluor 488 conjugate) with that of ZFNs (detected via immunostaining). Alexafluor-transferrin accumulated in the thin rim of cytoplasm surrounding the nucleus, whereas ZFN cargo accumulated within the nucleus, consistent with prediction (Figure 3G).
To investigate whether transferrin-ZFN treatment resulted in significant toxicity, we performed MTT viability assays comparing transferrin-ZFN treated, cDNA-transfected and control HEK-293/A658 cells. There was no significant difference between groups at times between 24 and 72 h post-treatment (Supplementary Figure S3).
Rapid intracellular turnover following uptake of transferrin ZFNs
Part of the rationale for developing a protein-based delivery system was to gain better control over the timing and duration of nuclease exposure. To further evaluate the kinetics of ZFN uptake and turnover, we incubated HEK-293/A658 cells with transferrin-ZFNs for 1 h, transferred to fresh medium and harvested at intervals for analysis. Maximum ZFN levels were seen within 0.5 h, at the earliest time point tested, and declined rapidly following withdrawal of transferrin-ZFN-containing medium. Levels were nearly undetectable at 3.0 h (Figure 4A).
Figure 4.
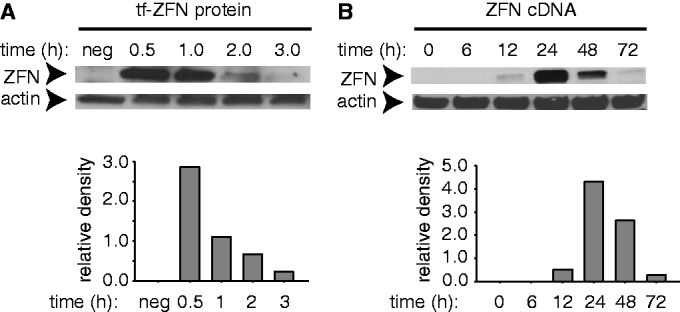
ZFN protein levels in HEK293/A658 cells as measured by immunoblotting. (A) Cells were incubated with 200 nM tf-ZFN1 for 60 min, washed and fresh medium without tf-ZFN1 was added. Cells were incubated for a further 0–3 h as indicated, after which proteins were extracted, resolved by SDS–PAGE and ZFN1 was detected by immunoblotting with anti-FLAG antibody. Lane marked ‘neg’ indicates cells not treated with tf-ZFN1. (B) Same as panel (A), except that cells were transfected with ZFN1/2 cDNA, incubated and analyzed at indicated times. Note difference in time scale in panels (A) and (B).
For comparison, we performed a parallel experiment in which cells were transfected with ZFN1/2-expressing plasmid (Figure 4B). The onset of expression was much slower, reflecting the need for transcription, mRNA processing and translation. Peak levels were not reached until 24 h post-transfection. The duration of nuclease exposure was also much longer, persisting at relatively high levels through 48 h post-transfection, but declining to an undetectable level at 72 h. Together, these results indicate that although both methods achieve comparable peak ZFN levels, protein delivery affords much better temporal control, increasing and declining for a far shorter period.
Transferrin-ZFNs cleave a predetermined genomic site in situ in living cells
To evaluate the ability of the transferrin-ZFN delivery system to induce cleavage at a genomic recognition site, we tested their activity in the U2OS 2-6-3 cell line, which harbors a ∼200 copy tandem double-strand break reporter array integrated at a single genomic site (20,25). A schematic showing the elements that make up the tandem repeat is shown in Figure 5A. Each tandem element has a CFP gene, which contains the ZFN target site and multiple copies of the lac operator. In addition, there are various transcriptional control elements, but in the experiments here, the inducer is not present, and the array is transcriptionally silent (20). The principle of the assay is to transfect with a plasmid encoding a LacI-ECFP fusion protein, which homes to the transgene locus, expose to ZFNs, fix and immunostain for 53BP1, a marker of DNA double-strand breaks that can be measured. On-target ZFN cleavage produces coincident 53BP1 and LacI-ECFP foci, whereas spontaneous endogenous double-strand breaks, or off-target ZFN cleavage, produce non-coincident foci.
We validated the in situ cleavage assay by co-transfection of LacI-EYFP, mCherry-53BP1 and ZFN1/2-encoding cDNAs. Cells that received the ZFN cDNA pair, but not control cells, showed coincident LacI-EYFP and 53BP1 foci (Supplementary Figure S4). The results demonstrate the ability of the assay to distinguish on-target cleavage, even in the presence of nonspecific background.
We then applied the assay to transferrin-ZFN-treated cells. The results showed that coincident foci were present in 19% of the transferrin-ZFN-treated cells (Figure 5B and C) but were not present in any of the control cells, indicating that transferrin-mediated ZFN delivery leads to on-target cleavage in a substantial fraction of cells.
Transferrin-ZFN treatment apparently led to some increase in the number of background foci (i.e. foci that are not coincident with LacI-ECFP), as seen in representative images in Figure 5. However, the number of background foci in the absence of treatment was too high, and their morphology was too diverse, to reliably quantify a treatment effect.
Transferrin-ZFNs stimulate gene correction
Gene correction, using ZFN-induced DSBs to stimulate homologous recombination with a wild-type donor template to correct a gene defect, is a major application of ZFN technology. We thus sought to investigate the ability of the transferrin-ZFN delivery approach to promote gene correction using reporter cell systems. Figure 6A shows a diagram of the reporter cassette in HEK-293/A658 cells, in which it is integrated at a random genomic site. ZFN cleavage is followed by homologous recombination repair using a donor template, which results in conversion of an inactive to an active GFP gene.
Figure 6.

Transferrin-ZFN mediated gene correction in HEK293/A658 cells. (A) Schematic diagram of the integrated GFP transgene in HEK293/A658 cells. A frameshift mutation has been introduced near the ZFN recognition site, leading to premature termination of translation (‘shown as STOP’). Treatment of the mutant cells with a GFP donor template that lacks the first 12 nt of the wild-type cDNA sequence, together with a GFP-targeting ZFN pair, induces homology-directed repair of the mutant locus and functional expression of GFP. (B) Representative examples of green fluorescent, GFP gene corrected HEK293/A658 cells after treatment with donor DNA and ZFN expression plasmid, middle column or tf-ZFN1/2, right column. (C) Representative flow cytometry plots demonstrating quantification of gene corrected, GFP-positive HEK293/A658 3 days after treatment with the transferrin-ZFN pair (left panel) or donor DNA alone (right panel). Treatment with donor DNA alone results in rare homologous recombination events at the transgene locus. (D) Results of independent experiments comparing the gene correction efficiency of ZFN cDNA and transferrin-ZFN-treated HEK293/A658 cells (n = 5 for transferrin-ZFN, ZFN cDNA, and no donor/no nuclease controls; n = 2 for donor/no nuclease control). Prior work has shown that there is no gene correction with nuclease in the absence of donor template (19).
Cells were co-transfected with donor template and treated, after 24 h, with the transferrin-ZFN protein pair. Alternatively, as a positive control, cells were transfected with a 1:9 mixture of ZFN1/2 cDNA expression vector and donor template. After a 3-day expression period, GFP-positive corrected cells were clearly seen by fluorescence microscopy in treated populations, but not in control populations (Figure 6B). Flow cytometry was used to quantify the gene correction rate. Figure 6C shows representative flow cytometry plots, and Figure 6D shows combined results from replicate experiments. Treatment with transferrin-ZFNs increases the frequency of gene correction by about an order of magnitude over the background level. The frequency approaches that seen with positive control populations that were transfected with ZFN1/2 cDNAs.
DISCUSSION
We have developed a method for targeted delivery of engineered nucleases based on receptor-mediated endocytosis. We demonstrate the ability to conjugate a transferrin ligand to a ZFN, with retention of enzymatic activity and specificity. Ligand-conjugated, but not control, ZFNs are taken up rapidly into a variety of cells, including primary human and murine HSPCs. Once internalized, ZFNs cleave their target sequence and promote gene correction in the presence of donor template.
A primary rationale for the present work was to develop an alternative option for delivery of engineered nuclease proteins into cells. One advantage of direct delivery of protein is improved temporal control. High levels of nuclease were present for only 1–2 h, which compares favorably to conventional methods based on vector-mediated cDNA expression, where nuclease remained present at high levels for about a day (i.e. from 24 to 48 h post-transfection). Prolonged expression may not be necessary for cleavage of the intended target site, as gene correction rates were only modestly lower with ligand-mediated protein delivery compared with cDNA transfection. In principle, shorter ZFN exposure should reduce the opportunity for cleavage and mutation at additional sites in the genome, although a direct test of this will require whole genome sequencing approaches that are beyond the scope of the present study.
We note that direct protein delivery may afford other advantages. For example, better temporal control over expression may permit manipulations of the target cell population that would not otherwise be feasible, such as delivery to synchronized cell populations. Additionally, as engineered nuclease technologies advance to clinical application, the ability to dispense with expression vectors containing strong exogenous transcriptional promoters could provide a safety and regulatory advantage.
Like other ‘supercharged’ proteins, ZFNs have some ability to enter cells via a non-specific process, without a targeting ligand. However, the results here demonstrate that transferrin conjugation significantly augments this intrinsic cell penetrating capability; indeed, we were unable to measure any uptake of non-conjugated ZFNs under our experimental conditions, which used concentrations that were about an order of magnitude lower than in the earlier study (11). In our experience, ZFNs have a tendency to aggregate and precipitate, both during expression in E. coli and when handled as purified protein at high concentration. Thus, the inherently lower protein concentrations required for targeted delivery, which makes use of high affinity ligand-receptor interaction, make the method more scalable and practical for widespread adoption.
Ligand conjugation is a well-studied approach for enhancing the delivery of conventional drugs and nanoparticles (15,16) and faces no evident barriers to clinical translation. Initial clinical applications for ligand-conjugated ZFNs might involve ex vivo treatment of isolated cells, such as HSPCs, followed by re-engraftment to the patient. In such applications, in vivo stability or clearance rates (which have not yet been investigated for transferrin-ZFNs) would not be limiting.
It will be of interest to investigate the ability to apply the method described here to other ligands and nuclease architectures. ZFNs are the most well-established class of engineered nucleases and, to our knowledge, are the only ones to have entered clinical trials. TAL effector nucleases and RNA guided endonucleases (e.g. CRISPR-Cas) appear promising in laboratory studies, and, in principle, a ligand-targeted delivery method should have many of the same advantages as demonstrated here for ZFNs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research reported in this publication was supported by the National Institutes of Health as an NIH Nanomedicine Development Center Award [PN2EY018244]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding for open access charge: US National Institutes of Health Award [PN2EY018244].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Shobha Yerigenahally for expert technical assistance. They thank our colleagues from the Nanomedicine Center for Nucleoprotein Machines for helpful discussions and the staff of the core laboratories of Georgia Regents University for invaluable technical assistance.
REFERENCES
- 1.Jensen NM, Dalsgaard T, Jakobsen M, Nielsen RR, Sorensen CB, Bolund L, Jensen TG. An update on targeted gene repair in mammalian cells: methods and mechanisms. J. Biomed. Sci. 2011;18:10. doi: 10.1186/1423-0127-18-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wirt SE, Porteus MH. Development of nuclease-mediated site-specific genome modification. Curr. Opin. Immunol. 2012;24:609–616. doi: 10.1016/j.coi.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 3.Gaj T, Gersbach CA, Barbas CF., III ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Segal DJ, Meckler JF. Genome engineering at the dawn of the golden age. Annu. Rev. Genomics Hum. Genet. 2013;14:135–158. doi: 10.1146/annurev-genom-091212-153435. [DOI] [PubMed] [Google Scholar]
- 5.Meyer M, de Angelis MH, Wurst W, Kuhn R. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc. Natl Acad. Sci. USA. 2010;107:15022–15026. doi: 10.1073/pnas.1009424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geurts AM, Cost GJ, Remy S, Cui X, Tesson L, Usal C, Menoret S, Jacob HJ, Anegon I, Buelow R. Generation of gene-specific mutated rats using zinc-finger nucleases. Methods Mol. Biol. 2010;597:211–225. doi: 10.1007/978-1-60327-389-3_15. [DOI] [PubMed] [Google Scholar]
- 7.Carbery ID, Ji D, Harrington A, Brown V, Weinstein EJ, Liaw L, Cui X. Targeted genome modification in mice using zinc-finger nucleases. Genetics. 2010;186:451–459. doi: 10.1534/genetics.110.117002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cannon P, June C. Chemokine receptor 5 knockout strategies. Curr. Opin. HIV AIDS. 2011;6:74–79. doi: 10.1097/COH.0b013e32834122d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 10.Certo MT, Ryu BY, Annis JE, Garibov M, Jarjour J, Rawlings DJ, Scharenberg AM. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods. 2011;8:671–676. doi: 10.1038/nmeth.1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF., III Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat. Methods. 2012;9:805–807. doi: 10.1038/nmeth.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cronican JJ, Beier KT, Davis TN, Tseng JC, Li W, Thompson DB, Shih AF, May EM, Cepko CL, Kung AL, et al. A class of human proteins that deliver functional proteins into mammalian cells in vitro and in vivo. Chem. Biol. 2011;18:833–838. doi: 10.1016/j.chembiol.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson DB, Cronican JJ, Liu DR. Engineering and identifying supercharged proteins for macromolecule delivery into mammalian cells. Methods Enzymol. 2012;503:293–319. doi: 10.1016/B978-0-12-396962-0.00012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson DB, Villasenor R, Dorr BM, Zerial M, Liu DR. Cellular uptake mechanisms and endosomal trafficking of supercharged proteins. Chem. Biol. 2012;19:831–843. doi: 10.1016/j.chembiol.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flygare JA, Pillow TH, Aristoff P. Antibody-drug conjugates for the treatment of cancer. Chem. Biol. Drug. Des. 2013;81:113–121. doi: 10.1111/cbdd.12085. [DOI] [PubMed] [Google Scholar]
- 17.Atkinson SF, Bettinger T, Seymour LW, Behr JP, Ward CM. Conjugation of folate via gelonin carbohydrate residues retains ribosomal-inactivating properties of the toxin and permits targeting to folate receptor positive cells. J. Biol. Chem. 2001;276:27930–27935. doi: 10.1074/jbc.M102825200. [DOI] [PubMed] [Google Scholar]
- 18.Yang J, Chen H, Vlahov IR, Cheng JX, Low PS. Evaluation of disulfide reduction during receptor-mediated endocytosis by using FRET imaging. Proc. Natl Acad. Sci. USA. 2006;103:13872–13877. doi: 10.1073/pnas.0601455103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 20.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, et al. From silencing to gene expression: real-time analysis in single cells. Cell. 2004;116:683–698. doi: 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Connelly JP, Barker JC, Pruett-Miller S, Porteus MH. Gene correction by homologous recombination with zinc finger nucleases in primary cells from a mouse model of a generic recessive genetic disease. Mol. Ther. 2010;18:1103–1110. doi: 10.1038/mt.2010.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang CC, Lodish HF. Murine hematopoietic stem cells change their surface phenotype during ex vivo expansion. Blood. 2005;105:4314–4320. doi: 10.1182/blood-2004-11-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pruett-Miller SM, Connelly JP, Maeder ML, Joung JK, Porteus MH. Comparison of zinc finger nucleases for use in gene targeting in mammalian cells. Mol. Ther. 2008;16:707–717. doi: 10.1038/mt.2008.20. [DOI] [PubMed] [Google Scholar]
- 24.Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 1999;15:34–39. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- 25.Kumaran RI, Spector DL. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J. Cell. Biol. 2008;180:51–65. doi: 10.1083/jcb.200706060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.