


Abstract
DNA methylation is an important epigenetic mark in eukaryotes, and aberrant pattern of this modification is involved in numerous diseases such as cancers. Interestingly, DNA methylation is reversible and thus is considered a promising therapeutic target. Therefore, there is a need for identifying new small inhibitors of C5 DNA methyltransferases (DNMTs). Despite the development of numerous in vitro DNMT assays, there is a lack of reliable tests suitable for high-throughput screening, which can also give insights into inhibitor mechanisms of action. We developed a new test based on scintillation proximity assay meeting these requirements. After optimizing our assay on human DNMT1 and calibrating it with two known inhibitors, we carried out S-Adenosyl-l-Methionine and DNA competition studies on three inhibitors and were able to determine each mechanism of action. Finally, we showed that our test was applicable to 3 other methyltransferases sources: human DNMT3A, bacterial M.SssI and cellular extracts as well.
INTRODUCTION
DNA methylation is an important epigenetic mark, which is essential among others for many biological processes, such as imprinting, X-inactivation, embryonic development, differentiation (1–4), maintenance of chromosomal stability (5) and gene transcription control in most eukaryotes [reviewed in (6–9)]. Consequently, abnormal patterns of DNA methylation are involved in various pathologies from neurodegenerative diseases (10,11) to cancers (12–14). Interestingly, unlike genetic mutations, epigenetic alterations are reversible and are therefore considered a promising therapeutic target (15).
In mammals, the enzymes responsible for DNA methylation are DNA methyltransferases (DNMTs), which catalyze the transfer of a methyl group from S-Adenosyl-L-Methionine (SAM) to the C5 position of cytosine in a CpG dinucleotide context (cytosine followed by a guanine). Three catalytically active DNMTs have been identified, DNMT1, DNMT3A and DNMT3B, whose features have been extensively described in several reviews (16–18). DNMT1 has mainly a maintenance role of DNA methylation pattern during replication because of its strong preference for hemimethylated DNA rather than unmethylated DNA (19–23), whereas DNMT3A and DNMT3B have a de novo methylation role (24,25). Nevertheless, it has been shown that DNMT1 collaborates with DNMT3A/3B during the de novo methylation process (26,27), whereas DNMT3s act also as maintenance methyltransferases in a DNMT1-deficient background (28,29) and in repetitive sequences (30–33). Hence, it is of interest to study the inhibition of both DNMT1 and DNMT3s. Here, we focused on the development of a universal and flexible DNMT assay.
Numerous enzymatic bioassays have been designed to find DNMT inhibitors. They are based on detection of the methylation reaction products, S-Adenosyl-L-Homocysteine (SAH) or methylated DNA [reviewed in (7)]. The quantification of SAH is often indirect and can suffer from different shortcomings. For example, SAH converting assays by coupled enzymes can reveal false positives that inhibit other enzymes than DNMTs, and therefore additional tests are needed to confirm the DNMT inhibitory activity of the hit compound (34,35). Similarly, the assay described by Graves et al. (36) uses an anti-SAH antibody that can cross-react with SAM. Therefore, we focused only on assays quantifying methylated DNA.
Previously, we developed a test to identify Dnmt3A/3L inhibitors (37). It is based on the use of an immobilized DNA duplex containing a single CpG site, which is cleaved by a restriction enzyme when not methylated. The duplex contains a fluorophore, which is lost on restriction cleavage when a compound inhibits DNA methylation. The assay is fully compatible with automation, and Medium Throughput Screenings have been performed on the murine Dnmt3A/3L catalytic complex (37,38), but the same assay gave poor results on DNMT1. In addition, the test is in heterogeneous phase, not allowing DNA-competition assays to investigate mechanisms of inhibition of the compounds. Thus, we preferred to develop a new test in homogeneous reaction.
To this aim, we switched to monitoring the incorporation of tritiated [3H] methyl groups into DNA. More precisely, DNMT transfers from [methyl-3H] SAM the radiolabeled methyl group into the DNA duplex, and the unreacted [methyl-3H] SAM can be separated from the radiolabeled DNA using standard methods such as gel filtration (39), filter-binding (40) or thin layer chromatography (41). The 3H–CH3-containing duplex can then be quantified by liquid scintillation. This radioactive assay can be applied to all DNMTs. However, this test has numerous drawbacks including significant radioactive wastes, high cost per point and low throughput. Consequently, we aimed at a new assay to quantify DNMTs inhibition, compatible with high-throughput screening (HTS). We chose a Scintillation Proximity Assay (SPA). In SPA, the scintillant is coated onto a microplate (Flashplate™) or incorporated into beads [Yttrium silicate (YSi) or polyvinyl toluene (PVT)]. Owing to the short distance that the β-particles emitted by tritium decay can travel in aqueous medium, only the bound molecules can excite the scintillant, which limits background noise and avoids purification step. In our assay, the methylation step is performed in homogeneous phase incorporating tritiated methyl groups into a biotinylated DNA duplex. The reaction is then transferred and stopped in a streptavidin coated Flashplate™ or in a microplate containing streptavidin coated beads (Figure 1).
Figure 1.

DNMT1 Flashplate™ SPA principle. Biotin is represented as a gray circle, tritium atoms as stars and the lightning represents the ability of a tritium atom to excite the plastic scintillator. In homogeneous phase, the hemimethylated duplex is methylated by the studied DNMT. The biotin is then captured and the methylation reaction is stopped on a streptavidine-coated Flashplate™. Only the bound 3H-CH3-DNA can excite the coated scintillant and emit a signal.
We characterized our system by determining the best SPA support, the methylation stop buffer, the streptavidin-biotin binding linearity range and the best duplex sequence. Subsequently, we were able to assess the inhibition activity against DNMT1 of reference compounds and proved the feasibility of SAM and DNA-competition assays. Finally, we carried out our test on 3 other DNMTs sources: human catalytic DNMT3A, bacterial M.SssI, and on cellular extracts as well.
MATERIALS AND METHODS
Materials and reagents
Two different concentrations of [methyl-3H] SAM (3TBq/mmol and 0.6TBq/mmol) were purchased from PerkinElmer (France) as well as Microscint™-20, OptiPlate™-24, OptiPlate™-96, streptavidin coated YSi beads, PVT beads and Flashplate™ PLUS Streptavidin 96-well scintillant coated microplates. Micro Bio-Spin® Columns with Bio-Gel® P-30 were purchased from Bio-Rad (France) and 384-well low-volume white round bottom polystyrene NBS™ microplate from Corning (France).
SAH, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, ethylenediaminetetraacetic acid (EDTA), bovine serum albumin, Tris–HCl (pH 7.4) and Tween®-20 were bought from Sigma-Aldrich (France). SAM chloride dihydrochloride from New England Biolabs (France) other chemicals from VWR (France).
All cell lines were obtained from the ATCC and grown at 37°C, 5% CO2. KG-1 cells were maintained in RPMI1640 (Lonza, France), supplemented with 10% fetal calf serum (Lonza, France), and K-562 cells were maintained in IMDM (Sigma, France), supplemented with 10% foetal calf serum (Lonza, France) and 4 mM l-Glutamine (Sigma, France).
Enzyme production
The sequence encoding the C-terminal domain (residues 624–912) of human DNMT3A (DNMT3A-C) was amplified by PCR from IMAGE clone (Origene) with the following primers: sense 5′-CCATGGCTCATATGAACCACGACCAGGAATTTGAC-3′ and anti-sense: 5′-CTCGAGAAGCTTTTACACACACGCAAAATACTC-3′. The amplicon was cloned into pCR®2.1 TOPO (Invitrogen™, Life Technologies, France). After amplification, the plasmid was digested by NdeI and HindIII, and the resulting fragment (884 pb) cloned into pET28a (Novagen®, Merck Chemicals, Germany). The final plasmid pET28a-DNMT3A(624–912) was transformed in Escherichia coli BL21(DE3, pLys) (Novagen®, Merck Chemicals, Germany) for the expression of the DNMT3A-C. Cells were amplified over night in LB medium supplemented with ampicilin and chloramphenicol antibiotics, and the protein was expressed by 4 h isopropyl β-D-1-thiogalactopyranoside induction. Cells were collected by centrifugation for purification according to Jeltsch et al. and Jia et al. (41,42). DNMT3A-C purity was assessed by Coomassie-stained SDS gels and estimated to be >90%. Finally, total proteins concentration was determined by Bradford assay as 1.11 µg/µL (28.3 µM).
His-DNMT1 (182 kDa, human) was cloned, expressed and purified as described by Lee et al. (43). The concentration of total proteins was determined by Bradford assay as 1.72 µg/µL (9.45 µM). M.SssI was purchased from New England Biolabs (France) at 4000 U/mL and was supplied with NEBuffer 2.
Duplex formation and cellular extracts
Oligonucleotides were purchased from Eurogentec (Belgium). Duplexes were formed by hybridization of the two complementary strands in sterilized water according to standard procedures.
Cellular extracts were obtained according to Fritsch et al. (44). Briefly, we used 10 billion cells (dried pellet), which were resuspended in a hypotonic buffer (10 mM Tris–HCl (pH 7.65), 1.5 mM MgCl2, 10 mM KCl) and disrupted with 20 strokes of a tight-fitting Dounce homogenizer. At the end of cell lysis, sucrose, spermidine and spermine-containing buffer was added to limit nuclei leak. The cytosolic fraction was separated from nuclei by 7 min centrifugation at 4°C at 9000 rpm. The soluble nuclear extract (SNE) was obtained by incubation of the nuclear pellet in a high salt buffer (900 mM NaCl, 20 mM Tris (pH 7.65), 25% glycerol, 1.5 mM MgCl2, 0.2 mM EDTA), to obtain a final concentration of 300 mM NaCl, for 30 min at 4°C and centrifugation at 10 000 rpm, 4°C for 10 min. The resulting pellet, which corresponds to chromatin fraction (nuclear chromatin extract (NCE)), was resuspended and digested with micrococcal nuclease (cat# N3755, Sigma, France). Soluble nuclear and chromatin fractions were then ultracentrifugated at 32 000 rpm for 1 h at 4°C, and protein concentration was determined by Bradford assay. The presence of DNMT1 and DNMT3A in both fractions was verified by a western blot (DNMT1 antibody NB100-264 from Novus Biological, UK, and DNMT3A antibody CST3598 from Cell Signaling, USA) as described in the Supplementary Information.
Enzymatic assays
The radioactive gel filtration assay was carried out in 50 µL of total reaction volume, containing the tested compound (up to 1% DMSO), 0.28 µM of [methyl-3H] SAM (3TBq/mmol), 0.2 µM of DNA duplex (Dup_0, Table 1) and 90 nM of His-DNMT1 in reaction buffer [20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (pH 7.2), 1 mM EDTA, 50 mM KCl, 25 µg/mL BSA (bovine serum albumin)]. The reaction was incubated at 37°C for 2 h, then the unreacted [methyl-3H] SAM and the enzyme were separated from 3H-CH3-containing DNA by passing through a Micro Bio-Spin® P-30 column. The filtrate was then transferred into a white opaque 24-well OptiPlate™, 1 mL of Microscint™-20 was added, and the microplate was read on TopCount NXT (PerkinElmer, France).
Table 1.
Sequences of tested duplexes.
| Duplex Name | Number of CpG | Sense | Anti-sense |
|---|---|---|---|
| Dup_0 | 8 | 5′-GATmCGCmCGATGmCGmCGAATmCGmCGATmCGATGmCGAT-3′ | 5′-ATCGCATCGATCGCGATTCGCGCATCGGCGATC-3′ |
| Zebularine-containing duplex | 8 | 5′-GATmCGCmCGATGmCGmCGAATmCGmCGATmCGATGmCGAT-3′ | 5′-ATZebGCATZebGATZebGZebGATTZebGZebGCATZebGGZ ebGATC-3′ |
| Dup_1 hmC | 8 | 5′-GATmCGCmCGATGmCGmCGAATmCGmCGATmCGATGmCGAT-3′ | BIOT-5′-ATCGCATCGATCGCGATTCGCGCATCGGCGATC-3′ |
| Dup_1 nmC | 8 | 5′-GATCGCCGATGCGCGAATCGCGATCGATGCGAT-3′ | BIOT-5′-ATCGCATCGATCGCGATTCGCGCATCGGCGATC-3′ |
| Dup_2 hmC | 1 | 5′-GCTATATATACGTACTGTGAACCCTACCAGACATGCACTG-3′ | BIOT-5′-CAGTGCATGTCTGGTAGGGTTCACAGTAmCGTATA TATAGC-3′ |
| Dup_2 nmC | 1 | 5′-GCTATATATACGTACTGTGAACCCTACCAGACATGCACTG-3′ | BIOT-5′-CAGTGCATGTCTGGTAGGGTTCACAGTACGTAT ATATAGC-3′ |
| Dup_3 | 1 | 5′-GCATATATATGAmCGATCCTGTAGGTCACTACCAGACATGCACTG-3′ | BIOT-5′-CAGTGCATGTCTGGTAGTGACCTACAGGATCGTCA TATATATGC-3′ |
| Dup_4 | 1 | 5′-GGAGGCCmCGCCTGCTGTAGGTCACTACCAGACATGCACTG-3′ | BIOT-5′-CAGTGCATGTCTGGTAGTGACCTACAGCAGGCGG CCTCC-3′ |
| Dup_5 | 1 | 5′-TCCTGTGAGCCTCCmCGCAGGTCACTACCAGACATGCACTG-3′ | BIOT-5′-CAGTGCATGTCTGGTAGTGACCTGCGGAGGCTC ACAGGA-3′ |
hmC, hemi-methylated cytosine; nmC, non-methylated cytosine; BIOT, biotin; Zeb, zebularine.
In the Flashplate™ assay, the reaction was performed in a 10 µL of total reaction volume in low-volume NBS™ 384-well microplates, containing the tested compound (up to 1% DMSO), 1 µM of a SAM/[methyl-3H] SAM (3TBq/mmol) mix in a ratio of 3-to-1 (isotopic dilution 1*:3), 0.3 µM of biotinylated DNA duplex (Dup_1 hmC for DNMT1, Dup_1 nmC for DNMT3A and M.SssI, Table 1), and 90 nM of DNMT1, 1 µM of DNMT3A or 100 nM of M.SssI. For DNMT1 and DNMT3A, the reaction was incubated at 37°C for 2 h in the aforementioned reaction buffer, whereas the M.SssI methylation reaction was carried out in 1X NEBuffer 2 for 1 h at 37°C. Eight µL are then transferred into a streptavidin 96-well scintillant coated Flashplate™ containing 190 µL of 20 µM SAH in 50 mM Tris–HCl (pH 7.4). The Flashplate™ was agitated at room temperature for 1 h, washed three times with 200 µL of 0.05% Tween®-20 in 50 mM Tris–HCl (pH 7.4), and read in 200 µL of 50 mM Tris–HCl (pH 7.4) on TopCount NXT. We performed all our measurements in 96-well Flashplate™, as our TopCount did not possess the module allowing lecture of 384-well microplates.
The YSi and PVT beads tests followed the same protocol. After the incubation at 37°C, 8 µL were transferred into a white 96-well OptiPlate™ containing 190 µL of 20 µM SAH in 50 mM Tris–HCl (pH 7.4) and 200 µg of YSi beads or 400 µg of PVT beads. The microplate was then read on TopCount.
For the Flashplate™ assay on cellular extracts, the methylation step was performed in a 10 µL of total reaction volume in low volume NBS™ 384-well microplates, containing the tested compound, 1 µM of [methyl-3H] SAM (3TBq/mmol), 0.3 µM of biotinylated DNA duplex (Dup_1 hmC or nmC, Table 1) and 1.5 µg/µL of K-562 SNE in the reaction buffer. The reaction was incubated at 37°C for 2 h, and 8 µL are then transferred into a streptavidin 96-well scintillant coated Flashplate™ containing 190 µL of 20 µM SAH in 50 mM Tris–HCl (pH 7.4). The Flashplate™ was agitated at room temperature for 1 h, washed three times with 200 µL of 0.05% Tween®-20 in 50 mM Tris–HCl (pH 7.4), and read in 200 µL of 50 mM Tris–HCl (pH 7.4) on TopCount. For the heat inactivated controls, the 5X solutions of 1.5 µg/µL were heated at 95°C for 10 min.
For every assay, 40 000 total counts were accumulated for each well, with a maximum time limit of counting fixed at 10 min per well. All signals were expressed in counts per minute (cpm).
Determination of IC50 values and Km values
The concentration at which 50% of inhibition is observed (IC50) was determined by analysis of a concentration range of the tested compound in duplicates. The negative and positive controls were defined as points without enzyme and points without any compound, respectively. The percentage of inhibition (%I) were calculated as following: %I = 100-100 × (cpmi − cpmneg)/(cpmpos − cpmneg), where cpmi is the inhibitor signal, cpmneg the negative control signal and cpmpos the positive control signal. The non-linear regression fittings with sigmoidal dose response (variable slope) were performed with GraphPad Prism 4.03 (GraphPad Software).
For Km determination, one substrate was varied while maintaining the other at a saturating concentration. SAM (0.6TBq/mmol) was varied from 0.25 to 20 µM with an isotopic dilution of 1*:1, whereas the DNA duplex concentration was kept at 1.0 µM. DNA duplex concentration was titrated from 0.05 to 1.0 µM, whereas SAM was hold at 15 µM (1*:2) total. All reactions were performed in duplicates. The Km value of each substrate was then determined by non-linear fitting of the data with the Michaelis–Menten equation on GraphPad Prism 4.03.
Competition studies
In SAM-competition assays, SAM (0.6TBq/mmol) was varied between 0.5 and 15 µM with an isotopic dilution of 1*:1 at a fixed DNA duplex concentration of 1.0 µM. For each SAM concentration, the tested compound concentration was adjusted between its IC10 and its IC80.
In DNA-competition assays, the DNA duplex concentration was varied between 0.05 and 1.0 µM, whereas SAM (0.6TBq/mmol) concentration was hold at 15 µM with an isotopic dilution of 1*:2. For each DNA duplex concentration, the tested compound concentration was adjusted between its IC10 and its IC80.
For each substrate concentrations, the IC50 of tested compound was calculated by non-linear regression fitting with sigmoidal dose response (variable slope). For each compound concentration, 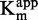 and
and 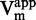 of each susbrate were approximated by non-linear fitting of the data with the Michaelis–Menten equation on GraphPad Prism 4.03.
of each susbrate were approximated by non-linear fitting of the data with the Michaelis–Menten equation on GraphPad Prism 4.03.
RESULTS AND DISCUSSION
Optimization of the Flashplate™ assay
The following steps of the assay (Figure 1) were optimized on DNMT1.
Methylation reaction stop
First, we assessed several stop methods to ensure that the reaction does not proceed during the streptavidin-biotin binding step. This issue was addressed on the previously developed radioactive gel filtration assay (39). Addition of MgCl2 (from 1 to 100 mM) or NaCl (from 0.5 to 1 M), known to inhibit DNMT (45), did not block the methylation reaction (data not shown). Acidification of the solution (4–5 decrease in pH) by adding concentrated HCl resulted in a drastic signal loss, probably due to DNA degradation. Heat inactivation was not investigated because it is poorly compatible with automation. Lastly, SAM (from 15 to 200 µM) or SAH (from 20 to 100 µM) were added with success, the first to compete with radiolabeled SAM, the latter being a well-known inhibitor of DNMTs as product of the methylation reaction. Finally, we chose to use as stop buffer 20 µM SAH in 50 mM Tris–HCl (pH 7.4) solution.
Comparison of SPAs supports
Second, as several SPA supports are commercially available, YSi beads, PVT beads and Flashplates™ were compared. To this purpose, the methylation reaction was performed with Dup_1 hmC (Table 1) according to the radioactive gel filtration protocol (39). Three aliquots of 35 µL were withdrawn and transferred into a Flashplate™ or into a 96-well microplate containing 200 µg of YSi beads or 400 µg of PVT beads, respectively. All the wells contained a final volume of 200 µL of a 20 µM SAH in 50 mM Tris–HCl (pH 7.4). After 1 h of streptavidin-biotin binding, the samples were read on a liquid scintillation counter. The best maximal signal was obtained with YSi beads at 165 000 cpm, against 125 000 cpm for Flashplate™ and 80 000 cpm for PVT beads. The signal-to-background ratio was way better for Flashplate™ (>400) than for YSi beads or PVT beads, both about 145 (Supplementary Figure S1A). In addition, the use of beads can induce more variability because of the difficulty to suspend them homogeneously. Moreover, YSi and PVT beads assays are twice more expensive than Flashplate™ test for equivalent performances. Therefore, Flashplate™ were the best compromise between sensitivity, signal-to-background ratio, cost and convenience. Besides, it allowed us to reduce the [methyl-3H] SAM amount and the total reaction volume.
Binding linearity and duplex choice
Third, we assessed the linearity of biotin-streptavidin binding in Flashplates™ by transferring aliquots of different volumes after the methylation reaction. For this optimization, the methylation reaction was performed according to the radioactive gel filtration protocol (39) with the use of Dup_1 hmC (Table 1). After 2 h of incubation, aliquots of different volumes were transferred into a Flashplate™ to a final 200 µL volume of 20 µM SAH in Tris–HCl (Supplementary Figure S1B). This binding step was linear up to 4 pmol of transferred biotinylated duplex. Therefore, we adjusted the volume to be transferred into the Flashplate™ according to the biotin concentration in the methylation reaction in order not to exceed this limitation.
Several duplexes were tested to identify the best DNMT1 target (Table 1). Dup_1 contained 8 CpGs and was used previously in the radioactive gel filtration assay (39). Sequences of Dup_2 and Dup_3 were described by Ceccaldi et al. (37), for DNMT3A and DNMT1, respectively, and displayed only one CpG site. Dup_4 and Dup_5 were designed after those used by Song et al. (23,46) for DNMT1 crystallization and in enzymatic assays, bearing one single CpG site.
The influence of the hemi or non-methylated status of the DNA duplex was confirmed. In agreement with the literature (19,21,22), DNMT1 had a strong preference for hemimethylated substrates, as illustrated by the difference in methylation of hemimethylated Dup_1 and hemimethylated Dup_2 compared with non-methylated Dup_1 and non-methylated Dup_2 (Figure 2). We compared the duplexes among them by calculating the CpGs concentration; thus, 0.3 µM of 8 CpG-containing duplex were compared with 2.4 µM of 1 CpG-containing DNA. Overall, for each duplex tested, we observed that the intensity of the signals at 2.4 µM concentration was lower than those measured at 0.3 µM concentration (Figure 2), suggesting a possible inhibition by an excess of substrate. Finally, we chose the hemimethylated duplex Dup_1, which gave the best results and had the advantage of displaying more than 1 CpG site and thus increasing the amount of transferred 3H–CH3 for the same amount of biotin.
Figure 2.

Comparison of different duplexes for DNMT1 methylation assay. Means ± standard deviation of duplicates are displayed. hmC stands for hemimethylated duplex and nmC stands for non-methylated duplex.
Optimization of the conditions for the methylation reaction by DNMT1
To optimize the DNMT1 Flashplate™ assay, we performed an enzymatic titration and studied the kinetics of the reaction at 37°C and at room temperature to limit evaporation. As 90 nM of enzyme gave the highest signal in the linear response range (Supplementary Figure S2A), it was chosen as concentration for the assay. Room temperature kinetics resulted in weak signals; therefore, the methylation reaction was performed at 37°C for 2 h, for which the response of the enzyme was both linear and maximal (Supplementary Figure S2B).
Next the Km of the two substrates SAM and DNA was measured. To determine the 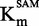 , we used the Michaelis–Menten model in a SAM concentration range spanning from 0.25 to 20 µM, whereas the DNA concentration was kept near saturating concentration at 1.0 µM (Figure 3A). The Km of DNA was determined by using the same fitting model with a DNA concentration range spanning from 0.05 to 1.0 µM and near saturating SAM concentration at 15 µM (Figure 3B). The approximated Km values were 4.4 ± 0.5 µM and 0.27 ± 0.03 µM for SAM and DNA, respectively. It is established that to maximize opportunities of identifying the full constellation of inhibitory molecules (competitive, non-competitive or uncompetitive), the optimal concentration of substrates is the Km value (47). We thus chose to conduct the assay at the Km value for biotinylated DNA and at Km/4 for [methyl-3H] SAM. This latter choice was dictated by the fact that it was too expensive to work at Km value for [methyl-3H] SAM, even with an isotopic dilution of 1*:3.
, we used the Michaelis–Menten model in a SAM concentration range spanning from 0.25 to 20 µM, whereas the DNA concentration was kept near saturating concentration at 1.0 µM (Figure 3A). The Km of DNA was determined by using the same fitting model with a DNA concentration range spanning from 0.05 to 1.0 µM and near saturating SAM concentration at 15 µM (Figure 3B). The approximated Km values were 4.4 ± 0.5 µM and 0.27 ± 0.03 µM for SAM and DNA, respectively. It is established that to maximize opportunities of identifying the full constellation of inhibitory molecules (competitive, non-competitive or uncompetitive), the optimal concentration of substrates is the Km value (47). We thus chose to conduct the assay at the Km value for biotinylated DNA and at Km/4 for [methyl-3H] SAM. This latter choice was dictated by the fact that it was too expensive to work at Km value for [methyl-3H] SAM, even with an isotopic dilution of 1*:3.
Figure 3.

Characterization of the DNMT1 assay and inhibition studies on reference compounds. (A) Determination of SAM Km at 1.0 µM of hemimethylated Dup_1. (B) Determination of DNA Km at 15 µM of [methyl-3H] SAM. (C) Two independent dose-response curves for zebularine-containing duplex, each in duplicates. (D) Two independent dose-response curves for sinefungin, each in duplicates. (E) Two independent dose-response curves for 47, each in duplicates. Km and coefficient of determination (R2) are displayed. Means of IC50 and coefficient of determination (R2) are displayed.
Z′ factor
Finally, this resulted in a new optimized DNMT1 inhibition assay on Flashplate™. To assess the reliability of our test, we determined the Z′ factor. This factor is a statistical parameter that evaluates the quality of an HTS assay. It is defined as 1-(3SDpos + 3SDneg)/(µpos-µneg) where SDpos is the standard deviation of positive controls, SDneg is the standard deviation of negative controls, µpos is the mean of positive controls, and µneg is the mean of negative controls (48). In our case, it resulted in Z′ = 0.88, which is characteristic of a good and reliable assay.
Tolerance to DMSO
As DMSO is widely used as solvent for chemical compounds in HTS, we assessed the DMSO tolerance of our Flashplate™ assay. Surprisingly, a slight stimulation (<10%) of DNMT1 activity was observed when adding 1 to 10% (v/v) of DMSO and then inhibition when adding 15 to 50% (v/v) (Supplementary Figure S3). Yokochi and Robertson hypothesized that the stimulation by DMSO is probably due to two types of interactions: DMSO-DNA and DMSO-SAM (45). We recommend not to exceed a final 1% (v/v) DMSO concentration. However, if needed (e.g. because of compound solubility), it is still possible to perform experiments in up to 10% (v/v) DMSO, with adjusting the corresponding controls.
Inhibition studies on DNMT1
Reference inhibitors
As the aim of our study was to evaluate the inhibitory activity of small chemical molecules, we validated it with three different inhibitors of the literature: sinefungin (49,50), zebularine-containing DNA (51) and 3-chloro-3-nitroflavanone 47 (37). Sinefungin is a bacterial analog of SAM and SAH and act as a SAM competitor (49,50). Zebularine, which is a nucleoside analog of cytidine, was used as DNA competitor, as it binds strongly but reversibly to the DNMT (51). In the zebularine-containing DNA, each cytidine in a CpG context on the anti-sense strand of Dup_1 was replaced by zebularine (Table 1). Finally, we previously identified compound 47 as a DNMT inhibitor (37).
For each compound, a classical sigmoidal dose-response curve spanning from 0 to nearly 100% of inhibition was obtained (Figure 3C–E). IC50 were approximated with narrow confidence interval and are reported in Table 2. The reproducibility of the assay was assessed by two independent experiments run each in duplicates (Figure 3C–E). Based on these good performances, the Flashplate™ assay was therefore considered validated for screening studies on DNMT1.
Table 2.
Inhibitory activities of reference compounds on DNMT1
| Inhibitors | DNMT1 |
|---|---|
| IC50 (µM) ± standard deviation | |
| Sinefungin | 80 µM ± 4 |
| Compound 47 | 44.5 µM ± 0.6 |
| Zebularine-containing duplex | 0.095 µM ± 0.004 |
Means of two independent experiments are displayed ± standard deviation. IC50 were assessed from dose-response curves of each compounds, for 0.3 µM of Dup_1 hmC (Table 1), 1µM SAM (isotopic dilution of 1*:3), 90 nM DNMT1.
SAM-competition studies
The assay was also developed with the aim to perform competition experiments with the 2 substrates, SAM and DNA. Thus, the assay ability to determine inhibitory mechanism was assessed on the reference inhibitors. Sinefungin was varied from IC10 to IC80 and, for each concentration, SAM concentrations spanned from 0.5 to 15 µM.
For each SAM concentration, we approximated the IC50 of sinefungin, and we plotted it against [SAM]/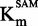 (Figure 4A). As displayed, the IC50 increased with SAM concentration, corresponding to a SAM-competitive inhibitor (47). To further confirm this, the
(Figure 4A). As displayed, the IC50 increased with SAM concentration, corresponding to a SAM-competitive inhibitor (47). To further confirm this, the 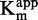 and
and 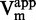 values for each sinefungin concentration were extrapolated using the Michaelis–Menten equation.
values for each sinefungin concentration were extrapolated using the Michaelis–Menten equation. 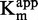 plotted against sinefungin concentration gave aligned points with a linear regression coefficient (R2) superior to 0.98 (Figure 4C), confirming the SAM-competitive behavior (52). Hence, both methods are consistent with literature indicating that sinefungin is a SAM competitor (49,50).
plotted against sinefungin concentration gave aligned points with a linear regression coefficient (R2) superior to 0.98 (Figure 4C), confirming the SAM-competitive behavior (52). Hence, both methods are consistent with literature indicating that sinefungin is a SAM competitor (49,50).
Figure 4.

SAM-competition assays on DNMT1 with sinefungin, zebularine-containing duplex and compound 47. Experiments with sinefungin are represented by open diamonds, zebularine-containing duplex by plain triangles and compound 47 by plain squares. (A and B) IC50 of each molecule as function of [DNA]/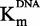 . (C and D)
. (C and D) 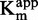 as function of [inhibitor]. (E and F) 1/
as function of [inhibitor]. (E and F) 1/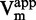 as function of [inhibitor]. (G and H) 1/
as function of [inhibitor]. (G and H) 1/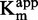 as function of [inhibitor]. For each linear regression, if the slope is significantly different from 0, then the regression is represented by a plain line, and the coefficient of determination (R2) is displayed along the corresponding line.
as function of [inhibitor]. For each linear regression, if the slope is significantly different from 0, then the regression is represented by a plain line, and the coefficient of determination (R2) is displayed along the corresponding line.
We then studied the zebularine-containing duplex. The IC50 seemed independent of the [SAM]/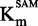 (Figure 4B), indicating a SAM non-competitive behavior (47). Moreover, Michaelis–Menten fitting resulted in an overall constant
(Figure 4B), indicating a SAM non-competitive behavior (47). Moreover, Michaelis–Menten fitting resulted in an overall constant 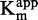 (Figure 4D) and a linearly increasing 1/
(Figure 4D) and a linearly increasing 1/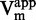 (R2 > 0.92, Figure 4F) when plotted against the inhibitor concentration. In addition, plotting of 1/
(R2 > 0.92, Figure 4F) when plotted against the inhibitor concentration. In addition, plotting of 1/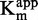 against the inhibitor concentration gave no linear dependency (Figure 4H), excluding a hypothetical uncompetitive behavior. These results are consistent with a SAM non-competitor as expected for a DNA competitor.
against the inhibitor concentration gave no linear dependency (Figure 4H), excluding a hypothetical uncompetitive behavior. These results are consistent with a SAM non-competitor as expected for a DNA competitor.
The Flashplate™ assay was able to discriminate between competitive and non-competitive inhibition for two reference inhibitors. Therefore, we applied the method to compound 47 to determine its mechanism of action. Plotting IC50 against [SAM]/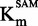 showed that compound 47 might be a SAM non-competitive or uncompetitive as the curve slightly decreased with [SAM]/
showed that compound 47 might be a SAM non-competitive or uncompetitive as the curve slightly decreased with [SAM]/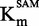 (Figure 4A) (47). Similarly to the zebularine-containing duplex,
(Figure 4A) (47). Similarly to the zebularine-containing duplex, 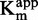 and 1/
and 1/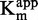 plots against inhibitor concentration did not give linear dependency (Figure 4C and G), whereas 1/
plots against inhibitor concentration did not give linear dependency (Figure 4C and G), whereas 1/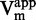 as a function of 47 concentration gave aligned points with R2 superior to 0.99 (Figure 4E). These data clearly indicated that compound 47 is a SAM non-competitive inhibitor.
as a function of 47 concentration gave aligned points with R2 superior to 0.99 (Figure 4E). These data clearly indicated that compound 47 is a SAM non-competitive inhibitor.
DNA-competition studies
In parallel, DNA competition experiments were carried out on DNA concentrations from 0.05 to 1.0 µM at a constant 15 µM SAM concentration with a 1*:2 isotopic dilution. First, we evaluated the zebularine-containing duplex, varying from IC10 to IC80. The IC50 increased with [DNA]/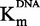 (Figure 5B), as the
(Figure 5B), as the 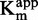 with inhibitor concentration (R2 > 0.97), confirming a DNA-competitive behavior as expected.
with inhibitor concentration (R2 > 0.97), confirming a DNA-competitive behavior as expected.
Figure 5.

DNA-competition assays on DNMT1 with sinefungin, zebularine-containing duplex and compound 47. Experiments with sinefungin are represented by open diamonds, zebularine-containing duplex by plain triangles and compound 47 by plain squares. (A and B) IC50 of each molecule as function of [DNA]/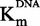 . (C and D)
. (C and D) 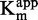 as function of [inhibitor]. (E and F) 1/
as function of [inhibitor]. (E and F) 1/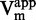 as function of [inhibitor]. (G and H) 1/
as function of [inhibitor]. (G and H) 1/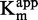 as function of [inhibitor]. For each linear regression, if the slope is significantly different from 0, then the regression is represented by a plain line, and the coefficient of determination (R2) is displayed along the corresponding line.
as function of [inhibitor]. For each linear regression, if the slope is significantly different from 0, then the regression is represented by a plain line, and the coefficient of determination (R2) is displayed along the corresponding line.
Then we studied sinefungin whose plot of IC50 against [DNA]/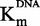 displayed a decreasing branch of hyperbola (Figure 5A), consistent with an uncompetitive inhibition (47). The Michaelis–Menten analysis confirmed this tendency, with a linear dependency of 1/
displayed a decreasing branch of hyperbola (Figure 5A), consistent with an uncompetitive inhibition (47). The Michaelis–Menten analysis confirmed this tendency, with a linear dependency of 1/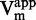 and 1/
and 1/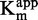 on the inhibitor concentration (R2 > 0.97, Figure 5E and G).
on the inhibitor concentration (R2 > 0.97, Figure 5E and G).
In agreement, we have identified a SAM-competitor that is DNA-uncompetitive (sinefungin) and a DNA-competitor that is SAM non-competitive (the zebularine-containing duplex). These results seem to suggest that DNMT1 proceeds according to an ordered sequential mechanism (53), where DNA binds first, followed by SAM, consistent with previous results (54,55). However, this remains a hypothesis, as we cannot exclude that DNMT1 was co-purified with some SAM and DNA.
As the method was validated on reference compounds, it was then applied to flavanone 47. Similar results as with sinefungin were obtained: IC50 against [DNA]/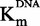 was compatible with both non-competitive or uncompetitive inhibition (Figure 5.A), whereas 1/
was compatible with both non-competitive or uncompetitive inhibition (Figure 5.A), whereas 1/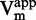 and 1/
and 1/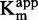 clearly identified an uncompetitive behavior (R2 > 0.97, Figure 5E and G).
clearly identified an uncompetitive behavior (R2 > 0.97, Figure 5E and G).
In agreement with literature, we have confirmed that sinefungin was a SAM-competitive and DNA-uncompetitive inhibitor (49,50), whereas the zebularine-containing duplex was qualified as a SAM non-competitor and DNA-competitior (51). Our assay was also able to identify the inhibitory mechanism of 3-chloro-3-nitroflavanone 47, which seemed to be SAM non-competitive and DNA-uncompetitive. Hence, the Flashplate™ assay is validated for screening and for mechanistic studies. To address the selectivity question, we tested it on two other C5 DNMTs (human DNMT3A and bacterial M.SssI) and on cellular extracts.
Applications to other DNMTs and cellular extracts
Application of the assay to nuclear extracts
The assay was also adapted to measure DNMT activities in NCE and SNE from leukemia K-562 cell line, known to be sensitive to demethylation (56,57). NCE and SNE were tested for DNA methylation on both the hemimethylated and unmethylated duplex (Dup_1 hmC and nmC, Table 1, Figure 6A). Due to low signals, we chose to test nuclear extracts in our assay without isotopic dilution at final concentration of 1 µM [methyl-3H] SAM. This loss in signal may be due to the dilution of the tritiated SAM by the cellular non-radioactive SAM.
Figure 6.

Application of the assay to K-562 nuclear extracts and other C5 DNMTs. (A) Increasing amounts of NCE and SNE from K-562 cells have been tested for DNA methylation activity using Dup_1 hmC and nmC (Table 1). The experiment was performed in duplicates. Means are represented with standard deviation. (B) Two independent dose-response curves for sinefungin on K-562 SNE, each in duplicates. (C) Two independent dose-response curves for sinefungin on DNMT3A-C, each in duplicates. (D) Two independent dose-response curves for sinefungin on M.SssI, each in duplicates. Means of IC50 and coefficient of determination (R2) are displayed.
The control of heat inactivated extracts did not display any activity (labeled ‘inact’ in Figure 6A), whereas NCE and SNE displayed a dose response on hemimethylated duplex. The fact that SNE signals were higher than NCE signals can be explained by the presence of more DNMT1 in SNE than in NCE, as shown in the corresponding western blot (Supplementary Figure S4). Interestingly, the signals recorded with unmethylated duplex were lower (Figure 6A), which correlates with the poor activity of DNMT1 on unmethylated duplex and the low quantity of DNMT3s in these nuclear extracts (Supplementary figure S4). Therefore, we decided to work at 1.5 µg/µL of K-562 SNE on hemimethylated Dup_1 (hmC Dup_1) to test the reference inhibitor sinefungin.
Sinefungin gave full dose-response curve from 0 to 100% of inhibition with good reproducibility and narrow confidence interval (IC50: 51 ± 6 µM, mean of two experiments each run in duplicates ± standard deviation, Figure 6B). This result proves that studying the inhibition of DNA methylation in cellular extract is achievable with the protocol developed here.
Application of the assay to other C5 DNMTs
The assay was easily adapted to human DNMT3A and M.SssI. As both enzymes are able to react on unmethylated DNA duplexes, they were tested on unmethylated Dup_1 (Dup_1 nmC, Table 1).
Titration of DNMT3A displayed a response linearity range between 0 and 1.5 µM, whereas M.SssI had a linear response up to 120 nM (data not shown). Therefore, we chose 1 µM of enzyme for DNMT3A and 100 nM for M.SssI.
Sinefungin was assayed on both enzymes and gave full dose-response curves from 0 to nearly 100% of inhibition with satisfactory confidence intervals and good reproducibility (IC50: 1.8 ± 0.6 µM on DNMT3A and 45 ± 7 µM on M.SssI, means of two experiments each run in duplicates ± standard deviation, Figure 6C and D).
Altogether, these data confirm that the assay can be applied to cellular extracts and other C5 DNMTs.
CONCLUSION
The existing in vitro DNMTs tests suffer from different shortcomings, whether measuring the SAH or the methylated DNA production. On one hand, SAH quantification is applied to both histone methyltransferases and DNMTs (34–36). However, they can reveal false positive due to cross-reaction between SAM and anti-SAH antibody or to inhibition of enzymes that convert SAH into ATP. On the other hand, measurement of methylated DNA is performed either in heterogeneous phase, preventing any competition study, or in radioactive homogeneous phase but with high costs per point and large radioactive waste.
We here described a DNMT1 Flashplate™ assay that was miniaturized in final 10 µL of reaction volume, then optimized and finally shows good reliability and sensitivity, with signal-to-background ratios up to 400 and a calculated Z′ factor of 0.88. This assay is able to monitor inhibition activities on DNMT1, DNMT3A and M.SssI of small molecules with satisfactory dose-response curves. It can also be applied to screen microorganism extracts (data not shown) and different source of enzymes such as purified DNMTs or cellular extracts. This assay is fully compatible with automation and can be easily adapted to HTS. In addition, it provides insights on the inhibition mechanisms of compounds, on SAM and DNA-competition studies, as we were able to confirm SAM-competitive and DNA-competitive inhibition of sinefungin and zebularine-containing DNA, respectively (49–51). Moreover, we were able to characterize the mechanism of action of 3-chloro-3-nitroflavanone 47, previously identified as a DNMT inhibitor (37), which appeared to be a SAM non-competitor and DNA-uncompetitor. Therefore, this assay can be useful either as a screening test in HTS or in mechanistic studies of a lead molecule.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre National de la Recherche Scientifique (CNRS) [ATIP to P.B.A.]; and Région Midi Pyrenées [Equipe d’Excellence to P.B.A. and FEDER CNRS/Région Midi Pyrenées to P.B.A.]. Funding for open access charge: CNRS, France.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Albert Jeltsch and his laboratory (Biochemistry Department Stuttgart University) for the protocol of DNMT3A purification. The authors thank Ahmed El Marjou (Institut Curie, Paris, France) for human DNMT1 production.
REFERENCES
- 1.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, Ueda Y, Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat. Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 2.Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300:455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 3.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 4.Takashima S, Takehashi M, Lee J, Chuma S, Okano M, Hata K, Suetake I, Nakatsuji N, Miyoshi H, Tajima S, et al. Abnormal DNA methyltransferase expression in mouse germline stem cells results in spermatogenic defects. Biol. Reprod. 2009;81:155–164. doi: 10.1095/biolreprod.108.074708. [DOI] [PubMed] [Google Scholar]
- 5.Bestor TH. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000;9:2395–2402. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 6.Espada J, Esteller M. DNA methylation and the functional organization of the nuclear compartment. Semin. Cell Dev. Biol. 2010;21:238–246. doi: 10.1016/j.semcdb.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Gros C, Fahy J, Halby L, Dufau I, Erdmann A, Gregoire JM, Ausseil F, Vispe S, Arimondo PB. DNA methylation inhibitors in cancer: recent and future approaches. Biochimie. 2012;94:2280–2296. doi: 10.1016/j.biochi.2012.07.025. [DOI] [PubMed] [Google Scholar]
- 8.Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fuso A, Nicolia V, Cavallaro RA, Scarpa S. DNA methylase and demethylase activities are modulated by one-carbon metabolism in Alzheimer's disease models. J. Nutr. Biochem. 2011;22:242–251. doi: 10.1016/j.jnutbio.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 11.Qureshi IA, Mehler MF. Advances in epigenetics and epigenomics for neurodegenerative diseases. Curr. Neurol. Neurosci. Rep. 2011;11:464–473. doi: 10.1007/s11910-011-0210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA. 2008;299:1345–1350. doi: 10.1001/jama.299.11.1345. [DOI] [PubMed] [Google Scholar]
- 13.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 14.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramchandani S, Bhattacharya SK, Cervoni N, Szyf M. DNA methylation is a reversible biological signal. Proc. Natl Acad. Sci. USA. 1999;96:6107–6112. doi: 10.1073/pnas.96.11.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng X, Blumenthal RM. Coordinated chromatin control: structural and functional linkage of DNA and histone methylation. Biochemistry. 2010;49:2999–3008. doi: 10.1021/bi100213t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12:206–222. doi: 10.1002/cbic.201000195. [DOI] [PubMed] [Google Scholar]
- 18.Svedruzic ZM. Mammalian cytosine DNA methyltransferase Dnmt1: enzymatic mechanism, novel mechanism-based inhibitors, and RNA-directed DNA methylation. Curr. Med. Chem. 2008;15:92–106. doi: 10.2174/092986708783330700. [DOI] [PubMed] [Google Scholar]
- 19.Bacolla A, Pradhan S, Roberts RJ, Wells RD. Recombinant human DNA (cytosine-5) methyltransferase. II. Steady-state kinetics reveal allosteric activation by methylated DNA. J. Biol. Chem. 1999;274:33011–33019. doi: 10.1074/jbc.274.46.33011. [DOI] [PubMed] [Google Scholar]
- 20.Bashtrykov P, Jankevicius G, Smarandache A, Jurkowska RZ, Ragozin S, Jeltsch A. Specificity of Dnmt1 for methylation of hemimethylated CpG sites resides in its catalytic domain. Chem. Biol. 2012;19:572–578. doi: 10.1016/j.chembiol.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 21.Pradhan S, Talbot D, Sha M, Benner J, Hornstra L, Li E, Jaenisch R, Roberts RJ. Baculovirus-mediated expression and characterization of the full-length murine DNA methyltransferase. Nucleic Acids Res. 1997;25:4666–4673. doi: 10.1093/nar/25.22.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pradhan S, Bacolla A, Wells RD, Roberts RJ. Recombinant human DNA (cytosine-5) methyltransferase. I. Expression, purification, and comparison of de novo and maintenance methylation. J. Biol. Chem. 1999;274:33002–33010. doi: 10.1074/jbc.274.46.33002. [DOI] [PubMed] [Google Scholar]
- 23.Song J, Rechkoblit O, Bestor TH, Patel DJ. Structure of DNMT1-DNA complex reveals a role for autoinhibition in maintenance DNA methylation. Science. 2011;331:1036–1040. doi: 10.1126/science.1195380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gowher H, Jeltsch A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J. Mol. Biol. 2001;309:1201–1208. doi: 10.1006/jmbi.2001.4710. [DOI] [PubMed] [Google Scholar]
- 25.Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 1998;19:219–220. doi: 10.1038/890. [DOI] [PubMed] [Google Scholar]
- 26.Fatemi M, Hermann A, Gowher H, Jeltsch A. Dnmt3a and Dnmt1 functionally cooperate during de novo methylation of DNA. Eur. J. Biochem. 2002;269:4981–4984. doi: 10.1046/j.1432-1033.2002.03198.x. [DOI] [PubMed] [Google Scholar]
- 27.Kim GD, Ni J, Kelesoglu N, Roberts RJ, Pradhan S. Co-operation and communication between the human maintenance and de novo DNA (cytosine-5) methyltransferases. EMBO J. 2002;21:4183–4195. doi: 10.1093/emboj/cdf401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walton EL, Francastel C, Velasco G. Maintenance of DNA methylation: Dnmt3b joins the dance. Epigenetics. 2011;6:1373–1377. doi: 10.4161/epi.6.11.17978. [DOI] [PubMed] [Google Scholar]
- 29.Xu F, Mao C, Ding Y, Rui C, Wu L, Shi A, Zhang H, Zhang L, Xu Z. Molecular and enzymatic profiles of mammalian DNA methyltransferases: structures and targets for drugs. Curr. Med. Chem. 2010;17:4052–4071. doi: 10.2174/092986710793205372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arand J, Spieler D, Karius T, Branco MR, Meilinger D, Meissner A, Jenuwein T, Xu G, Leonhardt H, Wolf V, et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012;8:e1002750. doi: 10.1371/journal.pgen.1002750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell Biol. 2003;23:5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dodge JE, Okano M, Dick F, Tsujimoto N, Chen T, Wang S, Ueda Y, Dyson N, Li E. Inactivation of Dnmt3b in mouse embryonic fibroblasts results in DNA hypomethylation, chromosomal instability, and spontaneous immortalization. J. Biol. Chem. 2005;280:17986–17991. doi: 10.1074/jbc.M413246200. [DOI] [PubMed] [Google Scholar]
- 33.Liang G, Chan MF, Tomigahara Y, Tsai YC, Gonzales FA, Li E, Laird PW, Jones PA. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell Biol. 2002;22:480–491. doi: 10.1128/MCB.22.2.480-491.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hemeon I, Gutierrez JA, Ho MC, Schramm VL. Characterizing DNA methyltransferases with an ultrasensitive luciferase-linked continuous assay. Anal. Chem. 2011;83:4996–5004. doi: 10.1021/ac200816m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ibanez G, McBean JL, Astudillo YM, Luo M. An enzyme-coupled ultrasensitive luminescence assay for protein methyltransferases. Anal. Biochem. 2010;401:203–210. doi: 10.1016/j.ab.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 36.Graves TL, Zhang Y, Scott JE. A universal competitive fluorescence polarization activity assay for S-adenosylmethionine utilizing methyltransferases. Anal. Biochem. 2008;373:296–306. doi: 10.1016/j.ab.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ceccaldi A, Rajavelu A, Champion C, Rampon C, Jurkowska R, Jankevicius G, Senamaud-Beaufort C, Ponger L, Gagey N, Ali HD, et al. C5-DNA methyltransferase inhibitors: from screening to effects on zebrafish embryo development. Chembiochem. 2011;12:1337–1345. doi: 10.1002/cbic.201100130. [DOI] [PubMed] [Google Scholar]
- 38.Ceccaldi A, Rajavelu A, Ragozin S, Senamaud-Beaufort C, Bashtrykov P, Testa N, Dali-Ali H, Maulay-Bailly C, Amand S, Guianvarc'h D, et al. Identification of novel inhibitors of DNA methylation by screening of a chemical library. ACS Chem. Biol. 2013;8:543–548. doi: 10.1021/cb300565z. [DOI] [PubMed] [Google Scholar]
- 39.Halby L, Champion C, Senamaud-Beaufort C, Ajjan S, Drujon T, Rajavelu A, Ceccaldi A, Jurkowska R, Lequin O, Nelson WG, et al. Rapid synthesis of new DNMT inhibitors derivatives of procainamide. Chembiochem. 2012;13:157–165. doi: 10.1002/cbic.201100522. [DOI] [PubMed] [Google Scholar]
- 40.Rubin RA, Modrich P. EcoRI methylase. physical and catalytic properties of the homogeneous enzyme. J. Biol. Chem. 1977;252:7265–7272. [PubMed] [Google Scholar]
- 41.Jeltsch A, Friedrich T, Roth M. Kinetics of methylation and binding of DNA by the EcoRV adenine-N6 methyltransferase. J. Mol. Biol. 1998;275:747–758. doi: 10.1006/jmbi.1997.1492. [DOI] [PubMed] [Google Scholar]
- 42.Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248–251. doi: 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide is a specific inhibitor of DNA methyltransferase 1. J. Biol. Chem. 2005;280:40749–40756. doi: 10.1074/jbc.M505593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fritsch L, Robin P, Mathieu JR, Souidi M, Hinaux H, Rougeulle C, Harel-Bellan A, Ameyar-Zazoua M, Ait-Si-Ali S. A subset of the histone H3 lysine 9 methyltransferases Suv39h1, G9a, GLP, and SETDB1 participate in a multimeric complex. Mol. Cell. 2010;37:46–56. doi: 10.1016/j.molcel.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 45.Yokochi T, Robertson KD. Dimethyl sulfoxide stimulates the catalytic activity of de novo DNA methyltransferase 3a (Dnmt3a) in vitro. Bioorg. Chem. 2004;32:234–243. doi: 10.1016/j.bioorg.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 46.Song J, Teplova M, Ishibe-Murakami S, Patel DJ. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012;335:709–712. doi: 10.1126/science.1214453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Copeland RA. Mechanistic considerations in high-throughput screening. Anal. Biochem. 2003;320:1–12. doi: 10.1016/s0003-2697(03)00346-4. [DOI] [PubMed] [Google Scholar]
- 48.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 49.Lin Y, Fan H, Frederiksen M, Zhao K, Jiang L, Wang Z, Zhou S, Guo W, Gao J, Li S, et al. Detecting S-adenosyl-L-methionine-induced conformational change of a histone methyltransferase using a homogeneous time-resolved fluorescence-based binding assay. Anal. Biochem. 2012;423:171–177. doi: 10.1016/j.ab.2012.01.019. [DOI] [PubMed] [Google Scholar]
- 50.Schluckebier G, Kozak M, Bleimling N, Weinhold E, Saenger W. Differential binding of S-adenosylmethionine S-adenosylhomocysteine and Sinefungin to the adenine-specific DNA methyltransferase M.TaqI. J. Mol. Biol. 1997;265:56–67. doi: 10.1006/jmbi.1996.0711. [DOI] [PubMed] [Google Scholar]
- 51.Champion C, Guianvarc'h D, Senamaud-Beaufort C, Jurkowska RZ, Jeltsch A, Ponger L, Arimondo PB, Guieysse-Peugeot AL. Mechanistic insights on the inhibition of c5 DNA methyltransferases by zebularine. PLoS One. 2010;5:e12388. doi: 10.1371/journal.pone.0012388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Copeland RA. Evaluation of Enzyme Inhibitors in Drug Discovery: A guide for Medicinal Chemists and Pharmacologists. Hoboken, New Jersey: John Wiley & Sons, Inc.; 2005. Reversible modes of inhibitor interactions with enzymes; pp. 48–81. [PubMed] [Google Scholar]
- 54.Flynn J, Reich N. Murine DNA (cytosine-5-)-methyltransferase: steady-state and substrate trapping analyses of the kinetic mechanism. Biochemistry. 1998;37:15162–15169. doi: 10.1021/bi9810609. [DOI] [PubMed] [Google Scholar]
- 55.Svedruzic ZM, Reich NO. DNA cytosine C5 methyltransferase Dnmt1: catalysis-dependent release of allosteric inhibition. Biochemistry. 2005;44:9472–9485. doi: 10.1021/bi050295z. [DOI] [PubMed] [Google Scholar]
- 56.Schnekenburger M, Grandjenette C, Ghelfi J, Karius T, Foliguet B, Dicato M, Diederich M. Sustained exposure to the DNA demethylating agent, 2′-deoxy-5-azacytidine, leads to apoptotic cell death in chronic myeloid leukemia by promoting differentiation, senescence, and autophagy. Biochem. Pharmacol. 2011;81:364–378. doi: 10.1016/j.bcp.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 57.Tsai HC, Li H, Van NL, Cai Y, Robert C, Rassool FV, Shin JJ, Harbom KM, Beaty R, Pappou E, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21:430–446. doi: 10.1016/j.ccr.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.