

Abstract
Motivation: Recently, mapping studies of expression quantitative loci (eQTL) (where gene expression levels are viewed as quantitative traits) have provided insight into the biology of gene regulation. Bayesian methods provide natural modeling frameworks for analyzing eQTL studies, where information shared across markers and/or genes can increase the power to detect eQTLs. Bayesian approaches tend to be computationally demanding and require specialized software. As a result, most eQTL studies use univariate methods treating each gene independently, leading to suboptimal results.
Results: We present a powerful, computationally optimized and free open-source R package, iBMQ. Our package implements a joint hierarchical Bayesian model where all genes and SNPs are modeled concurrently. Model parameters are estimated using a Markov chain Monte Carlo algorithm. The free and widely used openMP parallel library speeds up computation. Using a mouse cardiac dataset, we show that iBMQ improves the detection of large trans-eQTL hotspots compared with other state-of-the-art packages for eQTL analysis.
Availability: The R-package iBMQ is available from the Bioconductor Web site at http://bioconductor.org and runs on Linux, Windows and MAC OS X. It is distributed under the Artistic Licence-2.0 terms.
Contact: christian.deschepper@ircm.qc.ca or rgottard@fhcrc.org
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Recently, eQTL mapping studies (where gene expression levels are viewed as quantitative traits) have provided insight into the biology of gene regulation. Among eQTLs, it is customary to distinguish cis-eQTLs from trans-eQTLs. The former share the same locus as the expressed gene, whereas the latter are located on loci different from the expressed gene. Many eQTLs, particularly trans-eQTLs, form trans-eQTL hotspots where one single nucleotide polymorphism (SNP) is linked to the expression of several genes across the genome. Despite this observation, most available eQTL analysis tools treat genes as independent, and as such these methods are underpowered to detect trans-eQTL hotspots (Gilad et al., 2008). Likewise, some available packages [such as GGtools (Carey et al., 2009)] allow for data analysis and visualization of results, but are currently limited to univariate analyses.
We present an integrated hierarchical Bayesian model that jointly models all genes and SNPs to detect eQTLs. The iBMQ R/Bioconductor package incorporates genotypic and gene expression data into a single model while (i) coping with the high dimensionality of eQTL data (large number of genes), (ii) borrowing strength from all gene expression data for the mapping procedures and (iii) controlling the number of false positives to a desirable level.
2 METHODS
The main iBMQ function is written in C for computational efficiency and wrapped in convenient R code. iBMQ uses the OpenMP API to perform modeling operations in parallel, facilitating the analysis of large datasets. A sparse matrix representation enables efficient matrix calculations (see Supplementary Material). iBMQ adopts object-oriented programming, making use of existing S4 classes (e.g. eSet and SNPSet). The main functions are as follows:
eqtlMcmc: This function takes gene expression values (an eSet object) and genomic map data (a SNPSet object) as input, then generates posterior samples from our model (Scott-Boyer et al., 2012) via Markov chain Monte Carlo. Additional arguments include the number of Markov chain Monte Carlo iterations, number of burn-in iterations and whether sampled nuisance parameters should be saved to disk. The output is a matrix of marginal posterior probability of associations (PPAs), which is used for eQTL inference (See Supplementary Material for details). An eQTL for gene g at SNP j is declared significant if its corresponding PPA exceeds a given threshold.
calculateThreshold: This function calculates the PPA threshold for eQTL significance corresponding to a given false discovery rate based on the approach of Newton et al. (2004).
eqtlFinder: This function applies the calculated threshold to PPAs and identifies significant SNPs.
eqtlClassifier: Given the genomic position of each probe and SNP as input, this function classifies the eQTLs as either cis-eQTLs or trans-eQTL.
hotspotFinder: This function identifies single markers associated with several genes, and thus identifies trans-eQTL ‘hotspots’ (see Supplementary Material for details).
3 RESULTS
We applied iBMQ to data generated by Scott-Boyer and Deschepper (2013), available at GeneNetwork (accession number GN421). The data comprise 8725 genes and 977 markers in cardiac tissue from 24 AXB-BXA recombinant inbred strain mice, as measured using Illumina microarrays. We used 1 million iterations with 50 000 burn-in iterations as suggested in previous studies Scott-Boyer et al. (2012), which took ∼19 h using an Intel Xeon E5-2690 8-core processor. Using a false discovery rate threshold of 10%, iBMQ detected 1652 significant eQTLs, of which 278 were cis-eQTLs (where gene start is <1 Mb from eQTL peak) and 1357 were trans-eQTLs. The cis-eQTLs align along a diagonal in Figure 1. Among trans-eQTLs, iBMQ detected three clusters of ≥50 genes forming ‘trans-eQTL hotspots’, represented by vertically aligned dots. To verify whether the hotspots detected by iBMQ showed biological relevance and coherence, we tested whether corresponding groups of trans-eQTLs showed enrichment in genes from Gene Ontology (GO) term categories, using the DAVID Bioinformatics Resources (Table 1). In each case (i) there was significant enrichment for particular GO terms and (ii) iBMQ detected more trans-eQTL genes than the univariate R/QTL method (Broman et al., 2003). For the three trans-eQTLs hotspots, both methods detected significant enrichment for the following GO terms: GO:0012505 (cellular component), GO:0007167 (enzyme-linked receptor protein signaling pathway) and GO:0006955 (immune response). According to the method used, these GO terms ranked as either the top or the second most enriched category. Consequently, the significance of GO term enrichment was higher for trans-eQTL hotspots detected by iBMQ than for the corresponding hotspots detected by R/QTL. iBMQ appears to show greater sensitivity to detect trans-eQTL hotspots containing large number of genes.
Fig. 1.
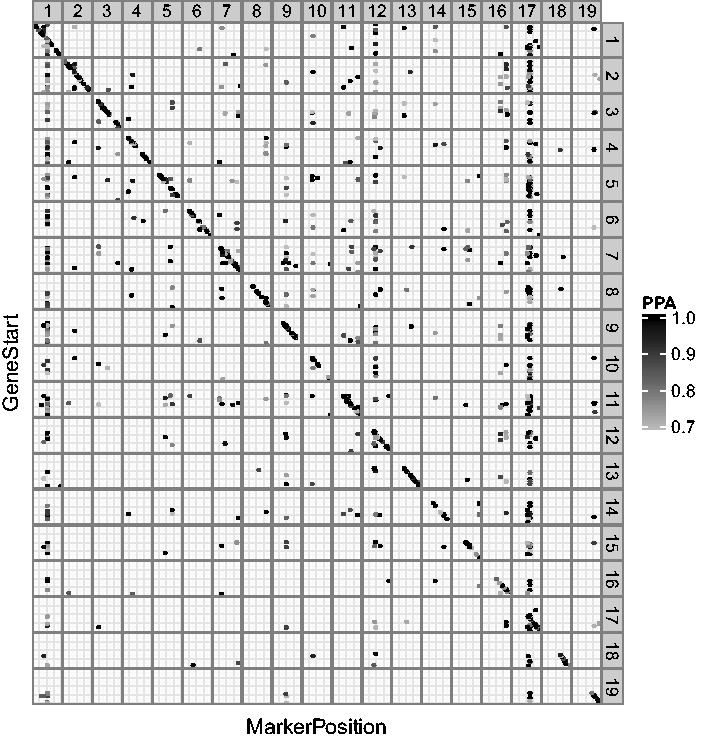
Genome-wide distribution of eQTLs found by iBMQ for mice cardiac tissue. The X-axis gives eQTL genomic position; the Y-axis gives the genomic positions of probe sets. Chromosome numbers are given in the gray box strips. cis-QTLs align along the diagonal line. Vertical bands represent groups of transcripts linked to one trans-eQTL. eQTL points are color-coded by PPA value (see PPA color scale). The Supplementary Material presents the code used in the analysis
Table 1.
Positions of iBMQ-detected trans-eQTL hotspots containing  50 genes, and comparisons with corresponding hotspots detected by the univariate R/QTL method
50 genes, and comparisons with corresponding hotspots detected by the univariate R/QTL method
| SNP | GO term | iBMQ |
R/QTL |
||
|---|---|---|---|---|---|
| #GO/Total | P-val | #GO/Total | P-val | ||
| 1@94.8 | 0012505 | 14/173 | 2.4e-4 | 5/27 | 4.0e-3 |
| 12@103.5 | 0007167 | 5/53 | 1.5e-3 | 3/24 | 6.9e-3 |
| 17@72.4 | 0006955 | 26/192 | 2.7e-13 | 6/49 | 2.5e-3 |
| 0017076 | 40/192 | 3.3e-7 | 10/49 | 0.015 | |
Note: In the first column, a position identified as 1@94.8 refers to a SNP at position 94.8 Mb on chromosome 1. Columns list the GO term ID, and for each method, the number of trans-eQTL genes belonging to the GO term category, the total number of genes in the hotspot and the enrichment P-value (see Supplementary Material for details).
Funding: The grant RP-146065 from ‘Fonds quebecois de recherche Nature et Technologies’ (to A.L. and C.F.D); NIH grant R01 HG005692 (to R.G. and G.I.).
Conflict of Interest: none declared.
Supplementary Material
REFERENCES
- Broman KW, et al. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;17:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- Gilad Y, et al. Revealing the architecture of gene regulation the promise of eQTL studies. Trends Genet. 2008;24:408–415. doi: 10.1016/j.tig.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton MA, et al. Detecting differential gene expression with a semiparametric hierarchical mixture method. Biometrics. 2004;5:155–176. doi: 10.1093/biostatistics/5.2.155. [DOI] [PubMed] [Google Scholar]
- Scott-Boyer MP, Deschepper CF. Genome-wide detection of gene co-expression domains showing linkage to regions enriched with polymorphic retrotransposons in recombinant inbred mouse strains. G3 (Bethesda) 2013;3:597–605. doi: 10.1534/g3.113.005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Boyer MP, et al. An integrated hierarchical bayesian model for multivariate eqtl mapping. Stat. Appl. Genet. Mol. Biol. 2012;11:4. doi: 10.1515/1544-6115.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey VJ, et al. Data structures and algorithms for analysis of genetics of gene expression with Bioconductor: GGtools 3.x. Bioinformatics. 2009;25:1447–1448. doi: 10.1093/bioinformatics/btp169. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.