

Abstract
BACKGROUND AND PURPOSE
At present there are no small molecule inhibitors that show strong selectivity for the Na+/Ca2+ exchanger (NCX). Hence, we studied the electrophysiological effects of acute administration of ORM-10103, a new NCX inhibitor, on the NCX and L-type Ca2+ currents and on the formation of early and delayed afterdepolarizations.
EXPERIMENTAL APPROACH
Ion currents were recorded by using a voltage clamp technique in canine single ventricular cells, and action potentials were obtained from canine and guinea pig ventricular preparations with the use of microelectrodes.
KEY RESULTS
ORM-10103 significantly reduced both the inward and outward NCX currents. Even at a high concentration (10 μM), ORM-10103 did not significantly change the L-type Ca2+ current or the maximum rate of depolarization (dV/dtmax), indicative of the fast inward Na+ current. At 10 μM ORM-10103 did not affect the amplitude or the dV/dtmax of the slow response action potentials recorded from guinea pig papillary muscles, which suggests it had no effect on the L-type Ca2+ current. ORM-10103 did not influence the Na+/K+ pump or the main K+ currents of canine ventricular myocytes, except the rapid delayed rectifier K+ current, which was slightly diminished by the drug at 3 μM. The amplitudes of pharmacologically- induced early and delayed afterdepolarizations were significantly decreased by ORM-10103 (3 and 10 μM) in a concentration-dependent manner.
CONCLUSIONS AND IMPLICATIONS
ORM-10103 is a selective inhibitor of the NCX current and can abolish triggered arrhythmias. Hence, it has the potential to be used to prevent arrhythmogenic events.
LINKED ARTICLE
This article is commented on by Terracciano and Hancox, pp. 765–767 of this issue. To view this commentary visit http://dx.doi.org/10.1111/bph.12299
Keywords: sodium/calcium exchanger, NCX, ORM-10103, EAD, DAD
Introduction
The Na+/Ca2+ exchanger (NCX) (Alexander et al., 2011) is considered to be a major regulator of Ca2+ homeostasis in the myocardium (Bers, 2000; 2002). In the forward mode, NCX is known to extrude Ca2+ from the cell to the extracellular space during diastole at relatively low free cytoplasmic Ca2+ concentrations and negative transmembrane potential. Because the extrusion of one Ca2+ is coupled with the entry of 3 Na+ into the cell, the forward mode of the NCX is accompanied by a net inward current; when the intracellular Ca2+ level is elevated, this can cause substantial depolarization, leading to early afterdepolarization (EAD) and delayed afterdepolarization (DAD) (Venetucci et al., 2007). EAD and DAD are generally thought to play important roles in arrhythmogenesis (Roden, 1996; Volders et al., 2000), especially under conditions of decreased K+ conductance, as in heart failure (Pogwizd et al., 2001). It has been speculated, therefore, that specific blockers of NCX have the potential to be antiarrhythmic in dysrhythmias associated with Ca2+ overload (Pogwizd and Bers, 2002; Pogwizd, 2003). This hypothesis has not yet been tested directly, as the NCX inhibitors available also decrease the L-type Ca2+ current (ICaL) which in turn is known to decrease the intracellular Ca2+ load, thereby indirectly changing the magnitude of NCX. The effects of NCX blockers KB-R7943 and SEA-0400 were recently reported. Both of these drugs are effective inhibitors of NCX, even in submicromolar concentrations, and they have been demonstrated to reduce the incidence of ischaemia/reperfusion-related arrhythmias induced by a Ca2+ overload (Elias et al., 2001) and to decrease the pharmacologically-induced EAD and DAD in canine ventricular preparations (Nagy et al., 2004). However, results from other studies have indicated that KB-R7943 and SEA-0400 also inhibit the ICaL (Tanaka et al., 2002; Birinyi et al., 2005), which makes the interpretation of their antiarrhythmic effect somewhat uncertain. In the present study, we investigated the effects of ORM-10103 (chemical structure shown in Figure 1), another newly developed and specific NCX inhibitor, on the NCX current, ICaL, INa, Na+/K+ pump (Ip) and K+ currents of canine ventricular myocytes, and on the formation of EAD and DAD in canine ventricular muscle and Purkinje fibres.
Figure 1.
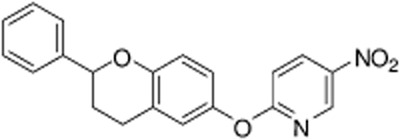
Chemical structure of ORM-10103.
Methods
All experiments were conducted in compliance with the Guide for the Care and Use of Laboratory Animals (USA NIH Publication No. 85-23, revised 1996) and conformed to Directive 2010/63/EU of the European Parliament. The protocols were approved by the Ethical Committee for the Protection of Animals in Research at the University of Szeged, Szeged, Hungary (Approval No. I-74-9-2009).
For voltage clamp and conventional microelectrode experiments, adult mongrel dogs of either sex weighing 8–16 kg were used. Following sedation (xylazine, 1 mg·kg−1, i.v.) and thiopental (30 mg·kg−1, i.v.)-induced anaesthesia, each heart was rapidly removed through a right lateral thoracotomy and immediately rinsed in oxygenated modified Locke's solution containing (in mM): Na+ 140, K+ 4, Ca2+ 1.0, Mg2+ 1, Cl− 126, HCO3− 25 and glucose 11. The pH of the solution, when gassed with 95% O2 and 5% CO2 at 37°C, ranged from 7.35 to 7.45.
Voltage clamp measurements
Cell isolation
Ventricular myocytes were enzymatically dissociated from the canine hearts. A portion of the left ventricular wall containing an arterial branch large enough to cannulate was then perfused in a modified Langendorff apparatus at a pressure of 60 cmH2O, with solutions in the following sequence: (i) isolation solution supplemented with CaCl2 (1.25 mM) for 10 min; (ii) isolation solution for another 10 min; (iii) isolation solution (150 mL) containing collagenase (type I, 0.33 mg·mL−1; Sigma Chemical, St. Louis, MO, USA) (10 min). Protease (type XIV, 0.04 mg·mL−1; Sigma Chemical) was added to the final perfusate and another 15–20 min of digestion was allowed. The isolation solution (Ca2+ free) was Eagle's Minimum Essential Medium, Joklik modification (Sigma Chemical), supplemented with (in mM) HEPES 10 and NaHCO3 4.4 (pH 7.2, adjusted with NaOH). Portions of the left ventricular wall, judged to be well-digested, were diced into small pieces in isolation solution supplemented with CaCl2 (1.25 mM) for 15 min. These tissue samples were then gently agitated in a small beaker to dislodge single myocytes from the extracellular matrix. The resulting cell suspension contained a mixture of subepicardial, midmyocardial and subendocardial myocytes. Throughout the entire isolation procedure, solutions were gassed with 100% O2, while their temperature was maintained at 37°C. Myocytes were allowed to settle to the bottom of the beaker for 10 min, after which half of the supernatant was replaced with fresh solution. This procedure was repeated three times. Myocytes placed in isolation solution supplemented with CaCl2 (1.25 mM) were maintained at 12–14°C before the experiment.
Experimental technique
One drop of cell suspension was placed in a transparent recording chamber mounted on the stage of an inverted microscope (TMS; Nikon, Tokyo, Japan), and individual myocytes were allowed to settle and adhere to the chamber bottom for at least 5 min before superfusion was initiated and maintained by gravity. Only rod-shaped cells with clear striations were used. HEPES-buffered Tyrode's solution (composition in mM: NaCl 144, NaH2PO4 0.33, KCl 4.0, CaCl2 1.8, MgCl2 0.53, glucose 5.5, HEPES 5.0, at pH of 7.4) served as the normal superfusate.
Micropipettes were fabricated from borosilicate glass capillaries (Clark Electromedical Instruments, Pangbourne, Reading, UK), using a P-97 Flaming/Brown micropipette puller (Sutter Co, Novato, CA, USA), and had a resistance of 1.5–2.5 MΩ when filled with pipette solution. The membrane currents were recorded with Axopatch-1D or Axopatch-200B amplifiers (Axon Instruments, Union City, CA, USA) by means of the whole cell configuration of the patch clamp technique. After the establishment of high (1–10 GΩ) resistance seals by gentle suction, the cell membrane beneath the tip of the electrode was disrupted by further suction or by applying 1.5 V electrical pulses for 1–5 ms. The membrane currents were digitized with a 333 kHz analogue to digital converter (Digidata 1200; Axon Instruments) under software control (pClamp 8.0; Axon Instruments). The results were analysed by using software programs purchased from Axon (pClamp 8.0). As the rundown of ICaL and IKs currents is commonly seen during the measurements, the current level was monitored during the initial equilibration period and also at the end of the measurements when a washout period of at least 10 min was applied in order to draw a distinction between drug effect and rundown of the current. The cells, in which excessive rundown was observed, were omitted from the analyses. Experiments were carried out at 37°C.
Measurement of NCX current
For the measurement of the Na+/Ca2+ exchanger current (NCX), the method of Hobai et al. (1997) was applied, in which special K+-free bath and pipette solutions were used (see below) in order to block the Na+, Ca2+, K+-currents and the Na+/K+ pump current. The I-V (current–voltage) relationship of Na+/Ca2+ exchanger current was measured through the use of ramp pulses at 20 s intervals. The ramp pulse initially led to depolarization from the holding potential of −40 mV to 60 mV with a rate of 100 mV·s−1, then to hyperpolarization to −100 mV, and depolarization back to the holding potential. The descending limb of the ramp was utilized to plot the I-V curve.
Compositions of solutions used (in mM)
K+-free bath solution: NaCl 135, CsCl 10, CaCl2 1, MgCl2 1, BaCl2 0.2, NaH2PO4 0.33, TEACl 10, HEPES 10, glucose 10 and ouabain 20 μM, nisoldipine 1 μM, lidocaine 50 μM, at pH 7.4.
Pipette solution: CsOH 140, aspartic acid 75, TEACl 20, MgATP 5, HEPES 10, NaCl 20, EGTA 20, CaCl2 10 (pH adjusted to 7.2 with CsOH).
The experimental protocol was as follows (Figure 2):
Figure 2.
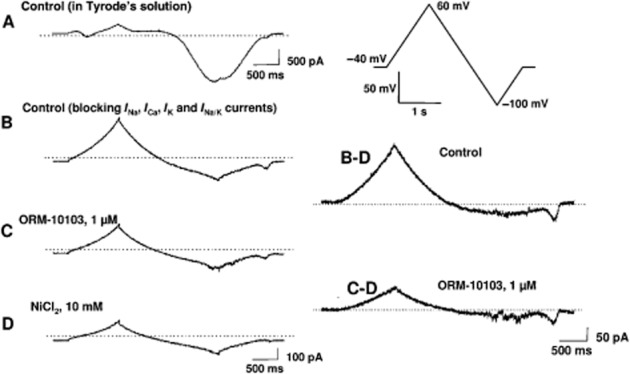
Determination of NCX current in canine ventricular myocytes. (A) Recording obtained with the voltage protocol shown in the inset with normal Tyrode's solution. (B) The current trace after blockade of Na+, Ca2+, K+ and Na+/K+ pump currents. (C) The current trace after superfusion with 1 μM ORM-10103. (D) The current trace at the end of the measurements after the application of 10 mM NiCl2. On the right, the control NCX current is shown, which is obtained by subtracting trace D from trace B. The NCX current in the presence of 1 μM ORM-10103 is obtained by subtracting trace D from trace C. Note the difference in the intensity-time calibration in the left and right panels.
The whole cell configuration was established in HEPES-buffered Tyrode's solution
The control I-V curve was recorded in the special K+-free bath solution after 8–10 min from establishing whole cell configuration
After 6–10 min of incubation, the I-V relationship was recorded in the presence of ORM-10103
At the end of the experiments, the Ni2+-insensitive current was measured by the application of 10 mM NiCl2
The NCX current was defined as the Ni2+-sensitive current, that is, the trace recorded in the presence of 10 mM NiCl2 subtracted from that measured in the absence of NiCl2. In separate experiments, the effect of ORM-10103 on the Ni2+-insensitive current was also tested.
Measurement of ICaL
The ICaL was recorded in HEPES-buffered Tyrode's solution supplemented with 3 mM 4-aminopyridine. A special solution was used to fill the micropipettes (composition in mM: CsOH 110, CsCl 20, TEACl 10, MgATP 5, EGTA 5, HEPES 10, GTP 0.1; pH was adjusted to 7.2 by aspartic acid). ICaL was evoked by 400 ms long depolarizing voltage pulses to various test potentials ranging from −35 to +55 mV. The holding potential was −80 mV. A short prepulse of −40 mV served to inactivate Na+ current. The amplitude of the ICaL was defined as the difference between the peak inward current at the beginning of the pulse and the current at the end of the pulse.
Measurement of K+ currents
The inward rectifier (IK1), transient outward (Ito), rapid (IKr) and slow (IKs) delayed rectifier potassium currents were recorded in HEPES-buffered Tyrode's solution. The compositions of the pipette solution (in mM) were the following: KOH 110, KCl 40, K2ATP 5, MgCl2 5, EGTA 5 and HEPES 10 (pH was adjusted to 7.2 by aspartic acid). Nisoldipine 1 μM was added to the external solution to eliminate ICaL. When IKr was recorded, IKs was inhibited by using the selective IKs blocker HMR 1556 (0.5 μM). During IKs measurements, IKr was blocked by 0.1 μM dofetilide. The currents were activated by applying depolarizing voltage pulses as shown by insets in Figure 7.
Figure 7.
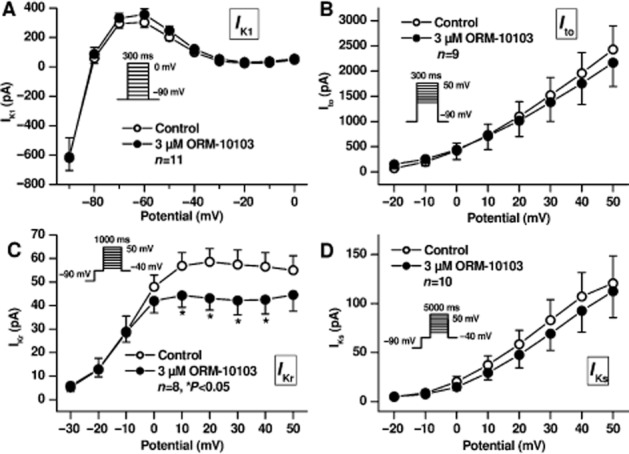
Effect of ORM-10103 on IK1 (A), Ito (B), IKr (C) and IKs (D). Insets show the applied voltage protocols. Values are means ± SEM, *P < 0.05.
Measurement of Na+/K+ pump current
Steady-state current at −30 mV was recorded in control conditions and in the presence of ORM-10103. After 5–7 min incubation with ORM-10103, the normal external solution (composition in mM: NaCl 135, CsCl 2, KCl 5, MgCl2 1, CdCl2 0.2, BaCl2 2, HEPES 5, glucose 10, pH 7.4 by NaOH) was replaced by K+-free solution. The Na+/K+ pump current (Ip) was defined as the difference between currents measured in 5 and 0 mM K+ containing solutions. The compositions of the pipette solution (in mM) were as follows: CsOH 100, NaCl 30, MgATP 5, MgCl2 2, TEACl 20, EGTA 5, HEPES 10 and glucose 10 (pH was adjusted to 7.2 by aspartic acid).
Conventional microelectrode measurements
The same adult mongrel dogs of either sex weighing 8–16 kg were used. Purkinje strands obtained from both ventricles and right ventricular papillary muscle tips were mounted individually in a tissue chamber superfused with oxygenated Locke's solution at 37°C. Each preparation was stimulated (Hugo Sachs Elektronik stimulator type 215/II, March-Hugstetten, Germany) initially at a constant cycle length of 1000 ms (frequency 1 Hz), with rectangular constant current pulses of 2 ms in duration. The current pulses were isolated from ground and delivered through bipolar platinum electrodes in contact with the preparations. At least 1 h was allowed for each preparation to equilibrate during continuous superfusion with modified Locke's solution, warmed to 37°C before the experimental measurements commenced. Transmembrane potentials were recorded with the use of conventional 5–20 MΩ, 3 M KCl-filled microelectrodes connected to the input of a high-impedance electrometer (Biologic Amplifier VF 102, Claix, France). The first derivative of transmembrane potential (dV/dtmax) was obtained electronically with a Biologic DV-140 (Claix, France) differentiator designed and calibrated to give a linear response over the range between 10 and 1000 V·s−1. In each experiment, the baseline action potential characteristics were first determined during continuous pacing at 1 Hz (on Purkinje fibres, continuous pacing at 2 Hz), and then when the pacing cycle length was sequentially varied from 300 to 5000 ms. The 25th action potential was measured at each cycle length, and the cycle length was then changed so that quasi-steady-state frequency–response relationships could be generated rapidly. The preparations were next superfused with the drug for 40–60 min before the pacing protocol was repeated and the parameters were measured again. Efforts were made to maintain the same impalement throughout each experiment. If an impalement became dislodged, however, electrode adjustment was attempted, and if the action potential characteristics of the re-established impalement deviated by less than 5% from those of the previous measurement, the experiment was continued. When this 5% limit was exceeded, the experiment was terminated and all the data involved were excluded from the analyses.
Recording of slow response action potentials
Adult male guinea pigs of either sex weighing 150–200 g were administered an anticoagulant, sodium heparin, and anaesthetized with 30 mg·kg−1 thiopental after sedation (xylazine, 1 mg·kg−1). The hearts were rapidly removed through a right lateral thoracotomy and immediately rinsed in ice-cold Krebs–Henseleit solution (contents in mM: NaCl 118.5, KCl 4.0, CaCl2 2.0, MgSO4 1.0, NaH2PO4 1.2, NaHCO3 25.0, glucose 10.0). The pH of this solution was set to 7.35 ± 0.05 when saturated with a mixture of 95% O2 and 5% CO2. After excision, the right or left ventricular papillary muscle preparations were immediately mounted in a 40 mL tissue chamber, and initially perfused with normal Krebs–Henseleit solution.
The preparations were stimulated continuously with an electrostimulator (PW-01; Experimetria Ltd., Budapest, Hungary) by means of constant rectangular voltage pulses, 1 ms in duration, delivered through a pair of bipolar platinum electrodes at a frequency of 1 Hz. Action potentials were recorded with a conventional microelectrode technique. Sharp microelectrodes, with a tip resistance of 10–20 MΩ when filled with 3 M KCl, were connected to the amplifier (Biologic Amplifier, model VF 102). The voltage output from the amplifier was sampled with an AD converter (NI 6025; Unisip Ltd., Budapest, Hungary). The action potential amplitude and maximum rate of depolarization were obtained with Evokewave v1.49 software (Unisip Ltd). Each preparation was allowed to equilibrate in normal Krebs–Henseleit solution at 37°C for at least 30 min.
Slow response action potentials were established in modified Krebs–Henseleit solution containing 25 mM KCl, supplemented with 100 μM BaCl2 to inhibit IK1 and with 1 μM forskolin to increase ICaL. The dV/dtmax values in the interval 5–20 V·s−1 and amplitudes of at least 60 mV were accepted; data from experiments in which these levels were not met were discarded.
The nomenclature of NCX used conforms to the BJP's Guide to Receptor and Channels (Alexander et al., 2011).
With the exception of ORM-10103 (from Orion Pharma, Espoo, Finland), nisoldipine (gift from Bayer AG, Leverkusen, Germany) and strophanthin-G (from BDH Chemicals Ltd, Poole, England), all chemicals were purchased from Sigma-Aldrich Fine Chemicals (St. Louis, MO, USA).
Statistics
All data are expressed as means ± SEM. Statistical analysis was performed with Student's t-test for paired data. The results were considered statistically significant when P was <0.05.
Results
Effects of ORM-10103 on the outward and inward NCX current
The experimental protocol applied to measure the NCX current is presented in Figure 2. Part A depicts a current recording in the presence of normal Tyrode's solution during the voltage ramp pulse illustrated in the top right corner, while part B illustrates a current recording after blocking Na+, Ca2+, K+ and Na+/K+ pump currents. In part C, 1 μM ORM-10103 was applied and the current was recorded with the same voltage pulse. Finally, 10 mM NiCl2 was added to the tissue bath to block the NCX current completely and the resulting recording (part D) was subtracted from the control recording (to give traces B–D) and from the ORM-10103 recording (to give traces C–D). It is clear from Figure 2 that the Ni2+-sensitive current, that is, the NCX current, was markedly reduced by 1 μM ORM-10103. In three separate experiments, after the Ca2+, Na+, K+, Na+/K+ pump currents and also INCX were blocked, application of 10 μM ORM-10103 did not change the Ni2+-insensitive current (not shown). These latter measurements indicate that the current reduced by ORM-10103 was indeed the NCX current and not a Ni2+-insensitive leakage current.
Figures 3 and 4 demonstrate that both the outward and the inward NCX currents were considerably reduced by ORM-10103 in a concentration-dependent manner. The effect in the outward direction was statistically significant in the potential range between 0 and 60 mV in the presence of ORM-10103 concentrations higher than 0.5 μM (Figure 4A, right panel). The blockade of the inward NCX current by ORM-10103 was also significant at concentrations higher than 0.5 μM (Figure 4A, left panel). The estimated EC50 values for the inward and outward NCX currents were 780 and 960 nM respectively (Figure 4B).
Figure 3.
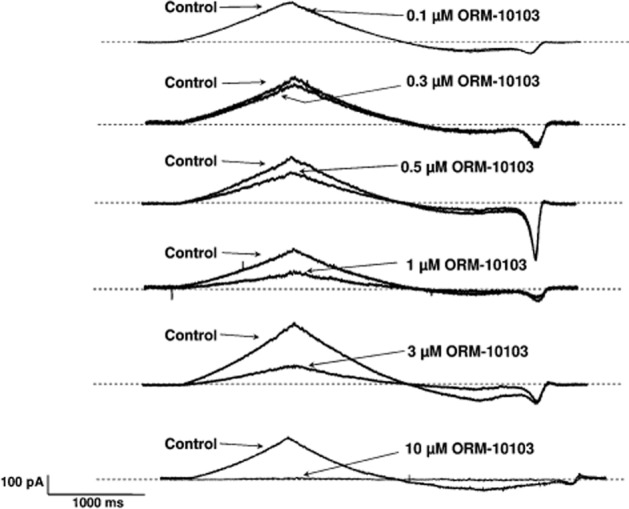
The concentration-dependent effect of ORM-10103 on the NCX current in canine ventricular myocytes. Each panel presents Ni2+-sensitive (NCX) current traces before and after superfusion of the cells with a concentration of ORM-10103 ranging from 0.1 to 10 μM.
Figure 4.
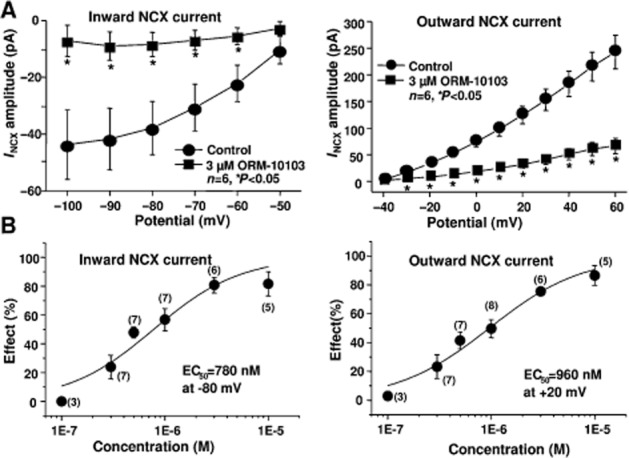
(A) The amplitude of the inward (left) and the outward (right) NCX currents in the absence or in the presence of 3 μM ORM-10103. Bars indicate ± SEM, *P < 0.05. (B) The concentration–response curve for ORM-10103 on the inward (left) and outward (right) NCX currents in canine ventricular myocytes at −80 and +20 mV respectively. Values are means ± SEM, n = 3–8.
Lack of effect of ORM-10103 on the L-type inward calcium, inward sodium and outward potassium currents
The possible effect of ORM-10103 on the L-type inward calcium current was also studied in canine ventricular myocytes. These experiments clearly revealed that even at a high (10 μM) concentration, ORM-10103 did not influence significantly ICaL (Figure 5A). After washout of the drug the current slightly decreased, further showing that the smaller average value measured after application of ORM-10103 is due to a slight rundown of the current. Nifedipine at 10 μM almost completely blocked the current, indicating that the recorded current was ICaL (Figure 5A). The effect of ORM-10103 on the inactivation kinetics was also investigated. The inactivation time constant was not significantly changed by 10 μM ORM-10103 (40.0 ± 4.6 ms vs. 35.0 ± 5.5 ms at 0 mV, n = 8, n.s.). The effect of ORM-10103 on the inward sodium current was assessed by measuring the dV/dtmax in the canine right ventricular papillary muscle, by use of the conventional microelectrode technique. At the high concentration of 10 μM, ORM-10103 did not change dV/dtmax significantly at stimulation cycle lengths in the range 300–5000 ms, suggesting that it has no effect on the inward sodium current (Figure 5B).
Figure 5.
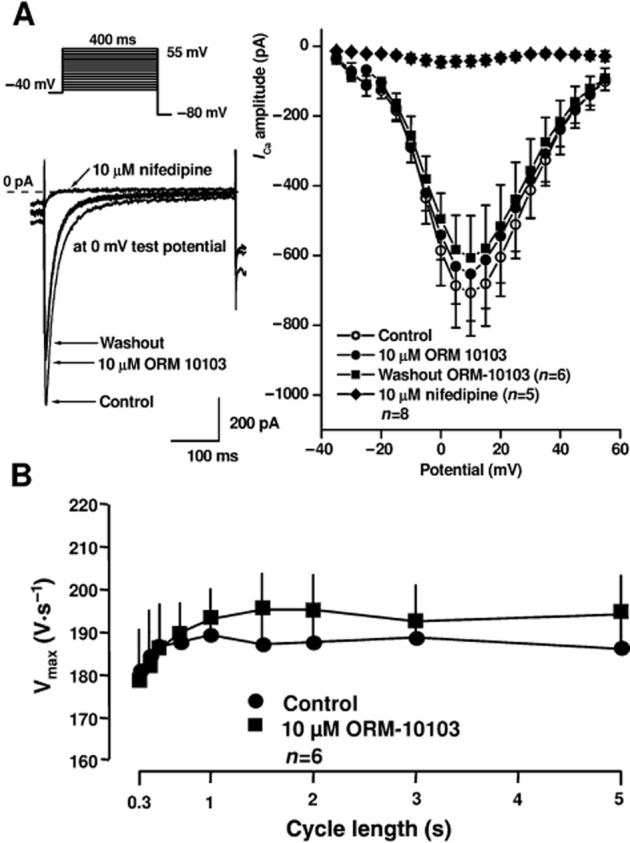
(A) Lack of effect of 10 μM ORM-10103 on the L-type ICa and INa assessed as dV/dtmax in canine ventricular myocytes. In the inset at the top, the voltage protocol is shown. The left panels indicate original current traces recorded in control conditions, in the presence of 10 μM ORM-10103, after washout of ORM-10103 and after application of 10 μM nifedipine. The right panel illustrates the current–voltage relationship of ICaL in the absence and presence of 10 μM ORM-10103 after washout of ORM-10103 and after application of 10 μM nifedipine. Values are means ± SEM. (B) Lack of effect of 10 μM ORM-10103 on the dV/dtmax indicative of INa in canine right ventricular papillary muscles. Values are means ± SEM.
As the ICaL measurements exhibited a slight rundown in some voltage clamp experiments, the effect of ORM-10103 was also studied on slow response action potentials recorded from guinea pig papillary muscles. As Figure 6 shows, ORM-10103 at 10 μM did not affect the amplitude (control: 81.2 ± 5.7 mV, ORM-10103: 81.6 ± 6.1 mV, n = 5, n.s.) or dV/dtmax (control: 12.4 ± 2.3 V·s−1, ORM-10103: 11.8 ± 2.6 V·s−1, n = 5, n.s.) of these slow response action potentials, suggesting a lack of effect of ORM-10103 on the ICaL. In the same preparation, the well-established calcium current blocker nisoldipine at 50 nM markedly reduced both the amplitude and dV/dtmax of the slow response action potentials.
Figure 6.
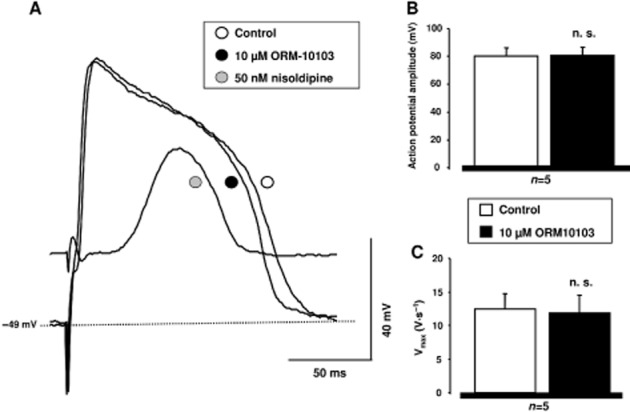
Lack of effect of ORM-10103 on slow response action potentials in guinea pig ventricular myocytes. (A) Shows original slow action potential recordings under control conditions, in the presence of 10 μM ORM-10103 and after application of 50 nM nisoldipine. (B) and (C) reveal the lack of effect of 10 μM ORM-10103 on the action potential amplitude and on the maximum rate of depolarization (dV/dtmax) respectively.
On recording outward potassium currents (n = 8–10), we found that IK1, Ito and IKs were not affected; however, IKr was slightly but significantly diminished (from 58.6 ± 5.6 pA to 43.1 ± 5.1 pA at 20 mV test potential, n = 8, P < 0.05) in the presence of 3 μM ORM-10103 (Figure 7).
The Na+/K+ pump current (Ip) was measured as described in the Methods section. The Ip current was 100.7 ± 14.6 pA in control conditions and it decreased to 86.7 ± 11.7 pA (13.9%, n = 6, P < 0.05) at the end of the 5–7 min incubation with 10 μM ORM-10103. During the measurements, a gradual slight decrease of the steady-state current was observed (Figure 8A). Therefore, a separate set of experiments was performed when the same protocol was applied but ORM-10103 was not added to the bath solution. Measuring the steady-state current using the same timescale, compared to the ORM-10103 experiments, a similar slight current decrease was recorded (119.7 ± 25.6 pA vs. 92.6 ± 27.6 pA, 22.7%, n = 3, P < 0.05, Figure 8B). Therefore, it was concluded that ORM-10103 does not influence Ip, the slight tendency of the current to decrease is not due to the effect of ORM-10103. In another experiment 10 μM strophanthin, known to block the Na+/K+ pump, was applied. Strophanthin effectively diminished the current and 0 K+ solution failed to decrease the current further showing that the measured current was the Na+/K+ pump current (Figure 8C).
Figure 8.
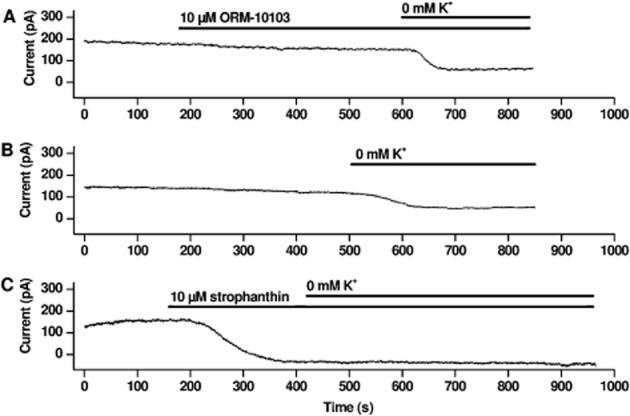
Lack of effect of ORM-10103 on Na+/K+ pump current. (A) Shows a representative experiment in which 10 μM ORM-10103 does not influence Ip; however, a slight gradual decrease in the current was detected. An original current trace in (B) indicates that the slight gradual current decrease, similar to that shown in (A), was also observed without addition of ORM-10103. (C) Strophanthin (10 μM) completely blocks Na+/K+ pump current. Ip was defined as the difference between currents measured in 5 and 0 mM K+ containing solutions. The current traces were recorded at a steady potential of −30 mV.
Effects of ORM-10103 on the EAD and DAD
The effects of ORM-10103 on EAD and DAD were studied in canine right ventricular papillary muscles and in canine cardiac Purkinje fibres, respectively, by applying the conventional microelectrode technique.
EAD was evoked in the papillary muscle preparation, stimulated at slow cycle lengths (1500–3000 ms) with a combination of 1 μM dofetilide and 100 μM BaCl2 (Figure 9A). Both 3 and 10 μM ORM-10103 clearly decreased the amplitude of the EAD. This effect was concentration-dependent and reversible upon washout of the ORM-10103 from the tissue bath containing dofetilide and BaCl2. Similar effects were seen in additional experiments (Figure 9A, right panel). ORM-10103 3 μM decreased the amplitude of the EAD from 19.3 ± 2.2 to 11.7 ± 2.4 mV (n = 6, P < 0.05). At 10 μM, the compound had a somewhat more pronounced effect, decreasing EAD from 19.4 ± 3.3 to 9.5 ± 4.0 mV (n = 4, P < 0.05).
Figure 9.
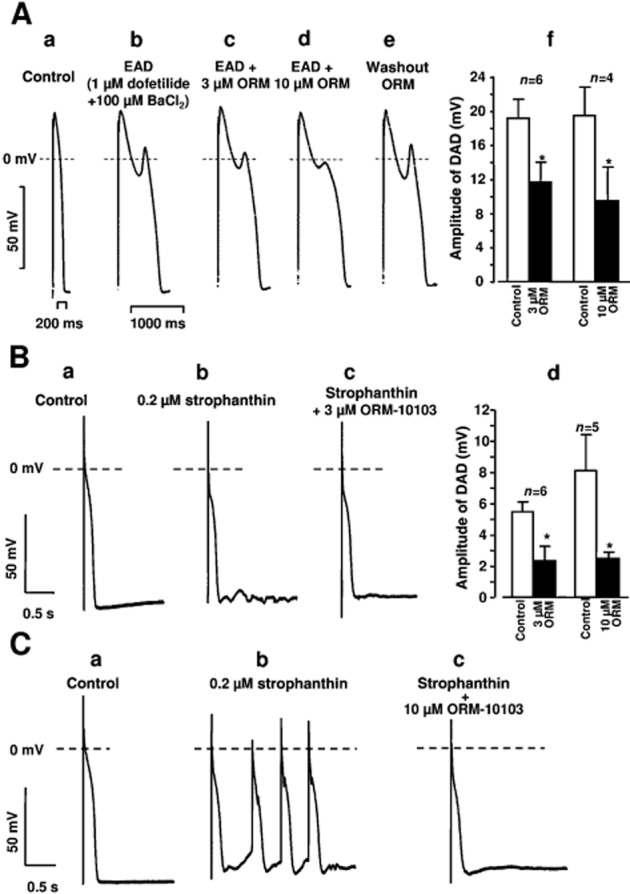
(A) Effects of 3 and 10 μM ORM-10103 (ORM) on the EAD evoked by 1 μM dofetilide + 100 μM BaCl2 in canine right ventricular papillary muscle. The stimulation cycle length was 2 s. Trace (a) is a control recording. Trace (b) reveals that EAD was elicited by 1 μM dofetilide + 100 μM BaCl2. In traces (c) and (d), 3 and 10 μM ORM-10103, respectively, were applied in the continuous presence of dofetilide and BaCl2. As shown in trace (e), after 30 min of washout of ORM-10103, the EAD amplitude was similar to that before the application of ORM-10103. In diagram (f), the effects of 3 and 10 μM ORM-10103 on the EAD amplitude are seen. Bars represent means ± SEM. (B) The effect of 3 μM ORM-10103 on the DAD amplitude in canine right ventricular Purkinje fibres. DAD was evoked by a 40 stimulus train with a stimulation cycle length of 400 ms in the presence of 0.2 μM strophanthin. Trace (a) is a control recording, trace (b) indicates the induction of DAD by 0.2 μM strophanthin, and trace (c) demonstrates that 3 μM ORM-10103 almost completely abolished DAD in the continuous presence of strophanthin. Diagram (d) depicts the effects of 3 and 10 μM ORM-10103 on the amplitude of DAD. Bars represent means ± SEM. (C) Effect of ORM-10103 (10 μM) on strophanthin-induced automaticity in canine right ventricular Purkinje fibres. Trace (a) is a control recording. Spontaneous activity was recorded after a 40 stimulus train with a stimulation cycle length of 400 ms in the presence of 0.2 μM strophanthin (trace b). Application of 10 μM ORM-10103 in the presence of strophanthin abolished the spontaneous activity (trace c).
DAD was evoked in Purkinje fibre preparations superfused with 0.2 μM strophanthin for 40 min (Figure 9B). In these experiments, a train of 40 stimuli was applied with a cycle length of 400 ms in the train. The train was then followed by a stimulation-free period of 20 s to allow observation of DAD formation. Following the addition of 3 or 10 μM ORM-10103, the DAD amplitude was decreased (Figure 9B). This effect was also concentration-dependent; ORM-10103 at 3 μM decreased the DAD amplitude from 5.5 ± 0.6 to 2.4 ± 0.8 mV (n = 6, P < 0.05) while 10 μM ORM-10103 did so from 8.1 ± 2.3 to 2.5 ± 0.3 mV (n = 5, P < 0.05). In the two experiments, strophanthin evoked a run of extra beats after the termination of the stimulus train, which could be successfully abolished by the application of 10 μM ORM-10103 (Figure 9C).
Discussion
The main finding of this study is that ORM-10103 effectively inhibited the NCX current without affecting ICaL, and this effect was associated with decreases in the amplitudes of EAD and DAD evoked in canine ventricular papillary muscle and cardiac Purkinje fibres, respectively. In our experiments, NCX current is defined as Ni2+-sensitive current. ORM-10103 inhibited INCX in canine ventricular myocytes at relatively low concentrations, with an estimated EC50 of 780–960 nM. In the same cells, even at the high concentration of 10 μM, ORM-10103 did not influence ICaL measured by the patch clamp technique, or INa estimated as dV/dtmax by the conventional microelectrode measurements. Consequently, decreases in the inward currents and thereby diminution of the Ca2+ load via the ICaL and INa cannot explain the effects of ORM-10103 on the amplitudes of EAD and DAD. ORM-10103 did not induce marked changes in Ito, IK1 and IKs, but a slight but significant decrease in the IKr was observed in the presence of ORM-10103. It is of interest that a reduction in outward potassium currents resulting in prolongation of repolarization would increase rather than decrease the liability to afterdepolarizations (Morissette et al., 2005). Therefore, the possibility that the rapid delayed rectifier K+ current is involved in the mechanism whereby ORM-10103 decreases EAD and DAD is unlikely. The same applies to Ip. Diminishing Ip would induce rather than reduce DAD. However, our experiments clearly showed that ORM-10103 did not influence this current. The possible involvement of other transmembrane ionic currents, such as ICl, cannot be completely ruled out.
To date, two compounds, KB-R7943 and SEA-0400, which potently inhibit the NCX current even at submicromolar concentrations have been reported. In addition, KB-R7943 has been shown to abolish experimental arrhythmias (Watano et al., 1999; Elias et al., 2001), whereas SEA-0400 significantly decreases the pharmacologically-induced EAD and DAD in canine ventricular preparations at concentrations at which it does not interfere with ICaL (Nagy et al., 2004). However, other studies have demonstrated that KB-R7943 and SEA-0400 in micromolar concentrations do inhibit the ICaL (Tanaka et al., 2002; Birinyi et al., 2005), which makes the interpretation of their antiarrhythmic effect somewhat uncertain. Our present investigation has provided further evidence that specific NCX inhibition results in suppression of triggered arrhythmias in in vitro cardiac preparations.
The possible therapeutic implications of our study appear to be rather complex. It is tempting to speculate that the suppression of EAD and DAD may be antiarrhythmic in both the ventricles and the atria (Chen et al., 2000) during a Ca2+ overload, as in heart failure, digitalis intoxication, and at the beginning of atrial flutter and fibrillation, especially when K+ currents have been down-regulated (Yue et al., 1997; van Wagoner and Nerbonne, 2000) and the NCX current up-regulated (Studer et al., 1994). It has been proposed that, on reperfusion after myocardial ischaemia, Ca2+ influx occurs via NCX in the reverse mode contributing to Ca2+ overload and the release of Ca2+ from the sarcoplasmic reticulum and thereby causing cardiac arrhythmias (Levi et al., 1993). Accordingly, blockade of the reverse mode of the NCX current may be beneficial.
In conclusion, the present study has provided evidence of the strong NCX inhibitory activity of ORM-10103 and its potential to suppress elementary arrhythmogenic phenomena, such as EAD and DAD. Further research is clearly needed with both in vitro and in vivo methods in order to elucidate the potential therapeutic targets and, in a wider sense, the possible beneficial effect of specific NCX inhibition.
Acknowledgments
This work was supported by grants from the Hungarian Scientific Research Fund (OTKA CNK-77855, K-82079, NK-104331), the National Office for Research and Technology (NKFP_07_01-RYT07_AF and REG-DA-09-2-2009-0115), the National Development Agency (TÁMOP-4.2.2/B-10/1-2010-0012 and TIOP-1.3.1-10/1-2010-0007), the HU-RO Cross-Border Cooperation Programmes (HURO/0802/011_AF-HURO_CARDIOPOL) and the Hungarian Academy of Sciences.
Glossary
- DAD
delayed afterdepolarization
- dV/dtmax
maximum upstroke velocity of action potential
- EAD
early afterdepolarization
- ICaL
L-type calcium current
- IK1
inward rectifier potassium current
- IKr
rapid component of the delayed rectifier potassium current
- INa
sodium current
- INCX
Na+/Ca2+ exchanger current
- Ip
Na+/K+ pump current
- Ito
transient outward potassium current
- NCX
Na+/Ca2+ exchanger
- ORM-10103
5-nitro-2-(2-phenylchroman-6-yloxy)pyridine
Conflict of interest
There are no conflicts of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation−contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Birinyi P, Acsai K, Banyasz T, Toth A, Horvath B, Virag L, et al. Effects of SEA-0400 and KB-R7943 on Na+/Ca2+ exchange current and L-type Ca2+ current in canine ventricular cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol. 2005;372:63–70. doi: 10.1007/s00210-005-1079-x. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Chen SA, Chang MS, Lin CI. Arrhythmogenic activity of cardiac muscle in pulmonary veins of the dog: implication for the genesis of atrial fibrillation. Cardiovasc Res. 2000;48:265–273. doi: 10.1016/s0008-6363(00)00179-6. [DOI] [PubMed] [Google Scholar]
- Elias CL, Lukas A, Shurraw S, Scott J, Omelchenko A, Gross GJ, et al. Inhibition of Na+/Ca2+ exchange by KB-R7943: transport mode selectivity and antiarrhythmic consequences. Am J Physiol Heart Circ Physiol. 2001;281:H1334–H1345. doi: 10.1152/ajpheart.2001.281.3.H1334. [DOI] [PubMed] [Google Scholar]
- Hobai IA, Khananshvili D, Levi AJ. The peptide ‘FRCRCFa’, dialysed intracellularly, inhibits the Na/Ca exchange in rabbit ventricular myocytes with high affinity. Pflugers Arch. 1997;433:455–463. doi: 10.1007/s004240050300. [DOI] [PubMed] [Google Scholar]
- Levi A, Brooksby P, Hancox JC. One hump or two? The triggering of calcium release from the sarcoplasmic reticulum and the voltage dependence of contraction in mammalian cardiac muscle. Cardiovasc Res. 1993;27:1743–1757. doi: 10.1093/cvr/27.10.1743. [DOI] [PubMed] [Google Scholar]
- Morissette P, Hreiche R, Turgeon J. Drug-induced long QT syndrome and torsade de pointes. Can J Cardiol. 2005;21:857–864. [PubMed] [Google Scholar]
- Nagy ZA, Virág L, Tóth A, Biliczki P, Acsai K, Bányász T, et al. Selective inhibition of sodium-calcium exchanger by SEA-0400 decreases early and delayed afterdepolarization in canine heart. Br J Pharmacol. 2004;143:827–831. doi: 10.1038/sj.bjp.0706026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogwizd SM. Clinical potential of sodium−calcium exchanger inhibitors as antiarrhythmic agents. Drugs. 2003;63:439–452. doi: 10.2165/00003495-200363050-00001. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Bers DM. Calcium cycling in heart failure: the arrhythmia connection. J Cardiovasc Electrophysiol. 2002;13:88–91. doi: 10.1046/j.1540-8167.2002.00088.x. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Schlotthauer K, Li L, Yuan W, Bers DM. Arrhythmogenesis and contractile dysfunction in heart failure. Roles of sodium−calcium exchange, inward rectifier potassium current, and residual-adrenergic responsiveness. Circ Res. 2001;88:1159–1167. doi: 10.1161/hh1101.091193. [DOI] [PubMed] [Google Scholar]
- Roden DM. Antiarrhythmic drugs. In: Hardman JG, Limbird LE, Molinoff PB, Ruddon RW, editors. Goodman & Gilman's The Pharmacological Basis of Therapeutics. New York: McGraw-Hill; 1996. pp. 839–871. Chapter 35. [Google Scholar]
- Studer R, Reinecke H, Bilger J, Eschenhangen T, Bohm M, Hasenfuss G, et al. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res. 1994;75:443–453. doi: 10.1161/01.res.75.3.443. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Nishimaru K, Aikawa T, Hirayama W, Tanaka Y, Shigenobu K. Effect of SEA0400, a novel inhibitor of sodium−calcium exchanger, on myocardial ionic currents. Br J Pharmacol. 2002;135:1096–1100. doi: 10.1038/sj.bjp.0704574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wagoner DR, Nerbonne JM. Molecular basis of electrical remodeling in atrial fibrillation. J Mol Cell Cardiol. 2000;32:1101–1117. doi: 10.1006/jmcc.2000.1147. [DOI] [PubMed] [Google Scholar]
- Venetucci LA, Trafford AW, O'Neill SC, Eisner DA. Na/Ca exchange: regulator of intracellular calcium and source of arrhythmias in the heart. Ann N Y Acad Sci. 2007;1099:315–325. doi: 10.1196/annals.1387.033. [DOI] [PubMed] [Google Scholar]
- Volders PGA, Vos MA, Szabo B, Sipido KR, Marieke DE, Groot SH, et al. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc Res. 2000;46:376–392. doi: 10.1016/s0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- Watano T, Harada Y, Harada K, Nishimura N. Effect of Na+/Ca2+ exchange inhibitor, KB-R7943 on ouabain-induced arrhythmias in guinea-pigs. Br J Pharmacol. 1999;127:1846–1850. doi: 10.1038/sj.bjp.0702740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue L, Feng J, Gaspo R, Li GR, Wang Z, Nattel S. Ionic remodeling underlying action potential changes in a canine model of atrial fibrillation. Circ Res. 1997;81:512–525. doi: 10.1161/01.res.81.4.512. [DOI] [PubMed] [Google Scholar]