

Abstract
BACKGROUND AND PURPOSE
Oestrogen therapy is known to induce cardioprotection in post-menopausal metabolic syndrome (PMS). Hence, we investigated the effect of 17-β oestradiol (E2) on functional responses to angiotensin II and cardiovascular dysfunction in a rat model of PMS.
EXPERIMENTAL APPROACH
PMS was induced in ovariectomized rats by feeding a high-fat diet for 10 weeks. Isometric tension responses of aortic rings to angiotensin II were recorded using an isometric force transducer. TUNEL assay and immunoblotting was performed to assess apoptosis and protein expression respectively in PMS.
KEY RESULTS
Endothelial dysfunction in PMS was characterized by enhanced angiotensin II-induced contractile responses and impaired endothelial dependent vasodilatation. This was associated with an increased protein expression of AT1 receptors in the aorta and heart in PMS. PMS induced cardiac apoptosis by activating Bax and PARP protein expression. These changes were associated with a down-regulation in the expression of silent information regulation 2 homologue (SIRT1)/P-AMP-activated PK (AMPK) and increased H3 acetylation in aorta and heart. E2 partially suppressed angiotensin II-induced contractions, restored the protein expression of SIRT1/P-AMPK and suppressed H3 acetylation. The role of SIRT1/AMPK was further highlighted by administration of sirtinol and compound C (ex vivo), which enhanced angiotensin II contractile responses and ablated the protective effect of E2 on PMS.
CONCLUSION AND IMPLICATIONS
Our results provide novel mechanisms for PMS-induced cardiovascular dysfunction involving SIRT1/AMPK/ histone H3 acetylation, which was prevented by E2. The study suggests that therapies targeting SIRT1/AMPK/epigenetic modifications may be beneficial in reducing the risk of cardiovascular disorders.
Keywords: post-menopausal metabolic syndrome, SIRT1, AMPK
Introduction
The accumulating incidences of type II diabetes mellitus and various cardiovascular disorders worldwide has been attributed to lifestyle changes, and in particular, the consumption of a high fat-enriched Western diet. Insulin resistance, one of the features of metabolic syndrome refers to defective insulin action and development of hyperinsulinaemia (Wang et al., 2004). The vascular endothelium plays a protagonistic role in maintaining the vascular tone by balancing the release of vasodilator substances (i.e. NO and prostacyclins) and vasoconstrictor substances (i.e. endothelin, TXA2, ROS; Katz, 1997; Puddu et al., 2000). During the last few decades, corroborating data from experimental and clinical studies has revealed that insulin resistance is a predisposing factor for endothelial dysfunction, which further leads to the development of hypertension, coronary artery diseases and other cardiovascular diseases. In general, endothelial dysfunction refers to loss of NO availability, uncoupling of endothelial NOS (eNOS), lack of substrate or cofactors of eNOS and accelerated NO degradation by ROS (Cai and Harrison, 2000). Because endothelial dysfunction is an early event, it can be of prognostic value. Insulin resistance and oestrogen deficiency have been established as independent risk factors for cardiac hypertrophy and hence heart failure (Rutter et al., 2003; Pelzer et al., 2005). The myocardium is considered to be a site of action of oestrogens and sex hormones as they prevent left ventricular hypertrophy in animal and human models. In a rat model of post-menopausal cardiac hypertrophy induced by pressure overload the protective Akt-eNOS-NO pathway activated by oestrogen is compromised and contributes to cardiac decompensation (Bhuiyan and Fukunaga, 2010). Therefore, therapies targeting endothelial dysfunction and increasing NO availability can be effective at preventing numerous cardio-metabolic disorders.
The renin angiotensin system (RAS) is a major common denominator implicated in the pathogenesis of various cardiovascular disorders associated with insulin resistance (Touyz et al., 1999). A direct link between the activation of RAS and the occurrence of endothelial dysfunction in insulin resistance has been demonstrated (McFarlane et al., 2001). Angiotensin II (Ang II), the major peptide of RAS, has been shown to be involved in stimulation of oxidative stress, promoting smooth muscle cell growth, inhibiting apoptosis and altering the redox state of the blood vessel (Paravicini and Touyz, 2006). Also, activation of MAPKs, in response to Ang II, transmit extracellular stimuli by phosphorylating a variety of substrates including transcriptional factors and histone H3 and hence regulating hypertrophy in various cellular subtypes (Wang et al., 1998; Soloaga et al., 2003).
Exciting research has shown that epigenetic changes in chromatin can affect gene transcription, replication, genome stability and repair in response to environmental stimuli and changes in key chromatin histone patterns have been noted with various pathological conditions (Li, 2002). The best-known covalent modifications, methylation, acetylation and phosphorylation, have been characterized in various disease models. We have learned from our experience that histones (H3) are acetylated and phosphorylated in heart and kidney under insulin-resistant conditions and may contribute to the development of insulin resistance and associated complications (Gaikwad et al., 2010). As far as angiotensin receptors are concerned, we have shown that both AT1 and AT2 receptors are epigenetically regulated by histone H3 modifications in aortic vasculature (Karpe et al., 2012).
Gender plays a prominent role in regulating cardiovascular risk, as women suffer less frequently from cardiovascular diseases than their male counterparts (Ho and Mosca, 2002). This tendency arises because of the presence of oestrogen in women, and naturally decreases after menopause, a clinical state of oestrogen deficiency (Colditz et al., 1987; Rossi et al., 2002). This menopausal transition is characterized by a rapid decline in endogenous oestrogen levels and an elevation in androgen levels thus resulting in increased visceral adiposity index (Lee et al., 2009). This ‘hormonal-shift’ plays a pivotal role in the development of insulin resistance and endothelial dysfunction adding to further cardiovascular risk. Taking into account the protective action of oestrogen, hormone therapy by far remains the only effective way to limit this risk in addition to a classical preventive strategy based on physical activity and reduced calorie intake. Oestrogens act both on vascular smooth muscle cells and endothelial cells to increase the production and bioavailability of a potent vasodilator through eNOS (Chen et al., 1999; Chambliss and Shaul, 2002).
To the best of our knowledge, this is the first study that demonstrates all the characteristics of post-menopausal metabolic syndrome (PMS) in vivo in Sprague-Dawley rats. We hypothesized that 17-β oestradiol (E2) treatment alleviates the insulin resistance, endothelial dysfunction and cardiomyopathy associated with PMS. We also hypothesized that the covalent modifications of histone represent the mechanism behind the vasoprotective role of oestrogen in PMS.
Methods
Animals
All experimental protocols were approved by the Institutional Animal Ethics Committee (IAEC no: 11/26) and were performed in accordance with the guidelines of the Committee for the Purpose of Control and Supervision on Experiments on Animals (CPCSEA). Female Sprague-Dawley rats (8–9 weeks; 200 ± 10 g) were obtained from the Central Animal Facility of National Institute of Pharmaceutical Education and Research (NIPER). The total number of animals used in these experiments was 144. They were housed in groups of three per cage and maintained under standard environmental conditions: temperature 22 ± 2°C, humidity 50 ± 10% and 12 h light and dark cycles with food and water ad libitum. All animals were acclimatized to their environment for 1 week before commencement of the experiments. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Model development and surgical procedures
Female rats underwent ovariectomy (OVX) or sham (SHAM) operation under ketamine (80 mg kg-1)/xylazine (8 mg kg-1) anaesthesia (both drugs administered i.p.). The depth of anaesthesia was assessed by observing muscle movements, jaw tone, swallowing reflex, withdrawal reflex (toe, paw, ear pinch), palpebral and corneal reflex. We also monitored the physiological parameters during surgery such as heart rate, respiratory rate and blood pressure. Ovariectomy was performed as described previously (Mittal et al., 2009). Female rats were then randomly divided into six groups and were fed on respective diets for model development (Figure 1). The groups included: (1) control (CON) – fed normal chow, n = 6; (2) sham operated (SHAM) – fed normal chow, n = 6; (3) ovariectomized (OVX) – fed normal chow, n = 8; (4) high-fat diet – fed (HFD), n = 6; (5) ovariectomized rats fed on high-fat diet (HFD + OVX), n = 8; and (6) chronic treatment with E2 in HFD + OVX + E2 rats; n = 8.
Figure 1.
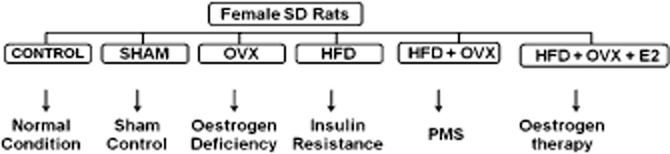
Protocol scheme: The female rats were divided into six groups as CON; SHAM; OVX; ovariectomized and fed high-fat diet HFD + OVX, and oariectomized rats fed on high-fat diet with 17-β oestradiol supplement (HFD + OVX + E2). 17-β Oestradiol was administered s.c. 80 μg·kg−1·day−1 in olive oil for 60 days. Animals were killed at the end of the study for further analysis.
Ovariectomized rats were fed on high-fat diet for a period of 10 weeks after surgery to develop a model comprising both characteristics of oestrogen deficiency and insulin resistance. This model clinically mimics PMS.
Drug and molecular target nomenclature conforms to the BJP's Guide to Receptors and Channels (Alexander et al., 2011).
Chronic treatment with E2
E2 was purchased from Sigma Chemicals (St Louis, MO, USA). Animals were allowed to recover from post-operative stress for a week after the surgery. E2 (80 μg·kg−1·day−1 in olive oil s.c.; Bebo et al., 2001) was then administered to animals in the HFD + OVX group for 60 days.
Morphometric parameters, biochemical estimations and measurement of serum oestradiol levels
Plasma was used for estimation of glucose, insulin, triglycerides, total cholesterol, high-density lipoprotein (HDL)-cholesterol CK-MB using commercially available spectrophotometric kits (Accurex Biomedical Pvt. Ltd., Mumbai, India). Plasma insulin and serum oestradiol levels were assayed, respectively, by use of a rat insulin elisa kit (DRG® International, Inc., Springfield, NJ, USA) and rat oestradiol EIA kit (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer's instructions.
Invasive BP measurement
Rats deprived of food overnight (fasted) were anaesthetized and the skin on the ventral side of the neck, right hind leg and chest were carefully shaved and disinfected. The carotid artery was differentiated from the nerve fibre, and cannulated with a PE catheter. After cannulation, the cannulation line was flushed with normal saline (0.1 mL) to prevent thrombosis. The cannula was then connected to a fluid filled pressure transducer (MLT844) attached to Powerlab data acquisition system (AD Instruments, Bella Vista, NSW, Australia) for measurement of BP.
Glucose and insulin tolerance tests (GTT and ITT)
Animals were kept on a 6 h fast for GTT and ITT, and a basal sample was taken, followed by oral d-glucose (2 g·kg−1) or bovine insulin (0.25 IU·kg−1, s.c.). Blood samples were collected at 5, 15, 30, 60, 90 and 120 min and plasma glucose concentration was determined to assess impairment in glucose tolerance and glucose disappearance with time (Vikram et al., 2010).
Aortic ring preparation and vascular reactivity studies
Briefly, after a proper model had been developed, animals were killed under ketamine-xylazine anaesthesia. The thoracic aorta was excised and placed in ice-cold oxygenated Krebs–Henseleit solution (KHS). Intact aortic rings (3–4 mm in length) were cut from the cleaned aorta. KHS comprised of (in mmol·L−1) NaCl: 118; KCl: 4.7; MgSO4.7H2O: 1.2; CaCl2.2H2O: 2.6; KH2PO4: 1.2; NaHCO3: 25; Glucose: 5.5; pH 7.4. Aortic rings were carefully mounted with the help of stainless steel hooks on the isometric force transducer in the water-jacketed organ bath chamber. A resting tension of 2 g was applied to each aortic ring. Each ring was allowed to equilibrate in 10 mL KHS (37°C) bubbled with carbogen (5% CO2 + 95% O2) for 120 min. KHS was changed every 15 min during this equilibration period. At the beginning of each experiment, aortic rings were primed with 80 mmol·L−1 KCl. Isometric tension responses were recorded using an isometric force transducer (BioDevices, Ambala, India). Cumulative concentration–response curves (CRCs) to Ang II (1 nmol·L−1 to 1 μmol·L−1); phenylephrine (PE; 1 nmol·L−1 to 1 μmol·L−1) and carbachol (CCh; 1 nmol·L−1 to 10 μmol·L−1) were constructed in all animal groups. To record CCh-mediated relaxations, aortic rings were precontracted with 100 nmol·L−1 PE and responses to increasing concentration of CCh were obtained. At the end of the experiment, aortic rings were blotted dry and their length and weights were measured to calculate tension normalized to cross-sectional area (mg·mm–2) as described previously (Karpe et al., 2012).
To determine the role of mediators involved in altered vascular reactivity in HFD + OVX + E2 group, we performed a mechanistic study using various antagonists of the physiological mediators. For this, aortic rings from HFD + OVX + E2 group were pre-incubated with L-NG-nitroarginine methyl ester (L-NAME), a non-selective competitive eNOS blocker, 100 μmol·L−1; sirtinol a SIRT1 blocker, 50 μmol·L−1 and compound C an AMPK blocker, 100 μmol·L−1 for 30 min and then CRCs to Ang II were constructed.
Nitrite determination
NO being unstable (rapidly oxidized to nitrite and nitrate) was estimated by the Griess method. Briefly, aortic rings were homogenized in ice-cold homogenizing medium (10 mmol·L−1 Tris-HCl; 0.1 mmol·L−1 EDTA; 10 mmol·L−1 sucrose; 0.8% NaCl, pH 7.4) and nitrite was estimated in the supernatant (Bitar et al., 2005; Karpe et al., 2012).
Detection of vascular O2− by lucigenin chemiluminescence
Vascular superoxide production was measured using lucigenin chemiluminescence as described previously (Rajagopalan et al., 1996). Briefly, aorta was harvested, cleaned of extraneous tissue, cut into rings of 5-mm length and was incubated in HEPES buffer (pH 7.4), which was continuously bubbled with 5%CO2/95% O2. Aortic rings were equilibrated at 37°C for 30 min. These were then transferred to glass tubes containing warm HEPES buffer and lucigenin (5 μmol·L−1) and equilibrated in the dark for 10 min at 37°C. Lucigenin chemiluminescence was recorded every 20 s for 10 min with a luminometer (Flexstation 3, Molecular Devices Corp., Menlo Park, CA, USA). Chemiluminescence was expressed as counts s-1. Chemiluminescence was also measured in tubes containing buffer and lucigenin without aortic rings, and these values were subtracted from chemiluminescence obtained from aortic rings. After the experiment, aortic rings were recovered, rinsed and protein content was estimated. Vascular superoxide concentration was quantified as relative light units mg-1 of tissue.
Histological determination and TUNEL assay
Rats were anaesthetized and the aorta and heart were harvested, cleaned of adhering adventitious tissue, and were stored in 10% formal saline. Heart sections from the left ventricle were used for the determination of cell death using a TUNEL assay kit (Calbiochem, Darmstadt, Germany). The assay was performed according to the manufacturer's instructions.
Western immunoblotting
Protein isolation and Western blotting were performed as previously described (Gaikwad et al., 2010; Gupta et al., 2010). Briefly, frozen aortic tissues were thawed, minced and homogenized in lysis buffer. Protein samples were resolved using 7.5, 10.0 and 14.0% SDS-PAGE depending on the molecular weight of desired proteins. These were then transferred to nitrocellulose membranes (Amersham BioSciences, Piscataway, NJ, USA) and were analysed with appropriate antibodies (see below; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Antibodies (dilutions included in parentheses) used were: AT1 (1:1000), caspase 3 (1:1000), PARP (1:1000), Bax (1:2000), Bcl2 (1:1000), PCNA (1:1500), SIRT1 (1:2000), P-AMPK (1:3000) and AMPK (1:3000). Actin was used as the loading control (1:3500). Histones were extracted as described previously (Tikoo et al., 2008). Immunoblotting for histones was performed: H3 acetylation (1:2000) and Ser 10 phosphorylation (1:2000). The antigen-primary antibody complexes were incubated with HRP-conjugated secondary antibodies and visualized using enhanced chemiluminescence (ECL) system and ECL hyperfilm (Amersham, Buckinghamshire, UK). Blots were scanned and analysed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Results are expressed as mean ± SEM and n refers to number of animals in a particular group. Statistical analysis was performed using SigmaStat statistical software (Systat Software Inc., San Jose, CA, USA). Comparisons between two groups were evaluated using Student's t-test. Multiple group comparisons were performed using one-way anova. Post hoc analysis was performed using Tukey's test. Results were considered significant if P < 0.05.
Results
HFD + OVX: Mimicking PMS
Feeding a high-fat diet to ovariectomized rats for 10 weeks led to the development of an animal model displaying characteristics of PMS. Body weights of OVX, HFD and HFD + OVX rats were significantly higher than those of SHAM rats as depicted by the body weight gain (Table 1). Consistent with the known effects of high-fat diet feeding, rats from the HFD group displayed mild hyperglycaemia and hyperinsulinaemia. However, oestrogen deficiency further aggravate the pathological condition in these rats. Hence, HFD + OVX rats displayed hyperglycaemia and hyperinsulinaemia to a significant extent as compared with SHAM rats (Figure 2A & B). HFD + OVX rats also showed impaired glucose tolerance and enhanced insulin sensitivity as observed in GTT and ITT respectively (Figure 2C to F). Furthermore, a significant elevation in plasma triglycerides, total cholesterol, low-density lipoprotein (LDL)-cholesterol and a significant decrease in HDL-cholesterol was observed in HFD + OVX rats as compared with SHAM rats (Table 1). HFD + OVX rats receiving showed a modest decrease in plasma glucose, insulin levels, thus displaying partially improved glucose tolerance and insulin sensitivity. Also, the plasma lipid profile was found to be improved after E2 treatment. Taken together, these data indicate that this rat model possesses all of the characteristics of the clinical state PMS.
Table 1.
Tabular representation of morphometric and biochemical parameters
| CONTROL | SHAM | OVX | HFD | HFD + OVX | HFD + OVX + E2 | |
|---|---|---|---|---|---|---|
| Initial BW (g) | 206 ± 1.59 | 202 ± 1.49 | 209 ± 3.24 | 202 ± 2.20 | 201 ± 1.52 | 204 ± 2.87 |
| Final BW (g) | 270 ± 2.81 | 267 ± 2.70 | 342 ± 7.46*** | 323 ± 4.66*** | 403 ± 6.77*** | 239 ± 2.75### |
| Weight gain (g) | 64 ± 3.59 | 65 ± 2.58 | 133 ± 5.53*** | 121 ± 5.99*** | 202 ± 7.08*** | 35 ± 2.94### |
| Absolute HW (g) | 0.92 ± 0.01 | 0.89 ± 0.04 | 1.08 ± 0.06 | 0.93 ± 0.05 | 1.12 ± 0.04* | 0.86 ± 0.03## |
| HW/BW (× 10−3) | 0.33 ± 0.01 | 0.33 ± 0.01 | 0.29 ± 0.02 | 0.30 ± 0.02 | 0.30 ± 0.02 | 0.35 ± 0.01 |
| HW/Tibia length (g·cm−1) | 0.21 ± 0.01 | 0.20 ± 0.01 | 0.25 ± 0.01* | 0.25 ± 0.01 | 0.28 ± 0.02** | 0.22 ± 0.01# |
| Absolute kidney wt (g) | 1.61 ± 0.07 | 1.58 ± 0.11 | 1.74 ± 0.07 | 1.68 ± 0.02 | 1.70 ± 0.06 | 1.58 ± 0.08 |
| KW/BW (× 10−3) | 6.01 ± 0.29 | 5.68 ± 0.34 | 5.69 ± 0.47 | 5.67 ± 0.20 | 6.26 ± 0.25 | 6.81 ± 0.19 |
| Absolute liver weight (g) | 7.76 ± 0.23 | 7.51 ± 0.23 | 9.29 ± 0.57 | 8.03 ± 0.33 | 9.86 ± 0.73* | 9.37 ± 0.11 |
| LW/BW (× 10−3) | 26.51 ± 0.23 | 27.11 ± 0.59 | 30.04 ± 1.71 | 30.19 ± 0.98 | 34.58 ± 1.44** | 26.79 ± 0.15# |
| WAT (g) | 3.44 ± 0.46 | 3.54 ± 0.47 | 7.45 ± 0.69* | 7.96 ± 0.78** | 14.27 ± 1.37*** | 7.8 ± 1.09## |
| WAT/BW (× 10−3) | 12.56 ± 1.46 | 12.76 ± 1.63 | 24.28 ± 2.77 | 29.35 ± 3.10** | 48.16 ± 6.15*** | 27.43 ± 3.08# |
| BAT (g) | 0.59 ± 0.08 | 0.54 ± 0.05 | 1.20 ± 0.17 | 1.00 ± 0.23 | 1.11 ± 0.02 | 2.17 ± 0.18# |
| BAT/BW (× 10−3) | 2.19 ± 0.31 | 1.96 ± 0.20 | 4.74 ± 0.50 | 3.61 ± 1.06 | 4.47 ± 0.56 | 8.75 ± 0.22# |
| Absolute uterus weight (g) | 0.53 ± 0.04 | 0.49 ± 0.02 | 0.28 ± 0.02*** | 0.50 ± 0.03 | 0.21 ± 0.01*** | 0.60 ± 0.03### |
| UW/BW (× 10−3) | 1.66 ± 0.03 | 1.64 ± 0.06 | 0.81 ± 0.08*** | 2.22 ± 0.22 | 0.71 ± 0.05** | 2.03 ± 0.01### |
| Plasma triglycerides (mmol·L−1) | 0.36 ± 0.04 | 0.43 ± 0.04 | 0.53 ± 0.03 | 0.42 ± 0.05 | 0.62 ± 0.04** | 0.44 ± 0.02 |
| Plasma total cholesterol (mmol·L−1) | 1.67 ± 0.09 | 1.77 ± 0.06 | 2.06 ± 0.04 | 2.23 ± 0.11* | 3.61 ± 0.16*** | 2.91 ± 0.07## |
| Plasma HDL cholesterol (mmol·L−1) | 1.10 ± 0.06 | 1.06 ± 0.03 | 1.08 ± 0.02 | 0.98 ± 0.06 | 0.64 ± 0.03*** | 1.41 ± 0.07### |
| Plasma LDL cholesterol (mmol·L−1) | 0.38 ± 0.07 | 0.60 ± 0.06 | 0.81 ± 0.07 | 1.16 ± 0.28 | 2.46 ± 0.06*** | 1.12 ± 0.04# |
All the morphometric parameters were recorded after proper development of an oestrogen deficient condition, insulin resistance, PMS and oestrogen replacement. BW, body weight; HW, heart weight; KW, kidney weight; LW, liver weight; WAT, white adipose tissue; BAT, brown adipose tissue; UW, uterus weight. Each data point represents the mean ± SEM, n = 6–8 rats per group.
*P < 0.05; **P < 0.01; ***P < 0.001; #P < 0.05; ##P < 0.01; ###P < 0.001; * vs. SHAM; # vs. HFD + OVX.
Figure 2.
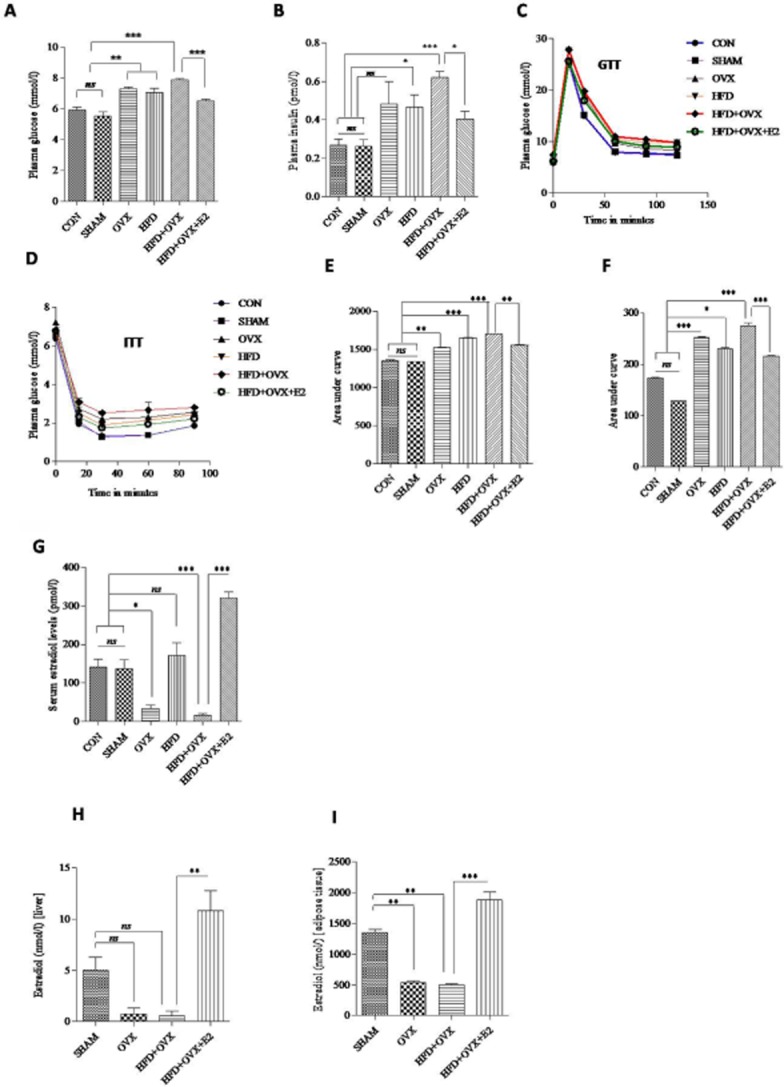
17-β Oestradiol (E2) therapy alleviates insulin resistance and mimics physiological serum oestradiol levels: E2 suppresses hyperglycaemia and hyperinsulinaemia in HFD + OVX rats as depicted by plasma glucose and insulin levels respectively (A & B). GTT and ITT with representative bar graphs showing area under curve (C–F) confirms improved insulin sensitivity after E2 treatment. After s.c. injection of E2, serum and tissue oestradiol levels were determined (2G to 2I). Each data point is represented as mean ± SEM, n = 6–8 rats per group. *P < 0.05; **P < 0.01; ***P < 0.001.
Taking in consideration the clause mentioned in the CPCSEA guidelines relating to the justification for using animals for experiments, the control group was not used further in the vascular reactivity studies and molecular studies because it mimics similar conditions to the SHAM group. Similarly, preliminary studies with the HFD + OVX + vehicle showed similar results to the HFD + OVX group. Hence, only the HFD + OVX group was used in all further studies.
Morphometric parameters, serum oestradiol levels and BP
Morphometric parameters are compiled in a tabular form in Table 1. Rats had an increased body weight after ovariectomy and high-fat diet feeding. Absolute heart weights were comparable among the groups. However, heart weight normalized to tibia length was significantly elevated in the HFD + OVX group, which is an indication of cardiac hypertrophy. Consistent with its known effects, chronic treatment with E2 restored the OVX-mediated decrease in uterine weight. White adipose tissue mass was significantly increased in the OVX, HFD and HFD + OVX rats. However, oestrogen treatment was found to decrease this mass. Similarly, brown adipose tissue mass was comparable among all groups except the HFD + OVX + E2 group where it was increased. Serum oestradiol levels were significantly increased, but remained within the physiological range in the HFD + OVX + E2 group of rats (87.51 ± 4.05 pg·mL−1) compared with HFD + OVX rats (7.92 ± 0.71 pg·mL−1). This shows that E2 treatment via the s.c. route delivered a dose of oestrogen that mimicked endogenous levels (Figure 2G). Furthermore, we estimated the level of 17-β estradiol in liver and adipose tissue in the four main groups to ascertain the bioavailability and success of administration of oestrogen s.c. in our animal model. We found increased levels of oestrogen in adipose tissue and liver (Figure 2H & I). Apart from this, the OVX, HFD and HFD + OVX rats showed a higher systolic and diastolic BP than the SHAM group. BP was reduced in the HFD + OVX + E2 group, whereas that in the HFD + OVX group was comparable with the SHAM group. Heart rate was comparable among all the groups (Supporting Information Table S1).
E2 attenuates endothelial dysfunction in HFD + OVX rats
It has been reported that agonist-mediated responses are modulated in ovariectomized and diabetic vasculature leading to vascular dysfunction (Rossi et al., 2002; Viswanad et al., 2006). However, no reports exist regarding responses to agonists in the PMS condition in vitro or in vivo. Hence, to evaluate endothelial dysfunction occurring in PMS, we performed vascular reactivity studies with Ang II (1 nM–1 μM) and PE (1 nM–1 μM) in the study groups. Ang II induced concentration-dependent sustained contractions in thoracic aortic segments of all animals, based on which cumulative response curves were generated (Figure 3A). Maximal response (Emax; mg·mm–2) was found to be significantly increased in OVX (346 ± 25), HFD (581 ± 60) and HFD + OVX (754 ± 50) group as compared with the SHAM group (168 ± 13). Apart from this, no appreciable change was observed in pD2 values to Ang II among all the groups (Supporting Information Table S2). Treatment with E2 partially attenuated the elevated responses to Ang II as evident from the Emax in mg·mm–2 values (351 ± 33) in HFD + OVX + E2 group as compared with HFD + OVX group (754 ± 50).
Figure 3.
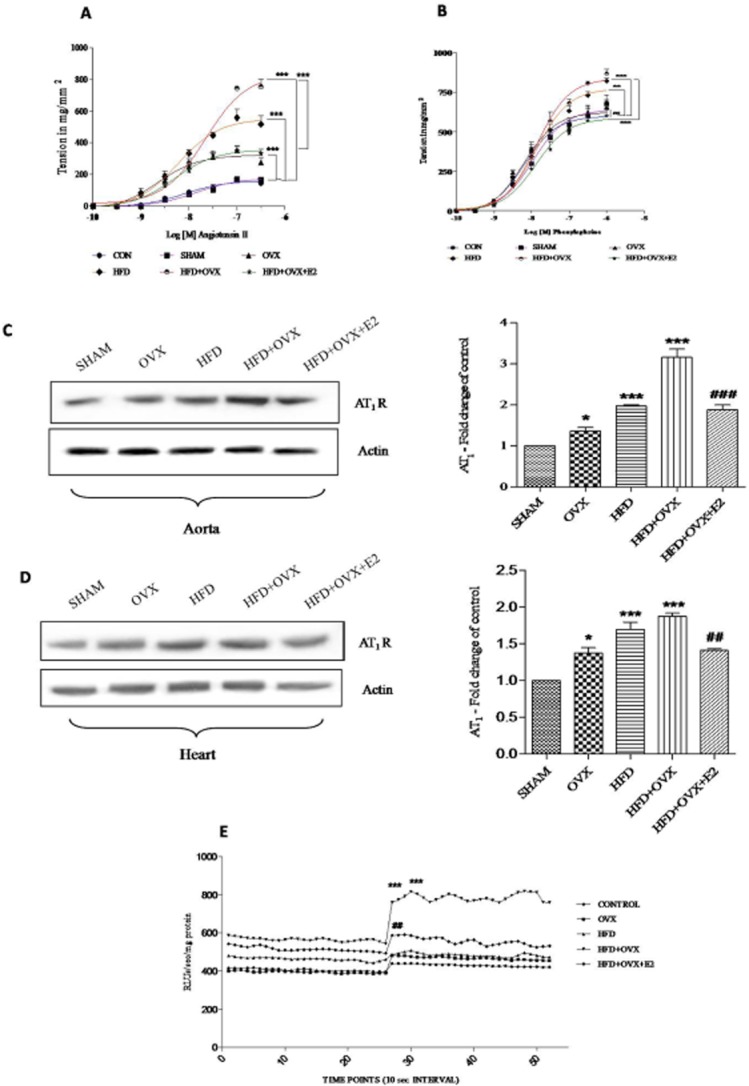
PMS alters Ang II-induced contractile responses in thoracic aorta: Cumulative concentration-responses curves (CRCs) to Ang II (A), cumulative CRCs to PE (B) showing enhanced contractile responses to Ang II and PE in PMS condition and partial alleviation by 17-β oestradiol. Protein expression of AT1 receptors was determined in aorta and heart and bar analysis of the blots (C & D). (E) Superoxide (O2-) production in response to Ang II in aortic rings detected by lucigenin chemiluminescence in all groups (E). Each data point is represented as mean ± SEM, n = 6–8 rats per group. Comparisons are shown by arrows. *P < 0.05; **P < 0.01; ***P < 0.001. # vs. HFD + OVX group.
In a similar setup of experiments, CRCs were also generated in response to PE (1 nm–1 μM; Figure 3B). PE also induced concentration-dependent contraction in thoracic aortic segments. In accord with the results mentioned earlier, Emax (mg·mm–2) was found to be significantly increased in the HFD (821 ± 20) and HFD + OVX (868 ± 23) groups as compared with the SHAM group (676 ± 58). However, no change in Emax was found in the OVX group (690 ± 13) as compared with the SHAM group (676 ± 58). pD2 values were comparable in all the groups. E2 was effective in decreasing the contractile responses to PE as evident by Emax values in mg·mm–2 (HFD + OVX + E2: 610 ± 33 vs. HFD + OVX: 868 ± 23) with no change in pD2 values. The protein levels of AT1 receptors were assessed using immunoblotting in aorta and heart (Figure 3C & D). HFD + OVX rats displayed a markedly elevated expression of AT1 receptors in aorta and heart. The altered expression of AT1 receptors was normalized by chronic treatment with E2 thus supporting the hormone therapy in PMS. Furthermore, lucigenin chemiluminescence data confirmed that enhancement of Ang II-induced oxidative stress via the activation of NAD(P)H oxidases in HFD + OVX rats, may mediate the endothelial dysfunction. This oxidative stress was suppressed by oestrogen therapy (Figure 3E).
We used CCh, a muscarinic agonist for relaxation studies. Vasorelaxation responses (% relaxation) to CCh were compromised significantly in OVX (67 ± 11.84), HFD (56 ± 14.92) and HFD + OVX (37 ± 2.07) groups as compared with the SHAM group (82 ± 2.71). This impairment of CCh-mediated vasorelaxation was partially prevented in the HFD + OVX + E2 group (% relaxation 76 ± 6.81; Figure 4A). This suggests that the endothelium was preserved, thus adding weight to the protective role of E2 for treating PMS. Additionally, aortic and heart nitrite levels (in μM) were significantly decreased in the OVX (49.89 ± 0.03), HFD (55.60 ± 3.09) and HFD + OVX (31.06 ± 1.86) groups compared with the SHAM (82.52 ± 1.18) group (Figure 4B & C). E2 markedly increased plasma nitrite levels (92.30 ± 2.60), supporting the hypothesis that vasodilatation of the thoracic aorta is induced by increased production and availability of NO and an improvement in endothelial function. Histological sections of thoracic aorta revealed that the arterial media thickness was increased in PMS (Figure 5A & B). This is indicative of vascular proliferation, which was normalized by E2 treatment.
Figure 4.
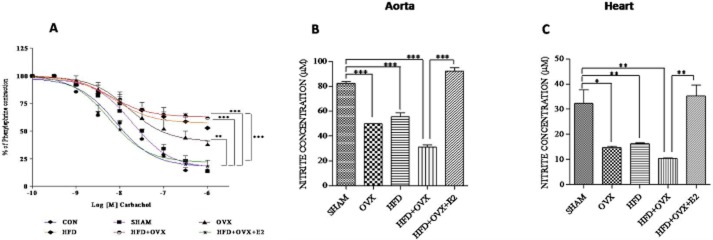
PMS attenuates endothelium-dependent relaxant responses in thoracic aorta and causes endothelial dysfunction: PMS impairs CCh-mediated vasorelaxation in thoracic aortic rings and 17-β oestradiol partially promotes vasorelaxation (A) and increases nitrite levels in aorta and heart suggesting elevated bioavailability of NO (B & C). Each data point is represented as mean ± SEM, n = 6–8 rats per group. Comparisons are shown by arrows. *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 5.
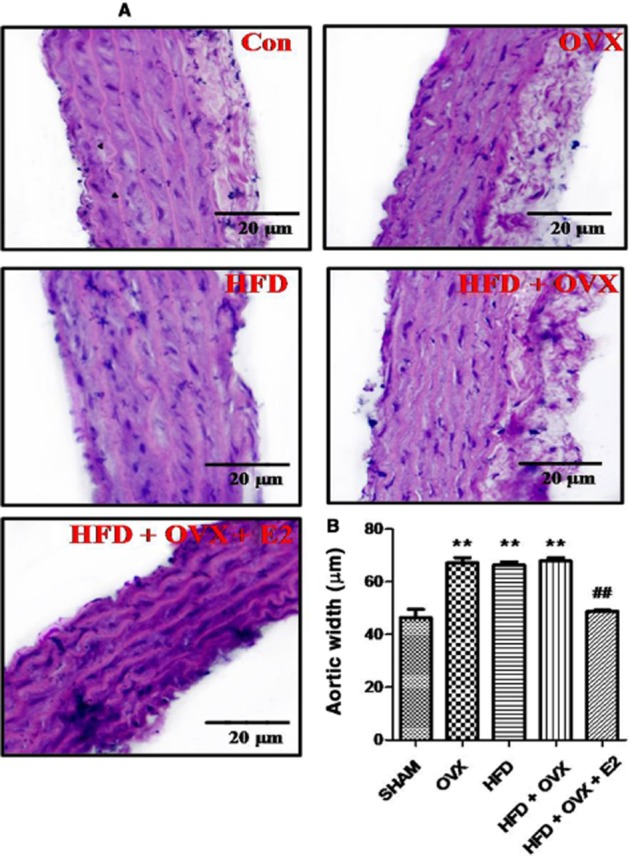
PMS alters the gross histology of thoracic aorta: PMS increases the aortic media thickness in HFD, OVX and HFD + OVX rats and 17- β oestradiol attenuates this effect. Sections of aorta were stained with H&E for gross microscopic changes in all groups (A). Quantitative interpretation of aortic media width in all groups (B). Each data point is represented as mean ± SEM, n = 6–8 rats per group. *P < 0.05; **P < 0.01; ***P < 0.001; * vs. SHAM; # vs. HFD + OVX group.
E2 prevents cardiac apoptosis induced by PMS
To assess cardiac apoptosis, we determined the expression of the apoptotic proteins Bax and PARP. There was a significant increase in the expression of Bax and the cleaved form of PARP in the OVX, HFD and HFD + OVX groups as compared with the SHAM group (Figure 6A). In contrast, the expression of anti-apoptotic Bcl2 and PCNA was found to be decreased in the PMS conditions (Figure 6B & C). Interestingly, this activation of apoptotic pathways was normalized by E2 treatment to a significant extent. The plasma LDH and creatine kinase – MB (CK–MB) levels were elevated in OVX, HFD and HFD + OVX groups as compared with SHAM group. This increase in LDH and CK-MB was normalized by E2 treatment. This indicates that E2 prevents cardiac damage and hence, cardiomyopathy (Figure 6D & E). To support this molecular data, we looked at gross histopathology and performed a TUNEL assay for confirmation of apoptosis and hence cardiomyopathy. Haematoxylin and eosin staining of heart sections revealed that the number of nuclei per section was decreased significantly in HFD + OVX heart compared to the SHAM group, thus indicative of hypertrophy (Figure 7A). Also in heart sections from OVX, HFD and HFD + OVX rats, the proportion of TUNEL positive cells were more than SHAM group (Figure 7B). E2 treatment partially prevented apoptosis as the number of TUNEL positive cells observed in the HFD + OVX + E2 group were less than those in the HFD + OVX group.
Figure 6.
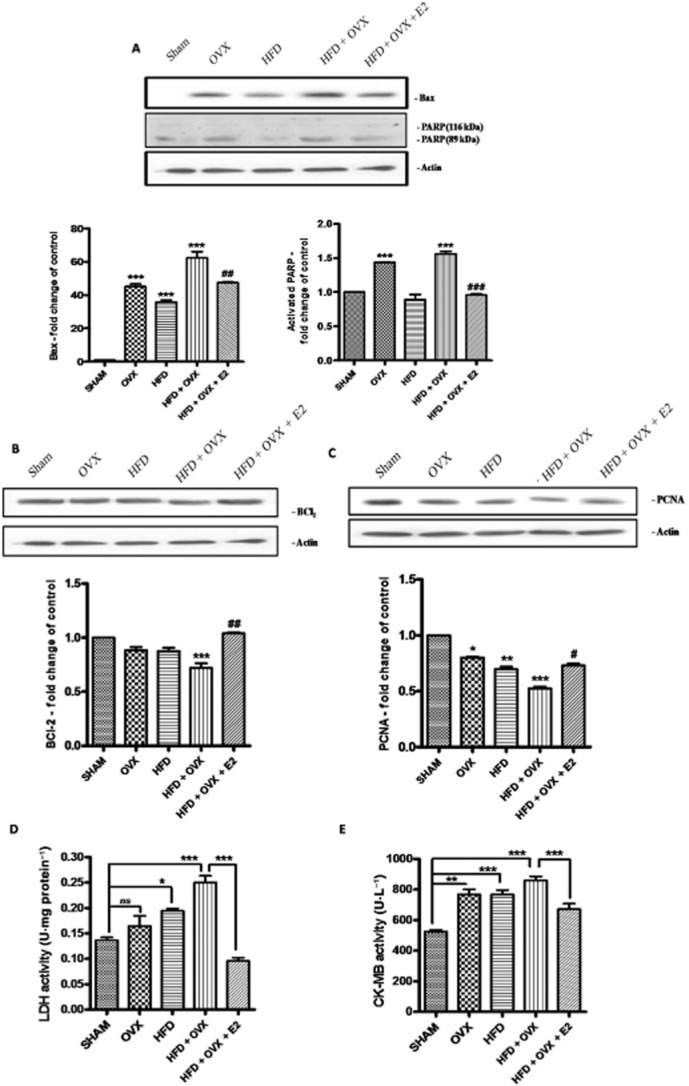
PMS induces the expression of apoptotic proteins in the heart: Expressions of protein involved in apoptosis were determined by immunoblotting. PMS alters of the expression of pro-apoptotic Bax and PARP (A), anti-apoptotic Bcl2 (B) and PCNA (C) in heart. LDH and CK-MB levels were also determined in all the groups and PMS was found to increase levels of LDH and CK-MB, which indicates signs of cardiomyopathy and 17-β oestradiol prevents the elevation (D & E). Each data point is represented as mean ± SEM, n = 6–8 rats per group. *P < 0.05; **P < 0.01; ***P < 0.001; * vs. SHAM; # vs. HFD + OVX group.
Figure 7.

17-β Oestradiol attenuates PMS-induced cardiac hypertrophy: Heart sections from all the groups were stained with haematoxylin and eosin to analyse gross microscopic changes (A). PMS induces apoptosis in heart as revealed by TUNEL assay. (A) Representative photomicrographs showing apoptotic cells having fragmented DNA (indicated by arrows). The graph illustrates the mean apoptotic cells per 100 cells (B). Each data point is represented as mean ± SEM, n = 6–8 rats per group. *P < 0.05; **P < 0.01; ***P < 0.001; #P < 0.05; ##P < 0.01; ###P < 0.001. * vs. SHAM; # vs. HFD + OVX group.
E2 attenuates Ang II-mediated contractile responses – the involvement of SIRT1/AMPK and eNOS
It is now known that SIRT1 and AMPK are important mediators in regulating eNOS phosphorylation to realize its cardio-protective role. Therefore, we determined the expression of these molecules in PMS. Western blotting analysis confirmed that the expression of both SIRT1 and P-AMPK were reduced in HFD + OVX aorta and heart. This indicates that in the PMS state there is a deficiency of SIRT1 and P-AMPK. Interestingly, E2 treatment prevented the loss of SIRT1 and P-AMPK (Figure 8A to D). We also found that the Ang II-mediated contractile responses were attenuated the in HFD + OVX + E2 group. However, the exact mechanisms behind this improvement in these functional contractile responses are largely unknown. To confirm and expand these mechanisms, we performed a more complete study involving pre-incubation of aortic segments with inhibitors of eNOS, SIRT1 and AMPK and then constructed CRCs in response to Ang II (Figure 8E). We used sirtinol as a SIRT1 inhibitor (Donato et al., 2011), L-NAME as an eNOS inhibitor (Karpe et al., 2012) and compound C as an AMPK inhibitor (Luciana et al., 2011; Meijer et al., 2013). Pre-incubation with L-NAME, sirtinol and compound C exhibited elevated Emax values as compared with the HFD + OVX + E2 group. This clearly indicates an interplay between eNOS, AMPK and SIRT1 in mediating protective effects of E2 in PMS. This is in corroboration with the results obtained in the vascular reactivity studies.
Figure 8.

Mechanism underlying protective role of 17-β oestradiol (E2) in alleviating endothelial dysfunction and modulation of global post-translational histone modifications: SIRT1 and P-AMPK are deficient in PMS and E2 upregulates their expression. Immunoblots of P-AMPK and SIRT1 in aorta (A & B) and heart (C & D). Cumulative CRCs to Ang II in endothelium-intact thoracic aortic rings pre-incubated with L-NAME (100 μM), compound C (100 μM) and sirtinol (50 μM) (E). Immunoblots and representative quantitative expression of H3 acetylation in aorta and heart of all groups (F). Each data point is represented as mean ± SEM, n = 6–8 rats per group. *P < 0.05; **P < 0.01; ***P < 0.001; #P < 0.05; ##P < 0.01; ###P < 0.001. * vs. SHAM; # vs. HFD + OVX group.
PMS alters histone post-translational modifications
Chromatin structure is crucial for gene transcription and repression. Multiple histone modifications favour gene activation or repression (Li, 2002; Karpe et al., 2012). We studied the total histone H3 acetylation in the aorta and heart of all the groups. H3 acetylation levels were higher in the OVX, HFD and more in HFD + OVX groups than in the SHAM group. This elevation in acetylation was suppressed by E2 treatment. This decrease in acetylation appears to be associated with the increased SIRT1 levels in the HFD + OVX + E2 group (Figure 8F). Hence, these histone modifications may play a role in regulating the expressions of key proteins in the PMS condition, thus mediating endothelial dysfunction and cardiomyopathy.
Discussion
Our data clearly show that chronic exposure to E2 improves the clinical features of PMS leading to alleviation of endothelial dysfunction and cardiac apoptosis. Furthermore, we showed that the beneficial effect occurs through the modulation of SIRT1/AMPK/H3 acetylation.
To our knowledge, the present study is the first to demonstrate all the clinical features of PMS in female Sprague-Dawley rats; this was model was obtained by combining oestrogen deficiency and HFD-induced insulin resistance. Recently, Tominaga et al. also demonstrated the development of PMS by feeding HFD to ovariectomized mice for 4 weeks (Tominaga et al., 2011). In their study, with the exception of glucose intolerance, no significant difference was observed in the lipid profile of the HFD + OVX group. Further, we noticed that OVX alone increased the insulin resistance and cardiac apoptosis compared to the group fed a HFD. In previous studies the clinical features of metabolic syndrome were observed in OVX rats and found to be comparable to those of HFD rats (Gorres et al., 2010; Tominaga et al., 2011). However, these studies were of short duration (4 weeks), which might explain the incomplete development of insulin resistance in ovariectomized female rats. In our study, a sustained deficiency in oestrogen (60 days) evoked the clinical features of metabolic syndrome rather than HFD alone. Therefore, our study shows the complete development of PMS as evident by biochemical parameters and glucose intolerance thus featuring all the metabolic disturbances that occur in metabolic syndrome.
The second major finding of the present study is that chronic 17-β oestadiol therapy alleviates endothelial dysfunction in PMS. Endothelial dysfunction is the result of a disturbance in the balance between vasodilator and vasoconstrictor effectors. Endothelial dysfunction can be characterized by altered Ang II-mediated contractile responses and compromised CCh-mediated vasorelaxation (Gibbons, 1997). We characterized the endothelial dysfunction in the PMS condition as evidenced by altered responses to Ang II and CCh and these alterations were alleviated by E2 treatment. The enhanced vascular reactivity to Ang II may be due to an up-regulation of AT1 receptors or through some other mechanism like functional coupling with L-type of calcium channels (Arun et al., 2005; Karpe et al., 2012). Recently, Nishida et al. demonstrated that NO is invoved in the heterologous down-regulation of AT1 receptors (Nishida et al., 2011). In our study, activation of NO by E2 might explain the suppressed contractile responses to Ang II and reduced AT1 receptor expression. Previous studies have demonstrated that increased production of Ang II accounts for the increased expression of NAD(P)H oxidase subunits in porcine cultured endothelial cells and cardiomyocytes further leading to apoptosis (Seshiah et al., 2002; Qin et al., 2006). We found that thoracic aortic rings isolated from the PMS group were supersensitive to Ang II and produced more free radicals in response to Ang II, indicating an enhanced expression of NAD(P)H oxidase or activity in the vasculature of PMS rats and this was normalized by E2 treatment.
The third finding of the study is that PMS-induced cardiac apoptosis is prevented by chronic E2 treatment. Both insulin resistance and early ovariectomy have been established as independent risk factors for cardiac apoptosis (Rossi et al., 2002; Yue et al., 2005). Cardiac apoptosis is recognized as a predictor of cardiac disorders like heart failure. Both extrinsic and intrinsic pathways of apoptosis have been implicated in cardiac apoptosis. The relative balance of pro-apototic (PARP, Bax) and antiapoptotic (Bcl2, PCNA) molecules determines the cell fate (Liou et al., 2010). In our study we examined the effect of PMS on cardiac apoptosis and found that PMS promotes pro-apoptotic signals and causes cardiac apoptosis, which in the long run may lead to myocardial infarction and heart failure.
Endothelial dysfunction is associated with decreased NO availability, which may be due to loss of NO production or loss of biological activity. In the endothelium, eNOS is the major NO forming enzyme and phosphorylation of eNOS at Ser1177 is positively correlated with increased NO bioavailability (Mount et al., 2007). Recently the increase in expression of P-AMPK and SIRT1 has been positively correlated with eNOS phosphorylation, NO bioavailability and endothelial function (Zhang et al., 2006; Mattagajasingh et al., 2007). Both endogenous SIRT1 and its overexpression protect cardiomyocytes from apoptosis (Alcendor et al., 2004). An energy sensor AMPK is also involved in maintaining homeostasis of cardiomyocytes in stress (Russell et al., 2004). Hence, both molecules are important for protecting the cardiovascular system. A deficiency in SIRT1 and P-AMPK in PMS may explain the occurrence of cardiovascular dysfunction and can be positively correlated with serum oestradiol levels. Oestrogen therapy increased the expression of SIRT1 and P-AMPK supporting the idea that E2 mediates cardioprotection in PMS (Dyck and Lopaschuk, 2006; Donato et al., 2011).
Next we tried to address the role of SIRT1 and AMPK by using commercially available inhibitors of SIRT1 and AMPK, sirtinol and compound C, respectively, in ex vivo studies. Incubation of sirtinol with mouse isolated aortic rings has been shown to attenuate the ACh-induced relaxations (Donato et al., 2011). Compound C, a cell-permeable pyrrazolopyrimidine derivative, acts as a potent competitive inhibitor of AMPK. In vitro assays indicate that compound C blocks AMPK activity with reported IC50 values ranging from 0.1 to 0.2 μM (Zhou et al., 2001). Meijer et al. have recently shown that perivascular adipose tissue modulates the insulin-induced vasoreactivity in muscle. Insulin has been shown to vasodilate muscle microcirculation through AMPK and compound C treatment reduced the insulin-induced vasodilatation in muscles of db/db mice (Meijer et al., 2013). Furthermore, Rossini et al. found that simavastatin increased eNOS phosphorylation through AMPK and reduced the contractility of rat isolated mesenteric resistance arteries. Compound C (ex vivo) prevented the effects of simavastatin on eNOS phosphorylation and contractility (Luciana et al., 2011). Berberine has been shown to activate AMPK L6 skeletal muscles strongly and this effect is prevented by compound C (Cheng et al., 2006). Both compound C and siRNA knockdown of AMPK demonstrate that glucagon regulates ACC1 and ACC2 activity through AMPK (Peng et al., 2012). Further, RNA interference and chemical inhibition of AMPK led to a decrease in MITF protein levels and this was associated with decreased cell viability (Borgdorff et al., 2013). Also the ability of resveratrol to induce AMPK to boost cellular ATP and mtDNA copy number was prevented by knockdown of the AMPKa1 subunit or by treatment with compound C, an AMPK inhibitor (Price et al., 2012). We showed that acute treatment (short incubation) with these inhibitors enhanced Ang II induced contractile responses and hence ablated the protective effect of oestradiol. Interestingly, compound C elevated the maximal contraction to Ang II to a greater extent than sirtinol and L-NAME. This can be explained if we assume that compound C inhibits AMPK and another unknown molecule and hence removes the observed protection of vascular reactivity effected by oestradiol through some other pathway.
There is accumulating evidence to support a role for epigenetic mechanisms in various diseases like cancer, neurological disorders and metabolic disorders including diabetes (Berdasco and Esteller, 2011). Diverse histone modifications suggest a complex covalent language and are coordinated to regulate the structure of chromatin (Pirola et al., 2010). An increase in the expression of SIRT1, hence a decrease in total acetylation, induced by E2 in aorta and heart may be of clinical importance as far as cardiovascular dysfunction is concerned. Inhibition of histone de-acetylation and subsequent increase in acetylation has been shown to decrease eNOS activity and possibly endothelial dysfunction (Rossig et al., 2002). An overexpression of SIRT1 promotes vasodilatation by de-acetylating eNOS, which explains the fundamental role SIRT1 has in regulating vascular tone (Mattagajasingh et al., 2007). Therefore, it is tempting to hypothesize that the relative balance of histone acetylation/de-actylation will influence the endothelial function in PMS. As far as cardiac hypertrophy is concerned, H3 acetylation and histone actyltransferases p300 are involved in regulating cardiomyocyte growth (Backs and Olson, 2006; Haberland et al., 2009). The transcriptional activity of p300 is enhanced in response to Ang II or pathophysiological stress, which promotes the transcription of GATA4, SRF and MEF2 in mediating hypertrophy (Backs and Olson, 2006; Sahar et al., 2007). Also we have learned from our previous studies that histone H3 phosphorylation is responsible for epigenetic regulation of AT1 and AT2 receptor expression in the thoracic and abdominal aorta respectively (Karpe et al., 2012). This raises an interesting question: can PMS-associated cardiovascular dysfunction be prevented by modulation of epigenetic histone modifications? However, further studies are needed to elucidate this and the findings will be of profound clinical importance.
Fabris et al., Han et al. and Pelzer et al. have all shown an effect of oestradiol on the OVX and HFD rats. However, our aim was to investigate the combined effect of HFD + OVX on cardiovascular dysfunction and plausible alleviation by oestradiol treatment. Oestradiol treatment effectively suppressed the increased expression of the cardiac apoptotic gene (Bax) and associated increase in AT1/ACE1 mRNA expression in ovariectomized rats (Pelzer et al., 2005; Fabris et al., 2011). Also, oestradiol treatment prevented the down-regulation of eNOS signalling and hence the vascular endothelial dysfunction in diabetic female rats (Han et al., 2012). Oestradiol treatment has also been shown to protect against HFD-induced glucose intolerance and insulin resistance. Oestradiol treatment improved insulin signalling (AKT) in skeletal muscle. (Riant et al., 2009; Camporez et al., 2013). These studies prove beyond doubt that oestradiol therapy is effective at protecting against the detrimental effects of OVX as well as HFD.
A wealth of observational and epidemiological data suggest that women taking hormone therapy (oestradiol or combination of oestradiol and progesterone) have a lower incidence of cardiovascular disease than their counterparts who do not receive hormone therapy (Miller et al., 2005). However, results from the Women's Health Initiative and other studies revealed that many of the effects of hormone therapy were not observed in these randomized clinical trials (Herrington et al., 2000; Manson et al., 2003). There are plausible explanations for these discrepancies between clinical and observational studies. One possible explanation is that the large amount of observational data are complicated by healthy user bias. Secondly, most of the women started the hormone therapy at or near menopause in observational studies while in randomized clinical trials, like the Women's Health Initiative, the women had undergone the menopause several years ago and their arteries had already undergone significant damage (Harman et al., 2005b; Manson et al., 2006). These unresolved clinical issues were addressed in a newly designed ‘Kronos Early Oestrogen Prevention Study’, a 5-year randomized trial which evaluated the effectiveness of low-dose oral oestrogen and transdermal oestradiol in preventing the progression of atherosclerosis in recent post-menopausal women (Harman et al., 2005a; Manson, 2013). Several intriguing effects of hormone therapy were observed in this trial. There was significant improvement in vasomotor functions, little progression in atheroscelorsis, improved lipid profile (reduced LDL and increased HDL) and insulin sensitivity (Manson, 2013). This new information supports the findings obtained in numerous observational studies as well as our results.
Conclusion
We demonstrated that combining oestrogen deficiency with a high-fat diet induces insulin resistance in rats, which then leads to the development of PMS and a novel animal model for studying the underlying mechanisms of this condition. Our results suggest that PMS is associated with aggravated Ang II signalling and cardiac apoptosis. We demonstrated, for the first time, that the SIRT1/AMPK/histone H3 pathway is involved in the protective effects of oestrogen therapy, which effectively reverses the features of PMS and also alleviates the cardiovascular dysfunction associated with this condition (Figure 9).
Figure 9.
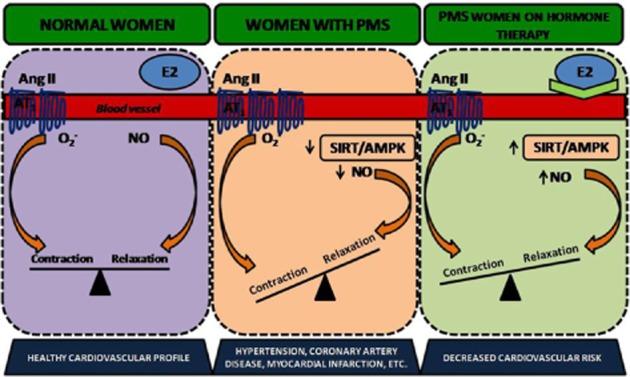
Schematic diagram of proposed pathway involved in mediating the protective role of oestrogen in treating PMS: PMS is associated with increased Ang II-induced contraction, increased ROS generation and decreased NO bioavailability, which is responsible for the compromised endothelial function. Apart from that, SIRT1 and P-AMPK are deficient in PMS. 17-β Oestradiol therapy in PMS condition is effective at generating NO via activation of the SIRT-1/AMPK pathway, thus mediating vasorelaxation, alleviating endothelial dysfunction and cardiomyopathy. Activation of SIRT-1/AMPK pathway thus serves as an excellent and effective approach to treat PMS and associated complications.
Glossary
- AMPK
AMP-activated PK
- Ang II
angiotensin II
- E2
17-β oestradiol
- PMS
post-menopausal metabolic syndrome
- SIRT1
silent information regulation 2 homologue
Conflict of interest
Authors declare no potential conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12290
Table S1 Effect of oestrogen therapy of BP and heart rate: All systemic parameters were recorded after proper development of oestrogen deficient condition, insulin resistance, Post-menopausal Metabolic Syndrome and oestrogen replacement. SBP-systolic; DBP-diastolic BP and HR: heart rate. Each data point is represented as mean ± SEM, n = 6–8 rats per group. (**) P < 0.01; (***) P < 0.001; (###) P < 0.001. (*) vs. SHAM; (#) vs. HFD + OVX.
Table S2 Tabular representation of apparent affinity constant (pD2-values): pD2 values for angiotensin II-mediated contractile responses, Phenylephrine-mediated contractile responses and carbachol-mediated relaxations in thoracic aorta. Each point is represented as mean ± SEM, n = 6–8 rats per group.
References
- Alcendor RR, Kirshenbaum LA, Imai S, Vatner SF, Sadoshima J. Silent information regulator 2, a longevity factor and class III histone deacetylase, is an essential endogenous apoptosis inhibitor in cardiac myocytes. Circ Res. 2004;95:971–980. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) 5th edition. Br J Pharmacol. 2011;164:S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun KHS, Kaul CL, Ramarao P. AT1 receptors and L-type calcium channels: functional coupling in supersensitivity to angiotensin II in diabetic rats. Cardiovasc Res. 2005;65:374–386. doi: 10.1016/j.cardiores.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Backs J, Olson EN. Control of cardiac growth by histone acetylation/deacetylation. Circ Res. 2006;98:15–24. doi: 10.1161/01.RES.0000197782.21444.8f. [DOI] [PubMed] [Google Scholar]
- Bebo BF, Fyfe-Johnson A, Adlard K, Beam AG, Vandenbark AA, Offner H. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. J Immunol. 2001;166:2080–2089. doi: 10.4049/jimmunol.166.3.2080. [DOI] [PubMed] [Google Scholar]
- Berdasco M, Esteller M. Hot topics in epigenetic mechanisms of aging: 2011. Aging Cell. 2011;11:181–186. doi: 10.1111/j.1474-9726.2012.00806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan MS, Fukunaga K. Characterization of an animal model of postmenopausal cardiac hypertrophy and novel mechanisms responsible for cardiac decompensation using ovariectomized pressure-overloaded rats. Menopause. 2010;17:213–221. doi: 10.1097/gme.0b013e3181b57489. [DOI] [PubMed] [Google Scholar]
- Bitar MS, Wahid S, Mustafa S, Al-Saleh E, Dhaunsi GS, Al-Mulla F. Nitric oxide dynamics and endothelial dysfunction in type 2 model of genetic diabetes. Eur J Pharmacol. 2005;511:53–64. doi: 10.1016/j.ejphar.2005.01.014. [DOI] [PubMed] [Google Scholar]
- Borgdorff V, Rix U, Winter GE, Gridling M, Muller AC, Breitwieser FP, et al. A chemical biology approach identifies AMPK as a modulator of melanoma oncogene MITF. Oncogene. 2013 doi: 10.1038/onc.2013.185. doi: 10.1038/onc.2013. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Camporez J, Jornayvaz F, Lee H-Y, Kanda S, Guigni BA, Kahn M, et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high-fat diet-induced hepatic and muscle insulin resistance. Endocrinology. 2013;154:1021–1028. doi: 10.1210/en.2012-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambliss KL, Shaul PW. Estrogen modulation of endothelial nitric oxide synthase. Endocrine Rev. 2002;23:665–686. doi: 10.1210/er.2001-0045. [DOI] [PubMed] [Google Scholar]
- Chen Z, Yuhanna IS, Galcheva-Gargova Z, Karas RH, Mendelsohn ME, Shaul PW. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J Clin Invest. 1999;103:401–406. doi: 10.1172/JCI5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Pang T, Gu M, Gao A-H, Xie C-M, Li J-Y, et al. Berberine-stimulated glucose uptake in L6 myotubes involves both AMPK and p38 MAPK. Biochim Biophys Acta (BBA)-Gen Subj. 2006;1760:1682–1689. doi: 10.1016/j.bbagen.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Colditz GA, Willett WC, Stampfer MJ, Rosner B, Speizer FE, Hennekens CH. Menopause and the risk of coronary heart disease in women. N Engl J Med. 1987;316:1105–1110. doi: 10.1056/NEJM198704303161801. [DOI] [PubMed] [Google Scholar]
- Donato AJ, Magerko KA, Lawson BR, Durrant JR, Lesniewski LA, Seals DR. SIRT1 and vascular endothelial dysfunction with ageing in mice and humans. J Physiol. 2011;589:4545–4554. doi: 10.1113/jphysiol.2011.211219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyck JRB, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabris B, Candido R, Bortoletto M, Toffoli B, Bernardi S, Stebel M, et al. Stimulation of cardiac apoptosis in ovariectomized hypertensive rats: potential role of the renin angiotensin system. J Hypertens. 2011;29:273–281. doi: 10.1097/HJH.0b013e328340d0d3. [DOI] [PubMed] [Google Scholar]
- Gaikwad A, Gupta J, Tikoo K. Epigenetic changes and alteration of Fbn1 and Col3A1 gene expression under hyperglycaemic and hyperinsulinaemic conditions. Biochem J. 2010;432:333–341. doi: 10.1042/BJ20100414. [DOI] [PubMed] [Google Scholar]
- Gibbons GH. Endothelial function as a determinant of vascular function and structure: a new therapeutic target. Am J Cardiol. 1997;79:3–8. doi: 10.1016/s0002-9149(97)00122-7. [DOI] [PubMed] [Google Scholar]
- Gorres BK, Bomhoff GL, Gupte AA, Geiger PC. Altered estrogen receptor expression in skeletal muscle and adipose tissue of female rats fed a high-fat diet. J Appl Physiol. 2010;110:1046–1053. doi: 10.1152/japplphysiol.00541.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta J, Gaikwad AB, Tikoo K. Hepatic expression profiling shows involvement of PKC epsilon, DGK eta, Tnfaip, and Rho kinase in type 2 diabetic nephropathy rats. J Cell Biochem. 2010;111:944–954. doi: 10.1002/jcb.22783. [DOI] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Li X, Zhou S, Meng G, Xiao Y, Zhang W, et al. 17β-estradiol antagonizes the down-regulation of ERα/NOS-3 signaling in vascular endothelial dysfunction of female diabetic rats. PLoS One. 2012;7:e50402. doi: 10.1371/journal.pone.0050402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman SM, Brinton EA, Cedars M, Lobo R, Manson JE, Merriam GR, et al. KEEPS: the Kronos early estrogen prevention study. Climacteric. 2005a;8:3–12. doi: 10.1080/13697130500042417. [DOI] [PubMed] [Google Scholar]
- Harman SM, Naftolin F, Brinton EA, Judelson DR. Is the estrogen controversy over? Deconstructing the Women's Health Initiative study: a critical evaluation of the evidence. Ann N Y Acad Sci. 2005b;1052:43–56. doi: 10.1196/annals.1347.004. [DOI] [PubMed] [Google Scholar]
- Herrington DM, Reboussin DM, Brosnihan KB, Sharp PC, Shumaker SA, Snyder TE, et al. Effects of estrogen replacement on the progression of coronary artery atherosclerosis. N Engl J Med. 2000;343:522–529. doi: 10.1056/NEJM200008243430801. [DOI] [PubMed] [Google Scholar]
- Ho JE, Mosca L. Postmenopausal hormone replacement therapy and atherosclerosis. Curr Atheroscler Rep. 2002;4:387–395. doi: 10.1007/s11883-002-0077-4. [DOI] [PubMed] [Google Scholar]
- Karpe PA, Gupta J, Marthong RF, Ramarao P, Tikoo K. Insulin resistance induces a segmental difference in thoracic and abdominal aorta: differential expression of AT1 and AT2 receptors. J Hypertens. 2012;30:132–146. doi: 10.1097/HJH.0b013e32834d9efe. [DOI] [PubMed] [Google Scholar]
- Katz SD. Mechanisms and implications of endothelial dysfunction in congestive heart failure. Curr Opin Cardiol. 1997;12:259–264. doi: 10.1097/00001573-199705000-00007. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CG, Carr MC, Murdoch SJ, Mitchell E, Woods NF, Wener MH, et al. Adipokines, inflammation, and visceral adiposity across the menopausal transition: a prospective study. J Clin Endocrinol Metab. 2009;94:1104–1110. doi: 10.1210/jc.2008-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. doi: 10.1038/nrg887. [DOI] [PubMed] [Google Scholar]
- Liou CM, Yang AL, Kuo CH, Tin H, Huang CY, Lee SD. Effects of 17beta-estradiol on cardiac apoptosis in ovariectomized rats. Cell Biochem Funct. 2010;28:521–528. doi: 10.1002/cbf.1687. [DOI] [PubMed] [Google Scholar]
- Luciana V, Mark W, Wenceslau CF, Al-abri M, Cobb C, Austin AC. Acute simvastatin increases endothelial nitric oxide synthase phosphorylation via AMP-activated protein kinase and reduces contractility of isolated rat mesenteric resistance arteries. Clin Sci. 2011;121:449–458. doi: 10.1042/CS20110259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane SI, Banerji M, Sowers JR. Insulin resistance and cardiovascular disease. J Clin Endocrinol Metab. 2001;86:713–718. doi: 10.1210/jcem.86.2.7202. [DOI] [PubMed] [Google Scholar]
- Manson JE. The kronos early estrogen prevention study. Womens Health. 2013;9:9–11. doi: 10.2217/whe.12.69. [DOI] [PubMed] [Google Scholar]
- Manson JE, Hsia J, Johnson KC, Rossouw JE, Assaf AR, Lasser NL, et al. Estrogen plus progestin and the risk of coronary heart disease. N Engl J Med. 2003;349:523–534. doi: 10.1056/NEJMoa030808. [DOI] [PubMed] [Google Scholar]
- Manson JE, Bassuk SS, Harman SM, Brinton EA, Cedars MI, Lobo R, et al. Postmenopausal hormone therapy: new questions and the case for new clinical trials. Menopause. 2006;13:139–147. doi: 10.1097/01.gme.0000177906.94515.ff. [DOI] [PubMed] [Google Scholar]
- Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer RI, Bakker W, Alta C-LAF, Sipkema P, Yudkin JS, Viollet B, et al. Perivascular adipose tissue control of insulin-induced vasoreactivity in muscle is impaired in db/db Mice. Diabetes. 2013;62:590–598. doi: 10.2337/db11-1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller VM, Clarkson TB, Harman SM, Brinton EA, Cedars M, Lobo R, et al. Women, hormones, and clinical trials: a beginning, not an end. J Appl Physiol. 2005;99:381–383. doi: 10.1152/japplphysiol.00248.2005. [DOI] [PubMed] [Google Scholar]
- Mittal G, Chandraiah G, Ramarao P, Ravi Kumar MNV. Pharmacodynamic evaluation of oral estradiol nanoparticles in estrogen deficient (ovariectomized) high-fat diet induced hyperlipidemic rat model. Pharm Res. 2009;26:218–223. doi: 10.1007/s11095-008-9725-x. [DOI] [PubMed] [Google Scholar]
- Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol. 2007;42:271–279. doi: 10.1016/j.yjmcc.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Nishida M, Ogushi M, Suda R, Toyotaka M, Saiki S, Kitajima N, et al. Heterologous down-regulation of angiotensin type 1 receptors by purinergic P2Y2 receptor stimulation through S-nitrosylation of NF-kB. Proc Natl Acad Sci U S A. 2011;108:6662–6667. doi: 10.1073/pnas.1017640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res. 2006;71:247–258. doi: 10.1016/j.cardiores.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Pelzer T, Jazbutyte V, Hu K, Segerer S, Nahrendorf M, Nordbeck P, et al. The estrogen receptor-alpha agonist 16 alpha-LE2 inhibits cardiac hypertrophy and improves hemodynamic function in estrogen-deficient spontaneously hypertensive rats. Cardiovasc Res. 2005;67:604–612. doi: 10.1016/j.cardiores.2005.04.035. [DOI] [PubMed] [Google Scholar]
- Peng IC, Chen Z, Sun W, Li Y-S, Marin TL, Hsu P-H, et al. Glucagon regulates ACC activity in adipocytes through the CAMKKβ/AMPK pathway. Am J Physiol Endocrinol Metab. 2012;302:E1560–E1568. doi: 10.1152/ajpendo.00504.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nat Rev Endocrinol. 2010;6:665–675. doi: 10.1038/nrendo.2010.188. [DOI] [PubMed] [Google Scholar]
- Price NL, Gomes AP, Ling AJY, Duarte FV, Martin-Montalvo A, North BJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puddu P, Puddu GM, Zaca F, Muscari A. Endothelial dysfunction in hypertension. Acta Cardiol. 2000;55:221–232. doi: 10.2143/AC.55.4.2005744. [DOI] [PubMed] [Google Scholar]
- Qin F, Patel R, Yan C, Liu W. NADPH oxidase is involved in angiotensin II-induced apoptosis in H9C2 cardiac muscle cells: effects of apocynin. Free Radic Biol Med. 2006;40:236–246. doi: 10.1016/j.freeradbiomed.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Kurz S, Manzel T, Tarpey M, Freeman BA, Griendling KK, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riant E, Waget A, Cogo H, Arnal J-F, Burcelin R, Gourdy P. Estrogens protect against high-fat diet-induced insulin resistance and glucose intolerance in mice. Endocrinology. 2009;150:2109–2117. doi: 10.1210/en.2008-0971. [DOI] [PubMed] [Google Scholar]
- Rossi R, Grimaldi T, Origliani G, Fantini G, Coppi F, Modena MG. Menopause and cardiovascular risk. Pathophysiol Haemost Throm. 2002;32:325–328. doi: 10.1159/000073591. [DOI] [PubMed] [Google Scholar]
- Rossig L, Li H, Fisslthaler B, Urbich C, Fleming I, Frastermann U, et al. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002;91:837–844. doi: 10.1161/01.res.0000037983.07158.b1. [DOI] [PubMed] [Google Scholar]
- Russell RR, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter MK, Parise H, Benjamin EJ, Levy D, Larson MG, Meigs JB, et al. Impact of glucose intolerance and insulin resistance on cardiac structure and function. Circulation. 2003;107:448–454. doi: 10.1161/01.cir.0000045671.62860.98. [DOI] [PubMed] [Google Scholar]
- Sahar S, Reddy MA, Wong C, Meng L, Wang M, Natarajan R. Cooperation of SRC-1 and p300 with NF-κB and CREB in angiotensin II-induced IL-6 expression in vascular smooth muscle cells. Arter Throm Vasc Biol. 2007;27:1528–1534. doi: 10.1161/ATVBAHA.107.145862. [DOI] [PubMed] [Google Scholar]
- Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD (P) H oxidase activity. Circ Res. 2002;91:406–413. doi: 10.1161/01.res.0000033523.08033.16. [DOI] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, et al. MSK2 and MSK1 mediate the mitogen-and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikoo K, Meena RL, Kabra DG, Gaikwad AB. Change in post-translational modifications of histone H3, heat shock protein 27 and MAP kinase p38 expression by curcumin in streptozotocin-induced type I diabetic nephropathy. Br J Pharmacol. 2008;153:1225–1231. doi: 10.1038/sj.bjp.0707666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga K, Yamauchi A, Egawa T, Tanaka R, Kawahara S, Shuto H, et al. Vascular dysfunction and impaired insulin signaling in high-fat diet fed ovariectomized mice. Microvasc Res. 2011;82:171–176. doi: 10.1016/j.mvr.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Touyz RM, El Mabrouk M, He G, Wu XH, Schiffrin EL. Mitogen-activated protein/extracellular signal regulated kinase inhibition attenuates angiotensin II mediated signaling and contraction in spontaneously hypertensive rat vascular smooth muscle cells. Circ Res. 1999;84:505–515. doi: 10.1161/01.res.84.5.505. [DOI] [PubMed] [Google Scholar]
- Vikram A, Jena G, Ramarao P. Pioglitazone attenuates prostatic enlargement in diet-induced insulin-resistant rats by altering lipid distribution and hyperinsulinaemia. Br J Pharmacol. 2010;161:1708–1721. doi: 10.1111/j.1476-5381.2010.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanad B, Srinivasan K, Kaul CL, Ramarao P. Effect of tempol on altered angiotensin II and acetylcholine-mediated vascular responses in thoracic aorta isolated from rats with insulin resistance. Pharmacol Res. 2006;53:209–215. doi: 10.1016/j.phrs.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Wang CCL, Goalstone ML, Draznin B. Molecular mechanisms of insulin resistance that impact cardiovascular biology. Diabetes. 2004;53:2735–2740. doi: 10.2337/diabetes.53.11.2735. [DOI] [PubMed] [Google Scholar]
- Wang Y, Huang S, Sah VP, Ross JJ, Brown JH, Han J, et al. Cardiac muscle cell hypertrophy and apoptosis induced by distinct members of the p38 mitogen-activated protein kinase family. J Bio Chem. 1998;273:2161–2168. doi: 10.1074/jbc.273.4.2161. [DOI] [PubMed] [Google Scholar]
- Yue TL, Bao W, Gu JL, Cui J, Tao L, Ma XL, et al. Rosiglitazone treatment in Zucker diabetic fatty rats is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion-induced myocardial injury. Diabetes. 2005;54:554–562. doi: 10.2337/diabetes.54.2.554. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lee TS, Kolb EM, Sun K, Lu X, Sladek FM, et al. AMP-activated protein kinase is involved in endothelial NO synthase activation in response to shear stress. Arterioscler Thromb Vasc Biol. 2006;26:1281–1287. doi: 10.1161/01.ATV.0000221230.08596.98. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.