

Abstract
BACKGROUND AND PURPOSE
4-Phenylbutyric acid (4-PBA) is a chemical chaperone that eliminates the accumulation of unfolded proteins in the endoplasmic reticulum (ER). However, its chaperoning ability is often weak and unable to attenuate the unfolded protein response (UPR) in vitro or in vivo. To develop more potent chemical chaperones, we synthesized six analogues of 4-PBA and evaluated their pharmacological actions on the UPR.
EXPERIMENTAL APPROACH
NRK-52E cells were treated with ER stress inducers (tunicamycin or thapsigargin) in the presence of each of the 4-PBA analogues; the suppressive effects of these analogues on the UPR were assessed using selective indicators for individual UPR pathways.
KEY RESULTS
2-POAA-OMe, 2-POAA-NO2 and 2-NOAA, but not others, suppressed the induction of ER stress markers GRP78 and CHOP. This suppressive effect was more potent than that of 4-PBA. Of the three major UPR branches, the IRE1 and ATF6 pathways were markedly blocked by these compounds, as indicated by suppression of XBP1 splicing, inhibition of UPRE and ERSE activation, and inhibition of JNK phosphorylation. Unexpectedly, however, these agents did not inhibit phosphorylation of PERK and eIF2α triggered by ER stress. These compounds dose-dependently inhibited the early activation of NF-κB in ER stress-exposed cells. 2-POAA-OMe and 2-POAA-NO2 also inhibited ER stress-induced phosphorylation of Akt.
CONCLUSION AND IMPLICATIONS
The 4-PBA analogues 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA strongly inhibited activation of the IRE1 and ATF6 pathways and downstream pathogenic targets, including NF-κB and Akt, in ER stress-exposed cells. These compounds may be useful for therapeutic intervention in ER stress-related pathological conditions.
Keywords: 4-phenylbutyric acid, chemical chaperone, endoplasmic reticulum (ER) stress, unfolded protein response (UPR), IRE1, ATF6, PERK–eIF2α
Introduction
The endoplasmic reticulum (ER) is an intracellular compartment responsible for post-translational modification and appropriate folding of newly synthesized proteins. Pathophysiological insults such as hypoxia, redox imbalance, change of calcium homeostasis and excessive protein synthesis disturb ER homeostasis, leading to the accumulation of unfolded and/or misfolded proteins in the ER, namely ER stress. ER stress-mediated tissue dysfunction is implicated in a wide range of diseases including cancers, infection, atherosclerosis, ischaemic injury, neurodegenerative disorders and metabolic diseases such as diabetes mellitus (Aridor and Balch, 1999). Hence, there is a need to find agents that attenuate ER stress and provide an effective treatment of ER stress-related disorders.
Chemical chaperones are a group of compounds that have the potential to stabilize proteins and/or facilitate protein folding. Among these, 4-phenylbutyric acid (4-PBA) is a well-known chemical chaperone. In several studies it has been demonstrated that 4-PBA acts as a chaperone to reverse the mislocalization and/or aggregation of proteins associated with human diseases (Burrows et al., 2000; Rubenstein and Zeitlin, 2000; Perlmutter, 2002). For example, 4-PBA reversed the accumulation of misfolded proteins in the ER, including ΔF508 cystic fibrosis transmembrane conductance regulator (Rubenstein and Zeitlin, 2000) and mutant α1-antitrypsin (Burrows et al., 2000). However, the chaperoning potential of 4-PBA is often weak and insufficient to attenuate ER stress, therefore, more efficient chemical chaperones are required for the effective attenuation of ER stress in vitro and in vivo.
There are three major transducers for the unfolded protein response (UPR) on the membrane of the ER: activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1) and PKR-like ER kinase (PERK) (Rutkowski and Kaufman, 2004). In response to ER stress, ATF6 translocates to the Golgi apparatus where it is cleaved by S1P and S2P, yielding a free cytoplasmic domain that functions as a transcription factor. Similarly, activated IRE1α catalyses the removal of a small intron from X-box-binding protein 1 (XBP1) mRNA. This splicing creates a translational frameshift in XBP1 to produce an active transcription factor. Activated ATF6 and XBP1 subsequently bind to the ER stress response element (ERSE) and the UPR element (UPRE), leading to the expression of target genes, which include 78 kDa glucose-regulated protein (GRP78). Activation of PERK leads to the phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) and the consequent induction of ER stress-responsive molecules including CCAAT/enhancer-binding protein-homologous protein (CHOP) (Walter and Ron, 2011).
In the present study, we aimed to develop potent chemical chaperones, which may be effective at attenuating pathogenic UPR. We synthesized six analogues of 4-PBA, evaluated their effects on individual UPR and identified three active compounds. Our results demonstrated that these three 4-PBA analogues strongly inhibit the induction of the IRE1 and ATF6 pathways and downstream pathogenic events including activation of NF-κB and phosphorylation of Akt in ER stress-exposed cells.
Methods
Reagents
2-Phenoxyacetic acid (2-POAA), 2-(4-methoxyphenoxy) acetic acid (2-POAA-OMe), sodium 2-(4-methoxyphenoxy) acetate (2-POAA-OMeNa), 2-(4-nitrophenoxy) acetic acid (2-POAA-NO2), 2-(pyridinium-1-yl) acetate (2-PyA) and 2-(2-naphathyloxy) acetic acid (2-NOAA) were synthesized by hydrolysis of corresponding acetic acid ethyl esters in 24–75% yields, which were obtained through a Williamson reaction or quaternization between phenolic compounds or pyridine and ethyl bromoacetate (Al-Amiery et al., 2011). The chemical structure and purity (95–99%) of these compounds were confirmed by 1H NMR analysis. The chemical structure of individual reagents is shown in Figure 1. 4-PBA was purchased from Calbiochem (San Diego, CA, USA), tunicamycin and thapsigargin were from Sigma-Aldrich (Tokyo, Japan), and TNF-α was from R&D Systems (Minneapolis, MN, USA). AB5 subtilase cytotoxin (SubAB) (Paton et al., 2006) was kindly provided by Dr James C. Paton (University of Adelaide, Australia). Valproic acid (VPA) was purchased from MP Biomedicals (Santa Ana, CA, USA), sodium butyrate (NaB) was from Alfa Aesar (Ward Hill, MA, USA), and suberoylanilide hydroxamic acid (SAHA) was from Cayman Chemical (Ann Arbor, MI, USA).
Figure 1.
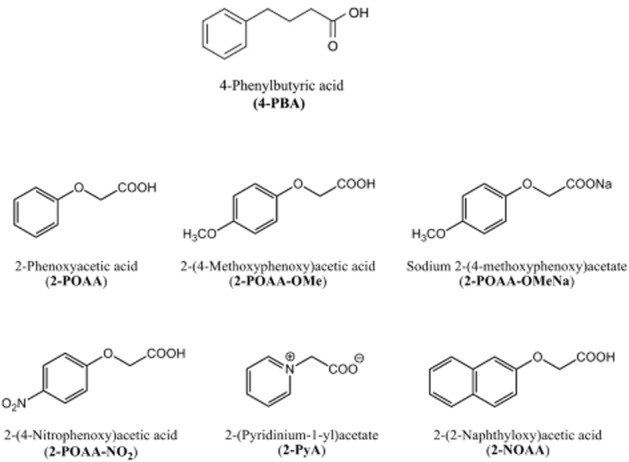
Chemical structure of 4-PBA and 4-PBA analogues. The chemical synthesis of 2-POAA, 2-POAA-OMe, 2-POAA-OMeNa, 2-POAA-NO2 and 2-NOAA was started from corresponding phenolic compounds that were reacted with ethyl bromoacetate in the presence of anhydrous potassium carbonate, followed by alkaline hydrolysis of the resultant esters. 2-PyA was synthesized by alkaline hydrolysis of ethyl 2-(pyridinium-1-yl) acetate that was prepared by the reaction of pyridine with ethyl bromoacetate.
Cell culture
The rat renal tubular epithelial cell line NRK-52E was purchased from American Type Culture Collection (Manassas, VA, USA). Cells were maintained in DMEM//Ham's F-12 (Gibco-BRL, Gaithersburg, MD, USA) supplemented with 5% FBS.
Establishment of stable transfectants
Using electroporation, NRK-52E cells were stably transfected with pcDNA3.1 (Invitrogen, Carlsbad, CA, USA) together with pUPRE-Luc (provided by Dr Laurie H. Glimcher, Harvard Medical School), pERSE-Luc (provided by Dr Laurie H. Glimcher) (Lee et al., 2003) or pNFκB-Luc (Panomics; Fremont, CA, USA) that introduces a luciferase gene under the control of UPRE, ERSE or the κB site respectively. Stable transfectants were selected by G418 (Sigma-Aldrich), and reporter clones of NRK/UPRE-Luc cells, NRK/ERSE-Luc cells and NRK/NFκB-Luc cells were established.
Transient transfection
To evaluate the cleavage of XBP1 mRNA, NRK-52E cells were transiently transfected with pCAX-F-XBP1ΔDBD-Luc (provided by Dr Takao Iwawaki, Gunma University, Japan) (Iwawaki and Akai, 2006) by electroporation. After 24 h, cells were seeded into 96-well plates, incubated for 24 h and subjected to stimulation. As a control, NRK-52E cells transfected with pSV40-Luc (pGL3-Control; Promega, Madison, WI, USA) were used. pSV40-Luc introduces a luciferase gene under the control of the simian virus 40 (SV40) promoter/enhancer.
Luciferase assay
Luciferase activity was evaluated by use of the Luciferase Assay System (Promega), according to the manufacturer's protocol.
Formazan assay
The number of viable cells was assessed by a formazan assay using Cell Counting Kit-8 (Dojindo Laboratory, Kumamoto, Japan).
Northern blot analysis
Total RNA was extracted by a single-step method, and Northern blot analysis was performed as described previously (Kitamura et al., 2011). cDNAs for GRP78 (provided by Dr Kazunori Imaizumi, Hiroshima University, Japan) (Katayama et al., 2001), CHOP (provided by Dr David Ron, University of Cambridge, UK) (Wang et al., 1998), GRP94 (provided by Dr Amy S. Lee, University of South California) (Gazit et al., 1999), 150 kDa oxygen-regulated protein (ORP150) (provided by Dr Satoshi Ogawa, Kanazawa University, Japan) (Ozawa et al., 1999), monocyte chemoattractant protein 1 (Rollins et al., 1988) and firefly luciferase were used to prepare radiolabelled probes. The expression of GAPDH and levels of 28S ribosomal RNA were used as loading controls.
Western blot analysis
Western blot analysis was performed as described previously (Kato et al., 2012). Primary antibodies used were: anti-phospho-eIF2α (Ser51) antibody from Cell Signaling Technology (Beverly, MA, USA), and anti-phospho-PERK (Thr980) antibody, anti-PERK antibody, anti-eIF2α antibody, anti-ATF4 (CREB-2) and anti-XBP1 antibody from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Levels of phosphorylated Akt and JNK were evaluated using PhosphoPlus Akt (Ser473) Antibody Kit and PhosphoPlus SAPK/JNK (Thr183/Tyr185) Antibody Kit, according to protocols provided by the manufacturer (Cell Signaling Biotechnology). As loading controls, levels of β-actin and lamin B1 were evaluated using anti-β actin antibody (Sigma-Aldrich) and anti-lamin B1 antibody (Invitrogen). Nuclear proteins were extracted using ProteoExtract Subcellular Proteome Extraction Kit (Merck KGaA, Darmstadt, Germany).
RT-PCR
Reverse transcription was performed using Omniscript Reverse Transcriptase (Qiagen, Valencia, CA, USA). Splicing of XBP1 mRNA was examined using the following primers: 5′-ACACGCTTGGGGATGAATGC-3′ and 5′-CCATGGGAAGATGTTCTGGG-3′ (Sigma-Aldrich), as described previously (Kato et al., 2012).
Analysis of histone deacetylase (HDAC) activity
HDAC activity was evaluated by using the Cyclex HDACs Deacetylase Fluorometric Assay Kit (Cyclex, Nagano, Japan) according to the manufacturer's instructions. Briefly, each compound was mixed with fluorescence-labelled substrate peptide, HDACs and lysyl endopeptidase, and incubated for 15 min. Fluorescence intensity was measured at an excitation wavelength of 355 nm and emission wavelength of 460 nm using a fluorescence microplate reader (Molecular Devices, Tokyo, Japan).
Statistical analysis
In reporter assays and microscopic analysis, experiments were performed in quadruplicate, and data are expressed as means ± SEM. Statistical analysis was performed using the non-parametric Mann-Whitney U-test to compare data in different groups. A P-value < 0.05 was considered to indicate a statistically significant difference.
Results
Suppression of ER stress response by 4-PBA analogues
NRK-52E cells were stimulated by an ER stress inducer thapsigargin (inhibitor of sarco/ER Ca2+ ATPase) in the absence or presence of 4-PBA analogues: 2-POAA, 2-POAA-OMe, 2-POAA-OMeNa, 2-POAA-NO2, 2-PyA and 2-NOAA. The cells were then subjected to Northern blot analysis of endogenous ER stress markers, GRP78 and CHOP. As shown in Figure 2B, D and F, 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA suppressed the induction of GRP78 and CHOP in a dose-dependent manner. Among these three compounds, 2-NOAA was most effective. In contrast, no suppressive effect was observed with the other 4-PBA analogues: 2-POAA, 2-POAA-OMeNa and 2-PyA (Figure 2A, C, E).
Figure 2.

Suppression of ER stress by 4-PBA analogues. (A–I) NRK-52E cells were pretreated with the indicated concentrations of 2-POAA (A), 2-POAA-OMe (B, G), 2-POAA-OMeNa (C), 2-POAA-NO2 (D, H), 2-PyA (E) or 2-NOAA (F, I) for 30 min, exposed to 500 nM thapsigargin (Tg) (A–F) or 2.5 μg·mL−1 of tunicamycin (Tm) (G–I) for 2 h and subjected to Northern blot analysis of GRP78 and CHOP. (J) Cells were pretreated with 2-NOAA or 4-PBA at indicated concentrations, exposed to Tg and subjected to Northern blot analysis. Expression of GAPDH is shown at the bottom as a loading control. (K, L) NRK-52E cells were exposed to Tm in the absence or presence of 2-POAA-NO2 for 48 h and subjected to phase-contrast microscopy (K) and quantitative assessment of vacuolated cells (L). Assays were performed in quadruplicate, and data are expressed as means ± SEM. Asterisks indicate statistically significant differences (P < 0.05).
To further confirm this result, cells were exposed to another ER stress inducer tunicamycin (inhibitor of protein glycosylation), and the effects of the 4-PBA analogues were re-tested. Consistent with the results in thapsigargin-treated cells, the induction of GRP78 and CHOP by tunicamycin was suppressed by 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA (Figure 2G–I), with 2-NOAA being the most effective. Again no suppressive effects were observed with the other 4-PBA analogues, 2-POAA, 2-POAA-OMeNa and 2-PyA.
Next, we compared the ability of 2-NOAA to attenuate ER stress with that of 4-PBA. Cells were treated with thapsigargin in the presence of increasing concentrations of 2-NOAA or 4-PBA and subjected to Northern blot analysis. The induction of GRP78 and CHOP by ER stress was suppressed by both 2-NOAA and 4-PBA in a dose-dependent manner. However, the suppressive effect of 2-NOAA was more potent than that of 4-PBA (Figure 2J). Similar, superior effects compared to 4-PBA, were also observed with 2-POAA-OMe and 2-POAA-NO2 (data not shown). The depression of ER stress markers by 2-POAA-NO2 was associated with a marked attenuation of cytoplasmic vacuolation (a typical morphological feature of renal tubular cells under ER stress in vitro and in vivo; Kitamura et al., 2011; Kato et al., 2012) (Figure 2K, L), which was not obvious after the treatment with 4-PBA (our unpublished observation).
Influence of 4-PBA analogues on IRE1 pathway
ER stress causes the induction of three major branches of the UPR: IRE1, ATF6 and PERK–eIF2α pathways. In the IRE1 pathway, activation of IRE1α at its endoribonuclease domain removes a small intron from XBP1 mRNA, leading to the production of functional XBP1 that then triggers activation of UPRE. Activation of IRE1α at its kinase domain also recruits TNF receptor-associated factor 2 (TRAF2) and thereby causes phosphorylation of JNK (Walter and Ron, 2011). To examine the effects of 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA on the IRE1 pathway, cells were treated with ER stress inducers in the presence of 4-PBA analogues and subjected to analysis of XBP1 splicing, activity of UPRE and phosphorylation of JNK. As shown in Figure 3A, the splicing of XBP1 mRNA induced by tunicamycin was suppressed by the treatment with 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA in a dose-dependent manner. Among these compounds, 2-NOAA was most effective. Similarly, the three agents inhibited the splicing of XBP1 elicited by thapsigargin dose-dependently and, again, the effect of 2-NOAA was most evident (Figure 3B). RT-PCR analysis also showed that splicing of XBP1 mRNA caused by ER stress was suppressed by the treatment with 2-NOAA (Figure 3C). The nuclear translocation of the XBP1 protein induced by ER stress was also blocked by this compound (Figure 3D). Consistent with these results, treatment with three compounds inhibited activation of UPRE by ER stress (Figure 3E). Quantitative analysis showed that 2-NOAA was the most effective at inhibiting the activation of UPRE by thapsigargin (IC50 = 1.59 mM) (Table 1). The inhibition induced by 2-POAA-OMe was less (IC50 = 1.92 mM) as was that induced by 2-POAA-NO2 (IC50 = 2.06 mM). The inhibitory effect of both agents was higher than that of 4-PBA (IC50 = 2.31 mM). This difference was not as a result of non-specific suppression of transcription or translation because the activity of luciferase driven by the SV40 promoter was not inhibited significantly by the treatment with these 4-PBA analogues (Figure 3F).
Figure 3.

Influence of 4-PBA analogues on the IRE1 pathway. (A, B) NRK-52E cells transfected with pCAX-F-XBP1ΔDBD-Luc were treated with Tm (A) or Tg (B) together with 4-PBA analogues for 9 h and subjected to the luciferase assay to evaluate the splicing of XBP1 mRNA. (C) NRK-52E cells were pretreated with 2-NOAA for 30 min, exposed to Tm for 4 h and subjected to RT-PCR analysis of XBP1 mRNA. XBP1(u), the unspliced form of XBP1; XBP1(s), the spliced form of XBP1. (D) Cells were pretreated with 5 mM 2-NOAA for 30 min and exposed to Tg for 2 h. Nuclear proteins were extracted from the cells and subjected to Western blot analysis of XBP1. The level of lamin B1 is shown at the bottom as a loading control. (E, F) NRK/UPRE-Luc cells (E) and NRK/SV40-Luc cells (F) were treated with Tg together with 4-PBA analogues for 9 h and subjected to luciferase assay. RLU, relative light unit. Assays were performed in quadruplicate. Data are expressed as means ± SEM, and asterisks indicate statistically significant differences (P < 0.05). (G, H) NRK-52E cells were pretreated with 4-PBA analogues for 30 min, treated with (G) or without (H) Tg for 15 min and subjected to Western blot analysis of phosphorylated JNK (p-JNK) and total JNK protein (JNK). (I) Cells were treated with 4-PBA analogues and subjected to Northern blot analysis of GRP78 and CHOP. (J, K) NRK/UPRE-Luc cells (J) and NRK/ERSE-Luc cells (K) were treated with 4-PBA analogues for 9 h and subjected to luciferase assay.
Table 1.
IC50 values of 4-PBA analogues and 4-PBA
| 2-POAA-OMe (mM) | 2-POAA-NO2 (mM) | 2-NOAA (mM) | 4-PBA (mM) | |
|---|---|---|---|---|
| ERSE activity | 1.88 | 1.93 | 0.48 | 2.59 |
| UPRE activity | 1.92 | 2.06 | 1.59 | 2.31 |
The suppression of the IRE1 pathway by 4-PBA analogues was further confirmed using JNK as an indicator. As shown in Figure 3G, thapsigargin-induced phosphorylation of JNK was inhibited by all three compounds. This suppressive effect was obvious and dose-dependent for 2-POAA-OMe and 2-POAA-NO2. Unexpectedly, the suppressive effect of 2-NOAA, the most potent inhibitor of GRP78 and CHOP, was weak. Paradoxically, the higher concentration of 2-NOAA was less effective. Subsequent experiments revealed that treatment with 2-NOAA, but not the other compounds, induced dose-dependent phosphorylation of JNK (Figure 3H). This effect was independent of ER stress because 2-NOAA, as well as 2-POAA-OMe and 2-POAA-NO2, did not induce the expression of GRP78 and CHOP (Figure 3I) or the activation of UPRE and ERSE in the absence of ER stress (Figure 3J, K).
Suppression of ATF6 pathway by 4-PBA analogues
Under ER stress conditions, ATF6 translocates to the Golgi apparatus where it is cleaved into an active transcription factor. Activated ATF6 moves into the nucleus and promotes ERSE activity, leading to expression of target genes including ER chaperones GRP78, GRP94 and ORP150 (Yoshida et al., 1998). To examine the effects of 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA on the ATF6 pathway, cells were treated with ER stress inducers in the presence of the 4-PBA analogues and subjected to Northern blot analysis. As shown in Figure 4A, B, ER stress-triggered induction of GRP78, GRP94 and ORP150 was suppressed by the treatment with these compounds. Of note, of the three agents, 2-NOAA was most effective.
Figure 4.

Suppression of ATF6 pathway by 4-PBA analogues. (A, B) NRK-52E cells were pretreated with 4-PBA analogues for 30 min, exposed to Tm (A) or Tg (B) for 2 h and subjected to Northern blot analysis of GRP78, GRP94 and ORP150. (C) NRK/ERSE-Luc cells were treated with 4-PBA analogues together with Tg for 9 h and subjected to luciferase assay. (D) NRK/ERSE-Luc cells were treated with Tg and 2-NOAA at indicated concentrations for 9 h and subjected to luciferase assay. Data are expressed as means ± SEM, and asterisks indicate statistically significant differences (P < 0.05).
To further confirm this result, the activity of ERSE was evaluated using reporter cells. Consistent with the results obtained by Northern blot, treatment with all three agents completely suppressed the activation of ERSE induced by ER stress (Figure 4C). This indicates that the ATF6 pathway may be a preferential target for the suppressive effects of the 4-PBA analogues. Indeed, a dose–response study showed that 2-NOAA significantly inhibited the activation of ERSE at 0.5 mM (Figure 4D). Consistent with the result of the Northern blot analysis, 2-NOAA was the most effective at inhibiting the activation of ERSE by thapsigargin (Table 1). The inhibitory effect of all three analogues was higher than that of 4-PBA.
Lack of inhibition of PERK and eIF2α phosphorylation by 4-PBA analogues
The PERK–eIF2α pathway is another branch of the UPR triggered by ER stress. We further examined the effects of the 4-PBA analogues on the PERK–eIF2α pathway. Cells were treated with thapsigargin in the absence or presence of 2-POAA-OMe, 2-POAA-NO2 or 2-NOAA and subjected to Western blot analysis of phosphorylated PERK. The results show that, unexpectedly, none of the 4-PBA analogues inhibited the phosphorylation of PERK in thapsigargin-treated cells (Figure 5A–C). Consistent with this result, treatment with these analogues did not suppress phosphorylation of eIF2α by ER stress (Figure 5D–F). To elucidate the mechanisms underlying this unexpected finding, we examined the effects of 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA on the activity of PERK and eIF2α under non-stress conditions. We found that treatment with the 4-PBA analogues induced dose-dependent phosphorylation of PERK (Figure 5G). 2-NOAA exhibited the most potent effect. Consistent with this, these analogues also induced phosphorylation of eIF2α (Figure 5H). These results suggest that the 4-PBA analogues differentially regulate the major branches of the UPR; that is, they selectively block activation of the IRE1 and ATF6 pathways without suppressing PERK and eIF2α.
Figure 5.

Lack of inhibition of PERK and eIF2α phosphorylation by 4-PBA analogues. (A–F) NRK-52E cells were pretreated with 4-PBA analogues for 30 min, exposed to Tg for 2 h, and subjected to Western blot analysis of phosphorylated PERK (p-PERK) (A–C) and phosphorylated eIF2α (p-eIF2α) (D–F). The levels of total PERK and eIF2α proteins are shown at the bottom as loading controls. (G, H) Cells were treated with 4-PBA analogues for 2.5 h and subjected to Western blot analysis.
Effects of the 4-PBA analogues on ER stress-triggered NF-κB activation and Akt phosphorylation
ER stress and the UPR contribute to pathological processes through several different mechanisms. One of the most important downstream targets is NF-κB that plays a role in the development of various inflammatory and malignant diseases (Zhang and Kaufman, 2008; Kitamura, 2009). Previous reports demonstrated that NF-κB activation is induced by ER stress, at least in part, through ATF6-mediated transient phosphorylation of Akt (Yamazaki et al., 2009). Hence, we determined the effects of the 4-PBA analogues on NF-κB activation triggered by ER stress. Treatment of reporter cells with thapsigargin induced activation of NF-κB, and this was suppressed by 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA in a dose-dependent manner (Figure 6A). Among the three agents, 2-NOAA was most effective. The suppressive effects of these compounds on NF-κB were further confirmed by Northern blot analysis (Figure 6B). Similar suppressive effects were also observed in the cells treated with a more selective inducer of ER stress, SubAB; treatment of reporter cells with SubAB induced activation of NF-κB, and this was suppressed by 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA dose-dependently (Figure 6C). As with thapsigargin-treated cells, 2-NOAA was the most effective. This result was further supported by Northern blot analysis of the reporter gene (Figure 6D). Of note, treatment with 4-PBA analogues alone did not affect basal NF-κB activity (Figure 6E). It is also worth noting that activation of NF-κB by TNF-α was not suppressed by the 4-PBA analogues, except for a modest inhibition by the highest concentration of 2-NOAA (Figure 6F).
Figure 6.

Effects of 4-PBA analogues on ER stress-triggered NF-κB activation and Akt phosphorylation. (A–D) NRK/NFκB-Luc cells were treated with Tg (A, B) or 20 ng·mL−1 of SubAB (C, D) together with indicated 4-PBA analogues for 8 h and subjected to luciferase assay (A, C) and Northern blot analysis of luciferase (B, D). Activity of luciferase was normalized by the number of viable cells estimated by formazan assay, and relative values (%) are shown. Data are expressed as means ± SEM, and asterisks indicate statistically significant differences (P < 0.05). In (B) and (D), the level of 28S ribosomal RNA is shown at the bottom as a loading control. (E) NRK/NFκB-Luc cells were treated with 4-PBA analogues for 8 h and subjected to luciferase assay. (F) NRK/NFκB-Luc cells were treated with 10 ng·mL−1 of TNF-α in the absence or presence of 4-PBA analogues for 8 h and subjected to luciferase assay. (G, H) NRK-52E cells were pretreated with 4-PBA analogues for 30 min, treated with (G) or without (H) Tg for 3 h and subjected to Western blot of phosphorylated Akt (p-Akt) and total Akt protein (Akt).
Activation of NF-κB by ER stress is, at least in part, through modulation of Akt phosphorylation. Akt is a multifunctional serine/threonine protein kinase that regulates cell metabolism, survival, and proliferation and immune cell functions including production of inflammatory cytokines. Hence, Akt has an important role in the development of cancers and inflammation (Lee et al., 2011; Cheung and Testa, 2013). We further tested whether activation of Akt by ER stress is influenced by the 4-PBA analogues. Western blot analysis showed that treatment with 2-POAA-OMe and 2-POAA-NO2 suppressed Akt phosphorylation induced by ER stress. However, 2-NOAA did not show any suppressive effects on Akt (Figure 6G). We found that the lack of suppression of Akt by this compound was because it has a stimulating effect on Akt. As shown in Figure 6H, 2-NOAA, but not the other analogues, increased the phosphorylation of Akt under non-stress conditions.
Discussion
Various pathological insults cause ER stress, which is involved in the pathogenesis of a wide range of diseases. Seeking substances that attenuate ER stress is an important challenge for therapeutic intervention in ER stress-related disorders. Hence, we synthesized analogues of 4-PBA and evaluated their inhibitory potential on ER stress. Of the six compounds tested, 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA were identified to be powerful, selective inhibitors of the IRE1 and ATF6 pathways and downstream pathogenic targets including NF-κB and Akt in ER stress-exposed cells. Our current findings are summarized in Table 2.
Table 2.
Effects of 4-PBA analogues on ER stress responses
| ER stress response | 2-POAA-OMe | 2-POAA-NO2 | 2-NOAA | |
|---|---|---|---|---|
| Non-stress condition | ||||
| IRE1 | Kinase | → | → | ↑ |
| Nuclease | → | → | → | |
| ATF6 | → | → | → | |
| PERK-eIF2α | ↑ | ↑ | ↑ | |
| NF-κB | → | → | → | |
| Akt | → | → | ↑ | |
| ER stress condition | ||||
| IRE1 | Kinase | ↓ | ↓ | ↓ |
| Nuclease | ↓ | ↓ | ↓ | |
| ATF6 | ↓ | ↓ | ↓ | |
| PERK-eIF2α | → | → | → | |
| NF-κB | ↓ | ↓ | ↓ | |
| Akt | ↓ | ↓ | → | |
↑ up-regulation.
↓ down-regulation.
→ no change.
In the compounds tested, the aromatic ring is bound to the carboxyl end group by an ether linkage (pyridinium in the case of 2-PyA) having shorter alkyl chain than that of 4-PBA. In particular, 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA contain the polar group and condensed ring respectively. Their suppressive effects on the UPR might be correlated with the nonionic carboxyl group and the electronic state changed by the polar group and condensed ring on the phenyl group.
ER stress transduces signals through the three major branches of the UPR: IRE1, ATF6 and PERK pathways. In theory, chemical chaperones reduce the level of ER stress and thereby attenuate activation of individual UPR branches under stress conditions. Indeed, all three compounds effectively suppressed the ATF6 pathway to the same extent. However, their effects on the IRE1 pathway varied. The kinase activity of IRE1α, indicated by JNK phosphorylation, was inhibited by 2-POAA-NO2 most effectively, with 2-POAA-OMe and 2-NOAA being less potent. In contrast, the endoribonuclease activity of IRE1α, indicated by XBP1 splicing and UPRE activation, was inhibited by 2-NOAA most effectively, whereas the suppressive effects of 2-POAA-NO2 and 2-POAA-OMe were modest. Interestingly, 2-NOAA, but not the others, induced activation of IRE1α kinase without activation of IRE1α endoribonuclease. Also 2-NOAA, but not the others, was able to activate Akt. Currently, it is unclear how 2-NOAA induces phosphorylation of JNK and Akt. However, like 2-NOAA, 4-PBA also has stimulating effects on JNK and Akt. For example, Roque et al. (2008) showed that 4-PBA induces the expression of IL-8 via activation of JNK in lung epithelial cells. Also Kim et al. (2012) reported that 4-PBA triggers phosphorylation of Akt in MCF-7 cells. Like butyrate, 4-PBA is a well-known HDAC inhibitor that activates a variety of signalling molecules (Davie, 2003). Previous reports have indicated the potential of HDAC inhibitors to activate JNK via generation of reactive oxygen species or deacetylation-mediated inhibition of mitogen-activated protein kinase phosphatase 1 (Yu et al., 2003; Chi and Flavell, 2008; Sharma et al., 2010). Similar mechanisms might be involved in the activation of JNK and Akt by 2-NOAA and 4-PBA. It is noteworthy that, in contrast to 2-NOAA and 4-PBA, 2-POAA-NO2 and 2-POAA-OMe did not activate JNK. Similarly, 2-NOAA activated Akt whereas 2-POAA-NO2 and 2-POAA-OMe did not. In addition to their enhanced ability to inhibit the UPR branches, the lack of JNK and Akt activation may be another important property of these compounds, which confirms their superiority over the prototype 4-PBA.
The current, most unexpected finding is that, although all three 4-PBA analogues effectively inhibited ER stress-induced expression of GRP78 and CHOP, they did not affect the phosphorylation of PERK and eIF2α induced by ER stress. Subsequent experiments revealed that these compounds per se up-regulated PERK and eIF2α phosphorylation. We found that a similar phenomenon was also observed in the cells treated with the prototype 4-PBA (Supporting Information Figure S1a, b). Currently, the underlying mechanism of this effect is not unknown. However, Kahali et al. recently identified GRP78 as a possible molecular target of some HDAC inhibitor; that is, treatment with SAHA led to GRP78 acetylation, dissociation and subsequent activation of its client protein PERK (Kahali et al., 2010). Downstream activation of eIF2α was also observed. A similar mechanism could be ascribed to phosphorylation of the PERK and eIF2α by the 4-PBA analogues and 4-PBA.
To examine whether or not the effects of 4-PBA analogues on PERK and eIF2α are mediated by inhibition of HDAC, the influences of these compounds on HDAC activity were evaluated in vitro. 4-PBA and NaB were used as positive controls. Like 4-PBA and NaB, 2-POAA-NO2 significantly inhibited HDAC activity. In contrast, 2-POAA-OMe and 2-NOAA, which were similarly potent at activating PERK and eIF2α, failed to inhibit HDAC (Supporting Information Figure S2). This result suggests that activation of PERK and eIF2α by the 4-PBA analogues is independent of HDAC activity. To further confirm this result, cells were treated with known HDAC inhibitors VPA, NaB or SAHA and subjected to analysis of phosphorylated PERK and eIF2α. Western blot analysis showed that none of these HDAC inhibitors triggered activation of the PERK–eIF2α pathway in NRK-52E cells (Supporting Information Figure S3), confirming an HDAC-independent action of the 4-PBA analogues. To date, there is no information regarding the mechanisms underlying the selective activation of PERK by 4-PBA and its analogues, but they could facilitate dimerization of PERK directly or dissociation of GRP78 from PERK, leading to its selective phosphorylation.
In general, activation of the PERK–eIF2α pathway contributes to the survival of cells under ER stress conditions, mainly due to translational suppression (Harding et al., 2000). However, the PERK–eIF2α pathway may also contribute to cellular damage by inducting activation of the ATF4–CHOP pathway (Fawcett et al., 1999; Ma et al., 2002). An interesting finding in the current study is that, although 4-PBA analogues activated the PERK–eIF2α pathway, the induction of CHOP by ER stress was markedly suppressed by treatment with these compounds. This was associated with suppression of nuclear translocation of ATF4 (Supporting Information Figure S4). Therefore, additional unknown mechanisms may exist and underlie the disruption of the link between phosphorylation of PERK/eIF2α and activation of ATF4 induced by the 4-PBA analogues; they could suppress transcription/translation of ATF4 or its nuclear transport. Further investigations are required to elucidate this issue, which would be the next step in our investigation.
ER stress contributes to inflammatory processes and tumourigenesis via activation of NF-κB and Akt. We found that all the 4-PBA analogues tested suppressed ER stress-induced NF-κB activation. This raises the possibility that 2-POAA-OMe, 2-POAA-NO2 and 2-NOAA may be useful for therapeutic intervention in ER stress-related, NF-κB/Akt-mediated pathologies including inflammations and cancers. Previous reports showed that some HDAC inhibitors inhibit activation of NF-κB triggered by various stimuli (Glauben et al., 2009). For example, SAHA can suppress NF-κB activation triggered by TNF-α, IL-1β, LPS, PMA and reactive oxygen species (Takada et al., 2006). However, in our experimental conditions, the suppressive effect of the 4-PBA analogues on NF-κB was not via HDAC inhibition but conceivably via attenuation of ER stress because they did not suppress activation of NF-κB triggered by TNF-α (Figure 6F).
ER stress is involved in the development of various diseases. The clinical utility of 4-PBA as a treatment for ER stress-related hereditary and acquired disorders has been suggested from many experimental studies (Maestri et al., 1996; Rubenstein and Zeitlin, 1998; Burrows et al., 2000). However, its clinical use is still limited because of its lack of potency at regulating UPR. Our present data suggest that the 4-PBA analogues described here may provide more efficient tools for the treatment of ER stress-related, NF-κB/Akt-mediated pathologies including chronic inflammation and malignant diseases.
Acknowledgments
We thank Dr Laurie H. Glimcher, Dr Kazunori Imaizumi, Dr Takao Iwawaki, Dr Amy S. Lee, Dr Satoshi Ogawa and Dr David Ron for providing us with plasmids. We also appreciate the kind gift of SubAB from Dr James Paton. This work was supported by Strategic Research Project Grant from University of Yamanashi to M. K. and H. S., and in part, by Grant-in-Aids for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (No. 20390235) to M. K.
Glossary
- 2-NOAA
2-(2-naphathyloxy) acetic acid
- 2-POAA
2-phenoxyacetic acid
- 2-POAA-NO2
2-(4-nitrophenoxy) acetic acid
- 2-POAA-OMe
2-(4-methoxyphenoxy) acetic acid
- 2-POAA-OMeNa
sodium 2-(4-methoxyphenoxy) acetate
- 2-PyA
2-(pyridinium-1-yl) acetate
- 4-PBA
4-phenylbutyric acid
- ATF6
activating transcription factor 6
- CHOP
CCAAT/enhancer-binding protein-homologous protein
- eIF2α
eukaryotic translation initiation factor 2α
- ER
endoplasmic reticulum
- ERSE
ER stress response element
- GRP78
78 kDa glucose-regulated protein
- HDAC
histone deacetylase
- IRE1
inositol-requiring enzyme 1
- NaB
sodium butyrate
- ORP150
150 kDa oxygen-regulated protein
- PERK
PKR-like ER kinase
- SAHA
suberoylanilide hydroxamic acid
- SubAB
AB5 subtilase cytotoxin
- SV40
simian virus 40
- TRAF2
TNF receptor-associated factor 2
- UPR
unfolded protein response
- UPRE
UPR element
- VPA
valproic acid
- XBP1
X-box-binding protein 1
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12306
Figure S1 Effects of 4-PBA on PERK–eIF2α pathway. (A, B) NRK-52E cells were pretreated with 4-PBA for 30 min, treated with (A) or without (B) 500 nM thapsigargin (Tg) for 2 h and subjected to Western blot analysis of phosphorylated PERK (p-PERK) and phosphorylated eIF2α (p-eIF2α). Levels of total PERK and eIF2α proteins are shown at the bottom as loading controls.
Figure S2 Effect of 4-PBA analogues on HDAC activity. Individual compounds (2.5 mM) were subjected to analysis of HDAC activity. The reaction mixtures were incubated for 15 min, and fluorescence was measured by a microplate reader. Data are presented as percentages of HDAC activity versus untreated control (100%) (means ± SE). *P < 0.05, NS, not statistically significant. 4-PBA (2.5 mM) and NaB (2.5 mM) were used as positive controls.
Figure S3 Lack of PERK and eIF2α phosphorylation by HDAC inhibitors. Cells were treated with VPA, NaB or SAHA for 2.5 h at indicated concentrations and subjected to Western blot analysis of p-PERK and p-eIF2α.
Figure S4 Effect of 2-NOAA on ER stress-induced translocation of ATF4. Cells were pretreated with 5 mM 2-NOAA for 30 min and exposed to Tg for 2 h. Nuclear proteins were extracted from the cells and subjected to Western blot analysis of ATF4. The level of lamin B1 is shown at the bottom as a loading control.
References
- Al-Amiery AA, Musa AY, Kadhum AAH, Mohamad AB. The use of umbelliferone in the synthesis of new heterocyclic compounds. Molecules. 2011;16:6833–6843. doi: 10.3390/molecules16086833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aridor M, Balch WE. Integration of endoplasmic reticulum signaling in health and disease. Nat Med. 1999;5:745–751. doi: 10.1038/10466. [DOI] [PubMed] [Google Scholar]
- Burrows JA, Willis LK, Perlmutter DH. Chemical chaperones mediate increased secretion of mutant α1-antitrypsin (α1-AT) Z: a potential pharmacological strategy for prevention of liver injury and emphysema in α1-AT deficiency. Proc Natl Acad Sci USA. 2000;97:1796–1801. doi: 10.1073/pnas.97.4.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013;13:234–244. doi: 10.2174/1568009611313030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H, Flavell RA. Acetylation of MKP-1 and the control of inflammation. Sci Signal. 2008;1:pe44. doi: 10.1126/scisignal.141pe44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003;133:2485S–2493S. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ. Complexes containing activating transcription factor (ATF)/cAMP-responsive element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response. Biochem J. 1999;339:135–141. [PMC free article] [PubMed] [Google Scholar]
- Gazit G, Lu J, Lee AS. De-regulation of GRP stress protein expression in human breast cancer cell lines. Breast Cancer Res Treat. 1999;54:135–146. doi: 10.1023/a:1006102411439. [DOI] [PubMed] [Google Scholar]
- Glauben R, Sonnenberg E, Zeitz M, Siegmund B. HDAC inhibitors in models of inflammation-related tumorigenesis. Cancer Lett. 2009;280:154–159. doi: 10.1016/j.canlet.2008.11.019. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R. Analysis of the XBP1 splicing mechanism using endoplasmic reticulum stress-indicators. Biochem Biophys Res Commun. 2006;350:709–715. doi: 10.1016/j.bbrc.2006.09.100. [DOI] [PubMed] [Google Scholar]
- Kahali S, Sarcar B, Fang B, Williams ES, Koomen JM, Tofilon PJ, et al. Activation of the unfolded protein response contributes toward the antitumor activity of vorinostat. Neoplasia. 2010;12:80–86. doi: 10.1593/neo.91422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama T, Imaizumi K, Honda A, Yoneda T, Kudo T, Takeda M, et al. Disturbed activation of endoplasmic reticulum stress transducers by familial Alzheimer's disease-linked presenilin-1 mutations. J Biol Chem. 2001;276:43446–43454. doi: 10.1074/jbc.M104096200. [DOI] [PubMed] [Google Scholar]
- Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1–JNK pathway. Cell Death Differ. 2012;19:310–320. doi: 10.1038/cdd.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HD, Jang CY, Choe JM, Sohn J, Kim J. Phenylbutyric acid induces the cellular senescence through an Akt/p21(WAF1) signaling pathway. Biochem Biophys Res Commun. 2012;422:213–218. doi: 10.1016/j.bbrc.2012.04.086. [DOI] [PubMed] [Google Scholar]
- Kitamura M. Biphasic, bidirectional regulation of NF-κB by endoplasmic reticulum stress. Antioxid Redox Signal. 2009;11:2353–2364. doi: 10.1089/ars.2008.2391. [DOI] [PubMed] [Google Scholar]
- Kitamura M, Kato H, Saito Y, Nakajima S, Takahashi S, Johno H, et al. Aberrant, differential and bidirectional regulation of the unfolded protein response towards cell survival by 3′-deoxyadenosine. Cell Death Differ. 2011;18:1876–1888. doi: 10.1038/cdd.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YG, Lee J, Byeon SE, Yoo DS, Kim MH, Lee SY, et al. Functional role of Akt in macrophage-mediated innate immunity. Front Biosci. 2011;16:517–530. doi: 10.2741/3702. [DOI] [PubMed] [Google Scholar]
- Ma Y, Brewer JW, Diehl Alan J, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Maestri NE, Brusilow SW, Clissold DB, Bassett SS. Long-term treatment of girls with ornithine transcarbamylase deficiency. N Engl J Med. 1996;335:855–859. doi: 10.1056/NEJM199609193351204. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Kuwabara K, Tamatani M, Takatsuji K, Tsukamoto Y, Kaneda S, et al. 150-kDa oxygen-regulated protein (ORP150) suppresses hypoxia-induced apoptotic cell death. J Biol Chem. 1999;274:6397–6404. doi: 10.1074/jbc.274.10.6397. [DOI] [PubMed] [Google Scholar]
- Paton AW, Beddoe T, Thorpe CM, Whisstock JC, Wilce MC, Rossjohn J, et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443:548–552. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- Perlmutter DH. Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr Res. 2002;52:832–836. doi: 10.1203/00006450-200212000-00004. [DOI] [PubMed] [Google Scholar]
- Rollins BJ, Morrison ED, Stiles CD. Cloning and expression of JE, a gene inducible by platelet-derived growth factor and whose product has cytokine-like properties. Proc Natl Acad Sci USA. 1988;85:3738–3742. doi: 10.1073/pnas.85.11.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roque T, Boncoeur E, Saint-Criq V, Bonvin E, Clement A, Tabary O, et al. Proinflammatory effect of sodium 4-phenylbutyrate in ΔF508-cystic fibrosis transmembrane conductance regulator lung epithelial cells: involvement of extracellular signal-regulated protein kinase 1/2 and c-Jun-NH2-terminal kinase signaling. J Pharmacol Exp Ther. 2008;326:949–956. doi: 10.1124/jpet.107.135186. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in ΔF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med. 1998;157:484–490. doi: 10.1164/ajrccm.157.2.9706088. [DOI] [PubMed] [Google Scholar]
- Rubenstein RC, Zeitlin PL. Sodium 4-phenylbutyrate downregulates Hsc70: implications for intracellular trafficking of ΔF508-CFTR. Am J Physiol. 2000;278:C259–C267. doi: 10.1152/ajpcell.2000.278.2.C259. [DOI] [PubMed] [Google Scholar]
- Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Sharma V, Koul N, Joseph C, Dixit D, Ghosh S, Sen E. HDAC inhibitor, scriptaid, induces glioma cell apoptosis through JNK activation and inhibits telomerase activity. J Cell Mol Med. 2010;14:2151–2161. doi: 10.1111/j.1582-4934.2009.00844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada Y, Gillenwater A, Ichikawa H, Aggarwal BB. Suberoylanilide hydroxamic acid potentiates apoptosis inhibits invasion and abolishes osteoclastogenesis by suppressing nuclear factor-κB activation. J Biol Chem. 2006;281:5612–5622. doi: 10.1074/jbc.M507213200. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–5017. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, et al. Activation of the Akt-NF-κB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol. 2009;183:1480–1487. doi: 10.4049/jimmunol.0900017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273:33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- Yu C, Subler M, Rahmani M, Reese E, Krystal G, Conrad D, et al. Induction of apoptosis in BCR/ABL+ cells by histone deacetylase inhibitors involves reciprocal effects on the RAF/MEK/ERK and JNK pathways. Cancer Biol Ther. 2003;2:544–551. doi: 10.4161/cbt.2.5.454. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455–462. doi: 10.1038/nature07203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.