

Abstract
BACKGROUND AND PURPOSE
AVE 0991 (AVE) is a non-peptide compound, mimic of the angiotensin (Ang)-(1–7) actions in many tissues and pathophysiological states. Here, we have investigated the effect of AVE on pulmonary remodelling in a murine model of ovalbumin (OVA)-induced chronic allergic lung inflammation.
EXPERIMENTAL APPROACH
We used BALB/c mice (6–8 weeks old) and induced chronic allergic lung inflammation by OVA sensitization (20 μg·mouse−1, i.p., four times, 14 days apart) and OVA challenge (1%, nebulised during 30 min, three times per·week, for 4 weeks). Control and AVE groups were given saline i.p and challenged with saline. AVE treatment (1 mg·kg−1·per day, s.c.) or saline (100 μL·kg−1·per day, s.c.) was given during the challenge period. Mice were anaesthetized 72 h after the last challenge and blood and lungs collected. In some animals, primary bronchi were isolated to test contractile responses. Cytokines were evaluated in bronchoalveolar lavage (BAL) and lung homogenates.
KEY RESULTS
Treatment with AVE of OVA sensitised and challenged mice attenuated the altered contractile response to carbachol in bronchial rings and reversed the increased airway wall and pulmonary vasculature thickness and right ventricular hypertrophy. Furthermore, AVE reduced IL-5 and increased IL-10 levels in the BAL, accompanied by decreased Ang II levels in lungs.
CONCLUSIONS AND IMPLICATIONS
AVE treatment prevented pulmonary remodelling, inflammation and right ventricular hypertrophy in OVA mice, suggesting that Ang-(1–7) receptor agonists are a new possibility for the treatment of pulmonary remodelling induced by chronic asthma.
Keywords: AVE 0991, angiotensin-(1–7), allergic lung inflammation, asthma, airway remodelling, cytokines, Mas receptor
Introduction
Asthma is a major chronic respiratory disease of childhood (Pedersen et al., 2011) involving airflow limitation and a variable decline in lung function related to the duration and severity of the disease, as well as, genetic and environmental interactions. The persistent airflow limitation, decreased lung function and airway responsiveness are related to structural changes in the airways, referred to as remodelling (Tagaya and Tamaoki, 2007; Cho, 2011; Durrani et al., 2011). Airway remodelling is characterized by the loss of epithelial integrity and denudation of the basement membrane, subepithelial fibrosis, hyperplasia and hypertrophy of smooth muscle cells (SMCs), hyperplasia and hypertrophy of submucosal glands with mucus production, obstruction and thickening of the airway walls, loss of cartilage integrity and angiogenesis (Bergeron et al., 2010). These structural changes are caused by different immune-related mediators or cell mechanisms (Manso et al., 2012). Eosinophils, T-lymphocytes, mast cells, epithelia, macrophages, airway SMCs and fibroblasts are the main cells involved in airway remodelling (Cho, 2011; Girodet et al., 2011).
The pathophysiological mechanisms underlying airway remodelling have not yet been fully clarified. However, angiotensin II (Ang II), the main effector of the renin-angiotensin system (RAS), is involved in the pathophysiology of airway remodelling (McKay et al., 1998; Du et al., 2004; Wang et al., 2008). Ang II induced hypertrophy and TGF-β1 mRNA expression in human airway SMCs, as well as increasing the release of TGF-β1, suggesting that Ang II participates in the growth of SMC, in the airway remodelling process (McKay et al., 1998). Expression of TGF-β1, a potent profibrogenic factor, is increased in asthmatic airways and it is a primary candidate for the initiation and persistence of airway remodelling in asthma (Yang et al., 2012). Studies have also indicated that the effects of Ang II on airway remodelling are mediated by the angiotensin AT1 receptor (receptor nomenclature follows Alexander et al., 2011). Valsartan, an AT1 receptor antagonist, inhibited the inflammatory cell influx and attenuated structural airway changes and inflammation in rats chronically exposed to ovalbumin (OVA; Wang et al., 2008). Further, valsartan suppressed the synthesis of collagen III and V by down-regulating TGF-β1 mRNA and protein expression (Du et al., 2004).
In addition to Ang II, Ang-(1–7) has become, in recent years, another RAS peptide of considerable interest because its effects counter those of Ang II, mainly in the cardiovascular system (Santos et al., 2005; Ferreira et al., 2012). Ang-(1–7) is principally synthesized by ACE2 and interacts with the GPCR Mas (Santos et al., 2003; 2005). In an acute model of OVA-induced allergic asthma, Ang-(1–7), via Mas, inhibited the increase in total cell counts of eosinophils, lymphocytes and neutrophils El-Hashim et al. (2012). In the airways, Ang-(1–7) also reduced perivascular and peribronchial inflammation, fibrosis and goblet cell hyperplasia/metaplasia. Moreover, Ang-(1–7) attenuated the OVA-induced increase in the phosphorylation of IκB-α and ERK 1/2, suggesting that Ang-(1–7) could mediate an anti-inflammatory pathway in allergic asthma.
Although there has been considerable progress in our understanding of the pathophysiology of asthma, the prevention and treatment of airway remodelling still constitute a significant problem. To date, long-term medication in asthma treatment mainly focuses on anti-inflammatory effects and there are no therapeutic interventions that can reverse established remodelling of the airways (Zhang and Li, 2011). AVE 0991 (AVE) is a synthetic compound that is a mimic of Ang-(1–7) and is considered an agonist at Mas receptors (Wiemer et al., 2002; Santos and Ferreira, 2006). AVE is capable of reproducing the anti-fibrotic, anti-proliferative and anti-inflammatory effects of Ang-(1–7) (Wiemer et al., 2002; Pinheiro et al., 2004; Santos and Ferreira, 2006; Lubel et al., 2009; da Silveira et al., 2010; Barroso et al., 2012; Marques et al., 2012). In the present study, we tested the hypothesis that treatment with AVE could attenuate inflammation and the structural and functional changes induced by airway remodelling in a murine model of OVA-induced chronic allergic lung inflammation.
Methods
Animals
All animal care and experimental procedures in this study were approved by the Ethics Committee for Animal Experimentation of the Federal University of Minas Gerais, Brazil (CETEA; Proc#253/10). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Seventy-one male BALB/c mice (6–8 weeks of age) weighing 20–25 g, were divided into four groups: Control (CTRL; n = 17) and AVE groups (n = 12) that were treated with saline injections (i.p.) and exposed to saline ‘challenge’; a group of OVA-sensitized and challenged mice (OVA; n = 21) and a group of OVA-sensitized and challenged mice treated with AVE (OVA + AVE; n = 21). The animals were housed under a 12 h light–dark cycle (lights at 0600 h) with free access to standard chow and water.
OVA immunization and challenge
Chronic allergic lung inflammation was induced, as previously described by Vieira et al. (2008). Briefly, as shown in Figure 1, mice received four doses of OVA (20 μg diluted in 0.5 mL of saline, i.p.) 14 days apart (0, 14, 28 and 42 days) and on the 21st day, the mice were exposed to nebulized OVA (1% in saline), for 30 min, three times·per week, for 30 days, as the challenge. Mice of the CTRL and AVE groups were subjected to four injections of saline (NaCl 0.9%, 0.2 mL, i.p.) and were nebulized with saline only, using the same regimen as the OVA mice. Nebulization was performed in an acrylic box (30 × 15 × 20 cm) coupled to an ultrasonic nebulizer. OVA was purchased from Sigma (St. Louis, MO, USA).
Figure 1.
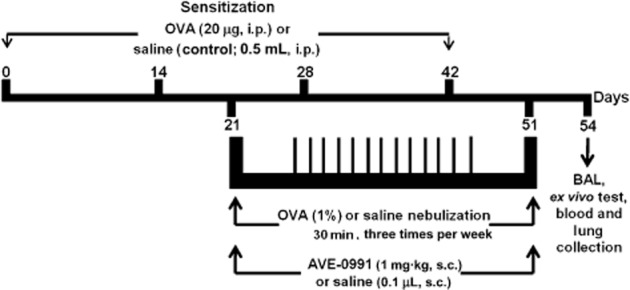
Experimental protocol: Mice received four i.p. injections of either OVA solution (20 μg per mouse) or saline (0.5 mL per mouse) on days 0, 14, 28 and 42. From day 21 to 51, mice were challenged with OVA nebulization (1%) or saline (three times per week, for 30 min). During the period of challenge, mice were treated with AVE (1 mg·kg−1, s.c.) or saline. The animals were killed 72 h after the last challenge (at day 54).
Treatment with AVE and A-779
Along with inhalation, half of the mice of CTRL and OVA groups were treated daily with AVE (1 mg·kg−1 diluted in 10 mM KOH and 1 mL of NaCl 0.9%, s.c.). AVE was a generous gift from Dr Juergen Puenter from Aventis Pharma.
In a smaller group of animals (n = 5), OVA + AVE animals also received the Mas receptor antagonist, A-779 (DAla7-Ang-(1-7). A-779 (1.5 μg h−1, 0.11 μl h−1; Bachem, Torrance, CA, USA) was continuously infused during the last 28 days of the protocol. Infusions were made with osmotic mini-pumps (Alzet, model 1004; DURECT Corporation, Cupertino, CA, USA) which were implanted subcutaneously in the interscapular region under anaesthesia (ketamine, 0.5 mg·kg−1 and xylazine, 0.43 mg·kg−1, i.p.; Robifarma Indústria Farmacêutica Ltda, Brazil).
Blood and lung samples
Seventy-two hours following the last challenge, some of the animals were anaesthetized with a mixture of ketamine (0.5 mg·kg−1) and xylazine (0.43 mg·kg−1; Robifarma Indústria Farmacêutica Ltda, Brazil), given i.p.. A midline neck incision was made and the trachea and jugular vein were exposed. After collecting blood from the jugular vein, the abdominal aorta and vena cava were cut. The trachea was then clamped and the lungs were collected in functional residual capacity. In some animals, the left lung and right ventricle were collected for morphometric analysis and the right lung was removed, snapped frozen in dry ice and kept at −80°C until assayed. The lungs of the remaining animals were used to evaluate airway responsiveness.
Levels of serum antibody
Anti-OVA IgE was measured as described by Saldanha et al. (2004). Serum samples were obtained from all groups following antigen exposure. Anti-OVA IgE antibodies were measured by capture-elisa using plates (Maxsorb, Nunc, Hampton, NH) coated with rat anti-mouse IgE (Southern Biotechnology Associates, Birmingham, AL), 50 μL of total serum and biotinylated OVA. The results are reported as arbitrary units (A.U.) using a positive reference serum assigned to be 1000 units.
Airway responsiveness test
Seventy-two hours following the last challenge with OVA, mice were killed and the primary bronchi of each animal was removed, cut in small rings (2–3 mm) and placed in vertical chambers (internal volume 10 mL) filled with Krebs–Henseleit solution (mmol·L−1): NaCl 110.8, KCl 5.9, NaHCO3 25.0, MgSO4 1.07, CaCl2 2.49, NaH2PO4 2.33 and glucose 11.51, oxygenated with carbogen gas (95% O2/5% CO2) at 37 ± 0.2°C under a tension of 1.0 g, for 1 h to equilibrate. Mechanical activity, recorded isometrically by a force transducer, was fed to an amplifier-recorder (Powerlab 4/20, ADInstruments, Inc., Colorado Springs, CO, USA) and to a personal computer equipped with an analogue-to-digital converter board (AD16JR; World Precision Instruments, Inc., Sarasota, FL, USA), using cell voltage monitoring system data acquisition/recording software (World Precision Instruments, Inc.). To investigate airway contractile response, carbachol was added in increasing cumulative concentrations (100 pmol−100 μmol·L−1) to the isolated bronchial rings and a concentration-response curve was obtained.
Bronchoalveolar lavage (BAL)
Seventy-two hours following the last nebulization, CTRL, OVA and OVA + AVE (n = 5–6) mice were anaesthetized with the mixture of ketamine and xylazine (0.5 mg·kg−1 and 0.43 mg·kg−1, respectively; i.p.), the trachea was exposed and the mice were placed with the head elevated 30° from the horizontal plane. Next, a 16-G cannula was inserted into the trachea and lungs were gently rinsed twice with 0.5 mL of saline. The BAL fluid collected was centrifuged (600× g for 8 min at 4°C) and the supernatant was frozen and stored at −80°C until assayed.
Airway and vessel morphometry
Lung tissue was fixed in formalin and embedded in paraffin. Sections (4 μm thick) were stained with haematoxylin and eosin for histological analysis. The ratio of the thickness of the muscle and bronchial epithelial layer was calculated by counting the number of points hitting muscle areas divided by the number of intercepts crossing the epithelial basement membrane (Vieira et al., 2008). The measurements were performed on five airway images (bronchiole) from each animal (four to five mice in each group). The muscle layer of five arterioles in each animal (four to five mice in each group) was also evaluated. A binary image was created and submitted to digital processing for the calculation of the thickness of the muscle layer of the arterioles. The external perimeter (EP), defined by the last layer of smooth muscle in the arterioles, was determined first, followed by the internal perimeter (IP) of the arterioles, delimited by the endothelium. The ratio between these perimeters (EP/IP) was considered to be the thickness index of the muscle layer. All measurements were obtained using the KS300 image analyzer (Carl Zeiss, Oberkochen, Germany).
Morphometric analysis of heart
The right ventricles were dehydrated by sequential washes with 70, 80, 90 and 100% ethanol and embedded in paraffin The tissue was cut longitudinally in 6 μm sections. at intervals of 30 μm and stained with haematoxylin and eosin for the analysis of cell morphology. Heart hypertrophy was evaluated by morphometry of cardiomyocytes; 350 cells were counted per group (n = 7), except for the group CTRL, in which 250 cells were counted (n = 5). Cardiac hypertrophy was assessed with the aid of a light microscope (BX 60, Olympus) with a ruler attached to the eyepiece, each section of which corresponded to 2.5 μm (Marques et al., 2011). Cardiomyocytes were examined with a 40× objective and 50 cells per animal were measured. Only cardiomyocytes with well-defined nuclei and cell boundaries were measured.
Expression of angiotensin receptor protein in the lung by Western blotting
Samples of protein (50 mg) extracted from the lung (n = 6 from each group) were applied to polyacrylamide gel/SDS 10% and then transferred to nitrocellulose membranes. The membranes were incubated with primary antibody for MAS (rabbit anti-angiotensin-(1–7) MAS receptor, 1:500, Alamone Labs, Jerusalem, Israel) or AT1 (rabbit anti-AT1 receptor, 1:1000, Alamone Lab) or AT2 (rabbit anti-AT2 receptor, 1:1000, Alamone Lab) receptors followed by a fluorescent secondary antibody [IRDye ® 680 conjugated goat (polyclonal) anti-Rabbit IgG (H + L) diluted to 1:10 000 (Li-COR Biosciences, Lincoln, NE, USA)]. Levels were normalized to GAPDH levels in the same sample. Staining was visualized and quantified in a Li-COR Odyssey Scanner.
Blood sample collection
Blood (1 mL) was collected from the jugular vein into chilled polypropylene tubes containing 1 mM p-hydroxymercury benzoate, 30 mM 1,1 o-phenanthroline, 1 mM PMSF, 1 mM pepstatin A and 7.5% EDTA (50 μL·mL−1 of blood). After centrifugation, plasma samples were frozen and stored at −80°C until assayed.
Measurement of Ang-(1–7) and Ang II
Peptides from plasma and lung homogenates were extracted onto a Bond-Elut phenyl silica cartridge (Varian, Inc., Santa Clara, CA, USA). The columns were pre-activated by sequential washes with 99.9% acetonitrile/0.1% heptafluorobutyric acid (HFBA) and 0.1% HFBA. Sequential washes with 10 mL of 99.9% acetonitrile/0.1% HFBA, 10 mL of 1% HFBA, 3 mL of 0.1% HFBA containing 0.1% BSA, 10 mL 10% acetonitrile/0.1% HFBA and 3 mL of 0.1% HFBA were used to activate the columns and increase peptide recovery. After the application of the sample, the columns were washed with 0.1% HFBA and 3 mL of 20% acetonitrile/0.1% HFBA. The adsorbed peptides were eluted with 3 mL of 99.9% acetonitrile/0.1% HFBA into polypropylene tubes rinsed with 1% BSA. After evaporation, peptide levels were measured by RIA, as described previously (Botelho et al., 1994; Santos et al., 2004).
Measurement of cytokine levels
The concentration of the cytokines, IL-4, IL-5 and IL-10, in BAL and lung homogenates were measured using commercially available antibodies and according to the procedures supplied by the manufacturer (R&D Systems, Minneapolis, MN, USA). Results are expressed as pg·mL−1 of plasma or pg·mg−1 of lung wet weight. The detection limit of the elisa assay was in the range of 4–8 pg·mL−1.
Data analysis
All results are expressed as mean ± SEM. Comparisons of angiotensin peptides levels, receptors expression and airway and vessel morphometry were performed using one-way anova followed by the Newman–Keuls post test. Kruskal–Wallis with Dunn's multiple comparison test was used to assess differences in right ventricular hypertrophy. Contractile response to carbachol in isolated mouse bronchial rings was evaluated using two-way anova followed by the Bonferroni post test. All analysis and graphics were performed with the Graphpad Prism software (GraphPad Software Inc., version 5.0; La Jolla, CA, USA). The level of significance was set to P < 0.05.
Results
In order to confirm the development of allergic lung inflammation, serum antibody, anti-OVA IgE, was measured. IgE was increased in the asthmatic group (154 ± 21 A.U.) in comparison with CTRL mice (11 ± 1.5 A.U.). In addition, OVA mice treated with AVE presented an attenuation of IgE levels (51 ± 8 A.U.).
Airway responsiveness test
As seen in Figure 2, a significant attenuation of the contractions of bronchial rings in response to carbachol was observed in OVA mice, suggesting that OVA treatment has induced structural alterations that diminished the bronchoconstriction induced by muscarinic receptor stimulation. OVA animals treated with AVE displayed an attenuation of this response. The response to carbachol in the group of mice treated with saline and AVE did not differ from the CTRL group.
Figure 2.
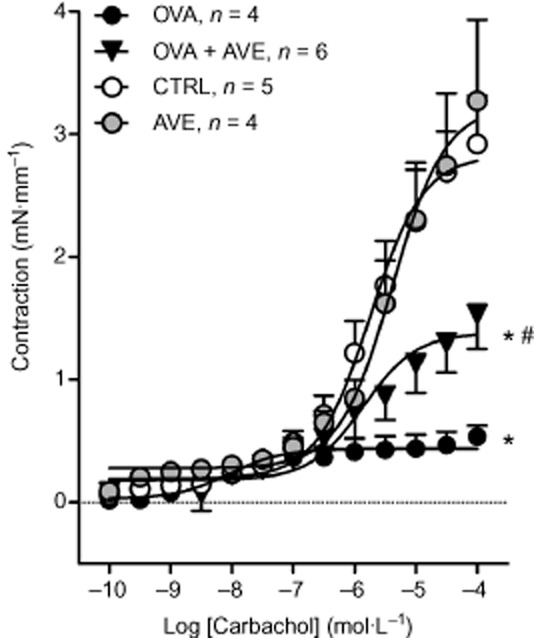
Contractile response induced by carbachol (100 pM–100 μM) in bronchial rings (2 mm) isolated from primary bronchi of mice sensitized and challenged with OVA (n = 4); mice sensitized and challenged with OVA and treated with AVE (1 mg·kg−1, s.c.; OVA + AVE; n = 6); CTRL (n = 5) and control mice treated with AVE (AVE; n = 4). *P < 0.05, significantly different from CTRL or AVE groups; #P < 0.05 significantly different from OVA group; two-way anova followed by Bonferroni's post test.
Airway and vascular remodelling
As summarised in Figure 3, The CTRL (Figure 3A) and AVE (Figure 3B) groups exhibited lung parenchyma with normal alveolar septa and bronchiolar wall thickness as well as aerated alveoli. The intra-acinar vessels exhibited a normal aspect. In contrast, the OVA group (Figure 3C) exhibited large interalveolar interstitial thickening, resulting mainly from the accumulation of inflammatory cells. Bronchiolar wall thickening was also observed, mainly due to hypertrophy and hyperplasia of the bronchial epithelium and the SMC layer. Moreover, a reduction in the alveolar lumen was found. The pulmonary vasculature exhibited an increase in size and number of vessels within the airway wall, contributing to the thickening of the wall. In OVA + AVE (Figure 3D), the lung parenchyma exhibited reduced inflammation in the interalveolar space, with a normal appearance of alveolar lumen and bronchioles. Morphometric analyses revealed that chronic treatment with AVE reversed airway (Figure 3E) and vessel remodelling (Figure 3F) in OVA-sensitized mice.
Figure 3.
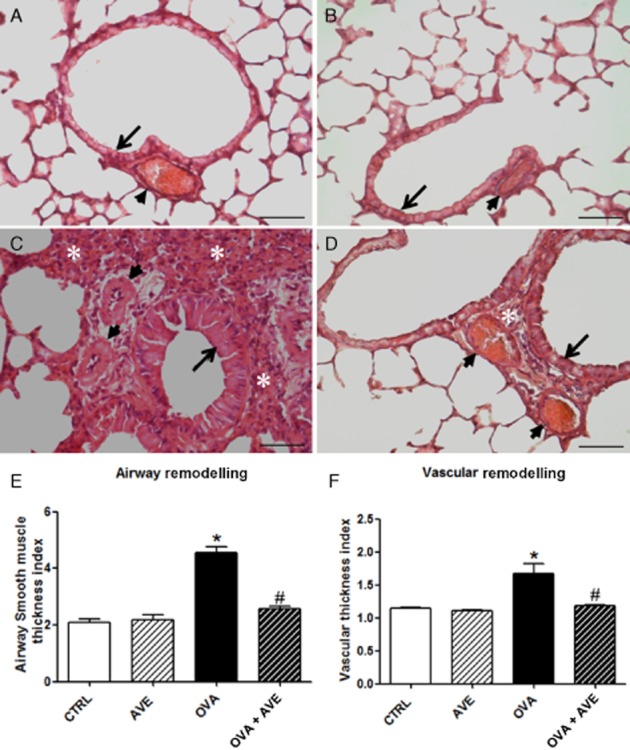
Representative histological images of airways (arrow) and pulmonary vessels (arrowhead) from CTRL (A), AVE (B), OVA (C) and OVA + AVE (D) groups (n = 5 each). A and B show lung parenchyma with normal aspect of the airway and vessels. C shows marked interalveolar interstitial thickening resulting mainly from the accumulation of inflammatory cells (white asterisk), bronchiolar wall thickening by hypertrophy and hyperplasia of bronchial epithelium and SMC layer, reduction in the alveolar lumen and vascular remodelling with increased size and number of vessels within the airway wall contributing to thickening of wall. Image D shows reduced inflammation in the interalveolar space with normal appearance of alveolar lumen and bronchioles of the lung parenchyma in OVA + AVE mice. In addition, reduced inflammation in the interalveolar space with normal appearance of alveolar lumen and bronchioles. (E) Thickness index of airway smooth muscle and bronchial epithelial layer was calculated by counting the number of points hitting muscle areas divided by the number of intercepts crossing the epithelial basement membrane. (F) Morphometric analysis of vascular thickness index calculated by the EP and IP ratio, EP/IP ratio. *P < 0.05 significantly different from CTRL and #P < 0.05 significantly different from OVA; one-way anova followed by Newman–Keuls post test. Bar = 20 μm.
Right ventricular hypertrophy
The OVA mice exhibited a significant increase in myocyte diameter of the right ventricle (Figure 4) in comparison with CTRL (Figure 4), suggesting that structural remodelling led to an increase in pulmonary vascular resistance and subsequent right ventricular hypertrophy. AVE treatment prevented right ventricular hypertrophy (Figure 4).
Figure 4.
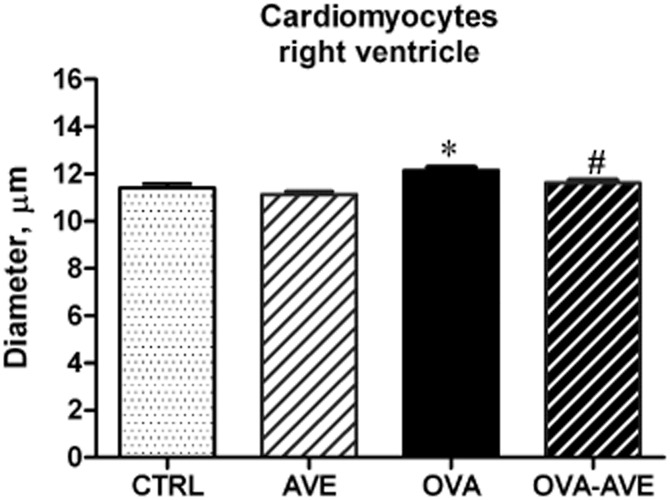
Effect of AVE treatment on right ventricular hypertrophy evaluated by measurement of transverse cardiomyocyte diameter (CTRL group, n = 5; OVA, AVE and OVA + AVE groups, n = 7 each). *P < 0.05, significantly different from CTRL; #P < 0.05, significantly different from OVA group; Kruskal–Wallis test followed by Dunn's test.
Cytokine concentrations in BAL and lung
Figure 5 shows IL-4, IL-5 and IL-10 in the BAL and lung of CTRL, OVA and OVA–AVE groups. As expected IL-5 was significantly increased in BAL (Figure 5A) and lungs (Figure 5B) of OVA mice in comparison with CTRL mice, while IL-4 was not changed (Figure 5C and D). AVE treatment of OVA-induced allergic pulmonary inflammation completely prevented IL-5 increase both in the BAL and lung (Figure 5A and B). Interestingly, AVE treatment also increased IL-10 levels in BAL, relative to the levels in CTRL mice (Figure 5E). No significant change was observed in IL-10 in the lung (Figure 5F) in OVA + AVE mice. The balance between these anti-inflammatory and pro-inflammatory cytokines, which can be evaluated by the ratio IL-10/IL-5, was increased in the BAL of OVA + AVE treated mice (Figure 5G) in comparison with CTRL or OVA mice. Furthermore, in the lung homogenates, this relationship was improved in OVA + AVE mice (Figure 5H) compared with OVA mice. In an additional group of animals (n = 5), OVA + AVE mice were treated simultaneously with the Mas receptor antagonist, A-779 (1.5 μg·h−1, s.c., for 28 days through ALZET osmotic mini-pumps). A-779 completely blocked the increase in IL-10 in the BAL (66.4 ± 3.3 vs. 89.1 ± 6.8 pg·mL−1 in OVA–AVE), but the decreased levels of IL-5 of OVA + AVE mice were not changed by A-779 treatment (68.6 ± 3.3 vs. 63.9 ± 1.5 pg·mL−1 in OVA).
Figure 5.
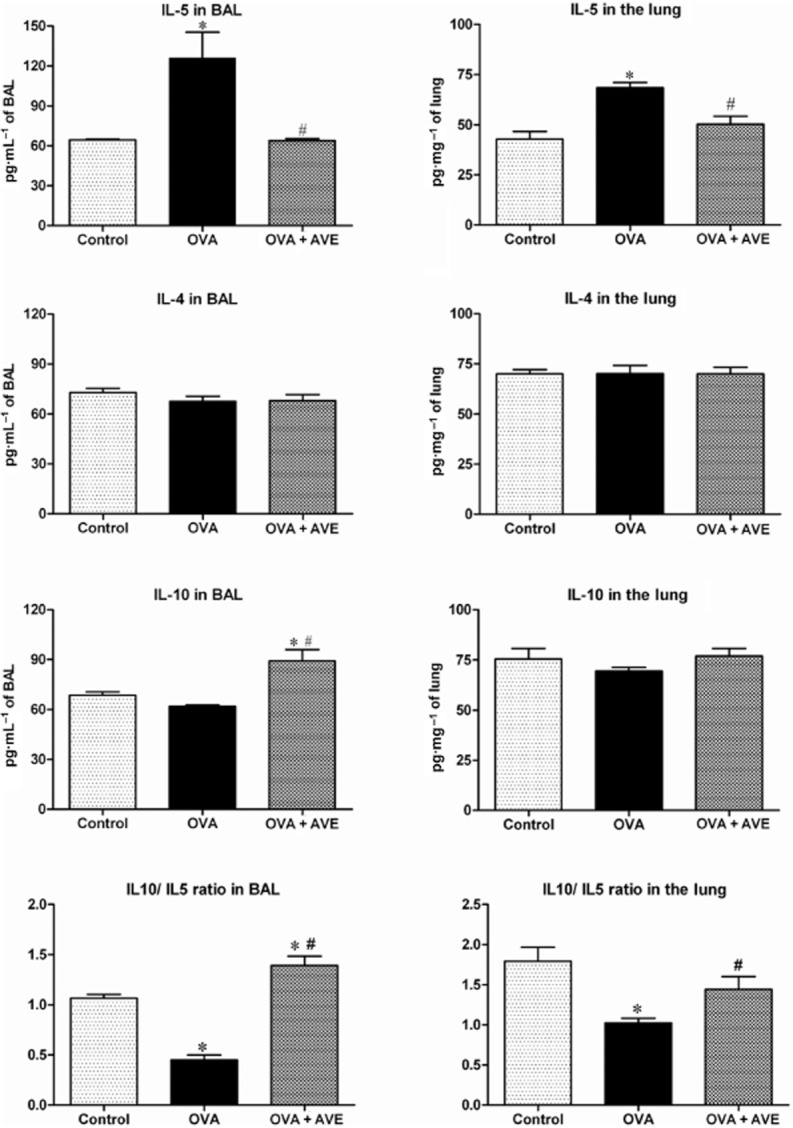
IL-4, IL-5 and IL-10 in the BAL and lungs of CTRL (n = 5), OVA sensitized and challenged (OVA; n = 5–6) and OVA sensitized and challenged treated with AVE (OVA + AVE; n = 4–6) groups. *P ≤ 0.05, significantly different from CTRL and #P ≤ 0.05, significantly different from OVA; one-way anova followed by Newman–Keuls post test.
Expression of angiotensin receptor proteins in the lung
As shown in Figure 6, Mas receptor expression in the OVA group of mice (Figure 6A) was significantly lower than in the CTRL mice. Treatment of OVA mice with AVE did not alter the decreased expression of Mas receptors (Figure 6A). CTRL mice treated with AVE presented levels of Mas similar to those in the CTRL group. In addition, no change in AT1 and AT2 receptor expression was observed in the different groups studied. These data suggest a decrease in the ACE2/Ang-(1–7)/Mas axis in this experimental model of chronic allergic lung inflammation.
Figure 6.

Expression of angiotensin receptor proteins in lung tissue, using Western blotting (n = 6 in each group). (A) Relative expression of Mas receptor; (B) Relative expression of AT1 receptor. (C) Relative expression of AT2 receptor. After incubation with primary and secondary antibody for Mas, AT1 and AT2 receptors, the membranes were stripped and incubated with primary and secondary antibodies to detect GAPDH; *P < 0.05, significantly different from CTRL or AVE groups; one-way anova followed by Newman–Keuls post test.
Pulmonary and circulating RAS profile
Figure 7 shows lung and plasma levels of angiotensin peptides. No significant difference was found in AVE mice in comparison with the CTRL group for any of the peptides in the plasma or in the lung. OVA mice exhibited a significant increase in plasma Ang II (Figure 7A) with no change in Ang-(1–7) levels (Figure 7B), indicating an overactivity of the circulating ACE-Ang II-AT1 axis in OVA-induced lung inflammation, compared with the CTRL group. No significant change in angiotensin peptides was observed in the lung of OVA mice (Figure 7D–F). AVE treatment of OVA mice did not alter the increased Ang II levels in the plasma (Figure 7A) but it did decrease Ang II levels in the lung (Figure 7D). Ang-(1–7) did not significantly change in plasma or lung of OVA + AVE mice. These changes resulted in a trend to alter the balance between Ang-(1–7) and Ang II in the lung (Figure 7F), suggesting that treatment of mice with chronic allergic lung inflammation with AVE alters the balance of angiotensin peptides in the lung, apart from its direct effects.
Figure 7.
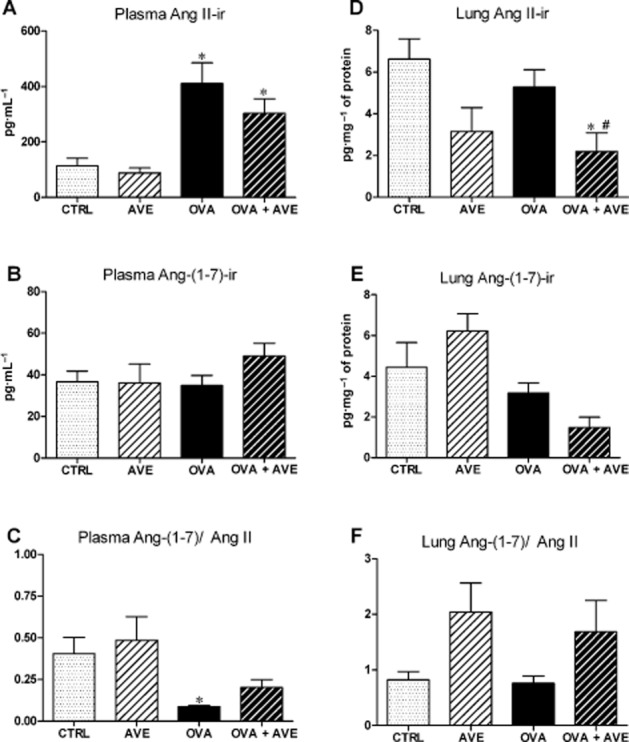
Levels of angiotensin peptides levels in plasma and lung measured by RIA. Plasma angiotensin II-immunoreactivity (Ang II-ir, pg·mL−1, n = 5–8; A); plasma Ang-(1–7)-ir (pg·mL−1, n = 5–8; B), plasma Ang-(1–7)/Ang II ratio (C); lung Ang II-ir (pg·mg−1 protein, n = 4–7; D); lung Ang-(1–7)-ir (pg·mg−1 protein, n = 4–7); lung Ang-(1–7)/Ang II ratio (F). *P < 0.05, significantly different from CTRL; one-way anova followed by Newman–Keuls post test.
Discussion and conclusions
The most important findings of the present study were the observations that AVE treatment prevented the development of airway and pulmonary vascular remodelling, attenuated right ventricle hypertrophy and pulmonary inflammation and improved contractile responses in bronchial rings to carbachol, in a model of chronic lung inflammation.
Recent investigations have changed our understanding of asthma from a purely inflammatory disease to a disease in which both inflammatory and structural components are equally involved (Al-Muhsen et al., 2011). Despite advances in the identification of key cellular and molecular mechanisms involved in airway remodelling, there are as yet no clear proposals regarding prevention, treatment or the best time to initiate treatment of such remodelling (Durrani et al., 2011). Ang-(1–7), via the receptor Mas, acts as an anti-inflammatory agent in an acute murine model of asthma (El-Hashim et al., 2012). Treatment of OVA-challenged mice with Ang-(1–7) for 4 days induced a significant dose-dependent decrease in the total cell count, eosinophils, lymphocytes, neutrophils and macrophages in the BAL. In addition, Ang-(1–7) treatment significantly reduced the perivascular and peribronchial inflammation, fibrosis and goblet cell hyper/metaplasia induced by OVA. In the present study employing AVE, which is a synthetic, non-peptide compound, mimicking the anti-fibrotic, anti-proliferative and anti-inflammatory effects of Ang-(1–7) (Wiemer et al., 2002; Pinheiro et al., 2004; Santos and Ferreira, 2006; Lubel et al., 2009; da Silveira et al., 2010; Barroso et al., 2012; Marques et al., 2012), we have advanced these observations by showing that AVE prevented the development of airway and pulmonary vascular remodelling and pulmonary inflammation, improving airway responsiveness in mice with chronic allergic lung inflammation.
Airway responsiveness to carbachol in a murine model of asthma has been used both in vitro (Chen et al., 2003) and in vivo (Milanese et al., 2001; Dimitropoulou et al., 2009) as a marker of airway SMC function. In the present study, airway responsiveness was tested ex vivo in bronchial rings from animals sensitized with OVA, with or without treatment with AVE. The OVA group exhibited significant, concentration–dependent, attenuation of bronchial ring contractions to carbachol, suggesting that these mice had reached an advanced degree of remodelling that prevented airway narrowing induced by the activation of muscarinic receptors. Depending on the extent and distribution of the remodelling, changes in the extracellular matrix can either enhance or prevent airway shortening (Palmans et al., 2000). Decreased shortening of asthmatic airway smooth muscle (ASM) can be dependent on the load against which the muscle contracts (Bramley et al., 1995). These authors also demonstrated that collagenase treatment of human lobar bronchial smooth muscle increased ASM shortening, suggesting that excessive extracellular matrix components increase the muscle resistive load to the shortening, independent of any changes in the muscle itself. In keeping with these findings, Milanese et al. (2001) showed that the thickening of the reticular basement membrane, which has been described as a characteristic feature of airway remodelling in asthma, was associated with less airway narrowing and air trapping, suggesting that reticular basement membrane thickening represents an additional load on ASM. Our data obtained in OVA-induced chronic asthma mice are compatible with these results. Furthermore, in the present study, we showed that treatment with AVE improved the contractile response to carbachol in bronchial rings, suggesting that AVE can decrease the accumulation of extracellular matrix components and/or reticular basement membrane thickening, that is induced by allergic inflammation.
Airway remodelling has been documented in all degrees of asthma severity and in both large and small airways. Invasive techniques for the evaluation of airway remodelling have shown structural alterations in the bronchial layers, as well as, inflammatory and fibrotic cell infiltration (Manso et al., 2012). Among the structural alterations, an increase in ASM mass is the most prominent feature of airway remodelling in asthma (Tagaya and Tamaoki, 2007). In addition to their physical property, human ASM cells produce and secrete a variety of inflammatory mediators that can contribute to the inflammatory state, airway responsiveness, and airway remodelling present in asthma (Xia et al., 2012). In the present study, AVE exerted a protective effect on airway remodelling, preventing the increase in thickness of ASM and the bronchial epithelial layer. As remodelling affects different components of the airways, AVE treatment may have modulated other factors beyond the SMC layer, such as fibrosis and inflammation. As observed by El-Hashim et al. (2012) treatment with Ang-(1–7) resulted in a significantly reduced peribronchial and perivascular fibrosis and inflammation in an OVA-challenged mouse model of acute allergic asthma.
Similar results were observed with pulmonary vascular remodelling. The thickness index of the vascular muscle layer was increased in allergic mice, while chronic treatment with AVE reversed this response. These findings confirm and extend previous observations that Ang-(1–7) has anti-proliferative effects in vascular SMC (Santos et al., 2005; Zhang et al., 2010). In agreement with these observations, the OVA group in the present study exhibited a significant right ventricular hypertrophy and treatment with AVE reversed this response. In earlier studies carried out at our laboratory, AVE prevented the development of hypertrophy and collagen deposition induced by isoproterenol treatment, in addition to an improvement in cardiac function (Ferreira et al., 2007). Right ventricular hypertrophy represents functional and structural adaptation in response to chronic pressure overload induced by different chronic and hypoxemic diseases. As the pulmonary circulation is a low-pressure system, with low vascular resistance to right ventricular output, factors that continuously and progressively increase afterload lead to right ventricular adaptation. As the increased thickness index of the vascular muscle layer was reversed by AVE, it is likely that the cardioprotective effect of AVE was related to prevention of an increase in pulmonary vascular resistance.
IL-5, a key cytokine in chronic inflammation, was increased in the OVA mice. Moreover, AVE treatment of OVA mice prevented lung and BAL increase in IL-5 and increased IL-10 in the BAL. These results suggest that AVE has a protective effect in the pulmonary remodelling probably by re-establishing the balance between pro- and anti-inflammatory cytokines in the lung. Studies in experimental models of chronic allergic lung inflammation have shown that OVA sensitization induces Th2 responses with the release of cytokines, particularly IL-5 (Bruijnzeel, 1994; Lampinen et al., 2004). IL-5 modulates TGF-β expression involved in the pulmonary remodelling and regulates the proliferation, differentiation and the trafficking of eosinophils to the lung (Carlson et al., 1993; Lampinen et al., 2004; Cho, 2011). The increase in eosinophils is implicated in the pulmonary lesions observed in allergic asthma (Kidd, 2003). In contrast to IL-5, IL-4 levels were not altered in OVA mice. This result, however, is in keeping with other studies showing that IL-4 may not play a significant role in chronic airway inflammation. Accordingly, IL-4-knockout (KO) mice developed increased epithelial hypertrophy, subepithelial fibrosis and airway inflammation in chronic models of asthma (Foster et al., 2000; Kumar et al., 2002).
IL-10, on the other hand, plays an important role downstream of the inflammatory cascade in the Th2 response to antigens (Kosaka et al., 2011). Our data indicate that AVE modulated the release of at least these cytokines, thereby contributing to a decrease in the pulmonary remodelling induced by inflammation. Furthermore, our data suggest that the increase in IL-10 induced by AVE was mediated by Mas receptors, as it was blocked by the Mas receptor antagonist, A-779. However, A-779 did not block the effect of AVE on IL-5. Because different pathways or cells are involved in the production of IL-10 and IL-5, it is possible that other receptors or receptor subtypes are involved in the effects of AVE on IL-5 release.
Previous studies by us (Pinheiro et al., 2004; Lemos et al., 2005) and others (Wiemer et al., 2002) had shown that different effects of Ang-(1–7) and AVE can be partially or even totally blocked by the AT2 receptor antagonist, PD123319, suggesting that Ang-(1–7) and AVE exert effects mediated through the AT2 or other receptors, sensitive to PD123319. We have recently identified and characterized a new peptide of the RAS, Ala-Arg-Val-Tyr-Ile-His-Pro, alamandine, in rats, mice and humans (Lautner et al., 2013), whose biological effects resemble those produced by Ang-(1–7), such as vasodilation, antifibrotic, antihypertensive and central effects. Alamandine can be formed, in vivo, directly from Ang-(1–7) by decarboxylation of its aspartate residue. More interestingly, we showed that alamandine binds to a Mas-related GPCR, member D (MrgD) and its effects are blocked by D-Pro7-Ang-(1–7), the MrgD ligand β-alanine and PD123319, but not by the Mas receptor antagonist, A-779. Moreover, alamandine induced vasorelaxation in aortic rings isolated from AT2 receptor KO mice and this effect was abolished by PD1233319, suggesting that PD123319 is also an MrgD antagonist/ligand. Thus, it is possible that some part of the effects of AVE could be ascribed to actions on the MrgD receptor. Further studies will be necessary to confirm this possibility.
The protective effects of AVE observed in the present study were not related to changes in the expression of Mas or AT1 or AT2 receptors in the lung. We found that Mas receptor expression was much lower (∼70%) in OVA mice, but it was not significantly altered in OVA + AVE mice. This result suggests that reduction in the activity of the ACE2/Ang-(1–7)/Mas receptor axis is involved in the pulmonary inflammation and remodelling in this model of chronic allergic lung inflammation. On the other hand, circulating levels of Ang II, but not the levels in lung tissue, were significantly increased in OVA-induced chronic allergic inflammation. These changes indicate a predominance of Ang II/AT1 actions in these mice. It is well known that Ang II stimulates proliferation and/or hypertrophy in a wide variety of cells, including in the human airway SMC, which suggests its participation in airway remodelling (McKay et al., 1998). Thus, the alterations in the profile of the RAS that we observed, did contribute to the pathophysiology of chronic allergic lung inflammation.
More interestingly, Ang II levels in the lung were decreased after treatment of OVA mice with AVE and, although this effect was not accompanied by changes in Ang-(1–7), there was a significant alteration in the balance between Ang-(1–7) and Ang II in the lungs of treated mice. Taken together, these results suggest that AVE treatment, in addition to its direct agonist effect, also decreased Ang II/AT1 actions in the lungs of mice with OVA-induced allergic inflammation. Collectively, these effects may have contributed to the attenuation of inflammation and pulmonary remodelling in our model of asthma in mice.
In summary, the results of the present study show, for the first time, that AVE, a Mas receptor agonist, prevented the development of airway and vascular pulmonary remodelling and pulmonary inflammation and improved contraction in bronchial rings from mice with OVA-induced chronic allergic lung inflammation. Moreover, right ventricular hypertrophy, which reflects increased pulmonary vascular resistance, was also prevented by AVE treatment. As airway remodelling is associated with a poor clinical outcome, these findings open a new possibility for the treatment of pulmonary remodelling induced by asthma and other chronic lung diseases, to include analogues of Ang-(1–7).
Acknowledgments
The authors are grateful to Jose Roberto da Silva and Luana Oliveira Prata for their skilful technical assistance. This study was funded by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and the Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG) through grants: INCT-Nanobiofar and Programa de Núcleos de Excelência (PRONEX).
Glossary
- ACE2
angiotensin-converting enzyme 2
- Ang II
angiotensin II
- Ang-(1-7)
angiotensin-(1-7)
- ASM
airway smooth muscle
- AVE
AVE 0991, agonist of Mas receptor
- BAL
bronchoalveolar lavage
- MRGD
Mas-related G-protein-coupled receptor D
- OVA
ovalbumin
- RAS
renin-angiotensin system
- SMC
smooth muscle cells
Conflict of interest
No conflicts of interest exist.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. J Allergy Clin Immunol. 2011;128:451–462. doi: 10.1016/j.jaci.2011.04.047. [DOI] [PubMed] [Google Scholar]
- Barroso LC, Silveira KD, Lima CX, Borges V, Bader M, Rachid M, et al. Renoprotective effects of AVE0991, a nonpeptide Mas receptor agonist, in experimental acute renal injury. Int J Hypertens. 2012;2012:808726. doi: 10.1155/2012/808726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron C, Tulic MK, Hamid Q. Airway remodelling in asthma: from benchside to clinical practice. Can Respir J. 2010;17:e85–e93. doi: 10.1155/2010/318029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botelho LM, Block CH, Khosla MC, Santos RAS. Plasma angiotensin(1–7) immunoreactivity is increased by salt load, water deprivation, and hemorrhage. Peptides. 1994;15:723–729. doi: 10.1016/0196-9781(94)90103-1. [DOI] [PubMed] [Google Scholar]
- Bramley AM, Roberts CR, Schellenberg RR. Collagenase increases shortening of human bronchial smooth muscle in vitro. Am J Respir Crit Care Med. 1995;152:1513–1517. doi: 10.1164/ajrccm.152.5.7582286. [DOI] [PubMed] [Google Scholar]
- Bruijnzeel PL. Eosinophil tissue mobilization in allergic disorders. Ann N Y Acad Sci. 1994;725:259–267. doi: 10.1111/j.1749-6632.1994.tb39808.x. [DOI] [PubMed] [Google Scholar]
- Carlson M, Peterson C, Venge P. The influence of IL-3, IL-5, and GM-CSF on normal human eosinophil and neutrophil C3b-induced degranulation. Allergy. 1993;48:437–442. [PubMed] [Google Scholar]
- Chen H, Tliba O, Van-Besien CR, Panettieri RA, Jr, Amrani Y. TNF-[alpha] modulates murine tracheal rings responsiveness to G-protein-coupled receptor agonists and KCl. J Appl Physiol. 2003;95:864–872. doi: 10.1152/japplphysiol.00140.2003. [DOI] [PubMed] [Google Scholar]
- Cho JY. Recent advances in mechanisms and treatments of airway remodeling in asthma: a message from the bench side to the clinic. Korean J Intern Med. 2011;26:367–383. doi: 10.3904/kjim.2011.26.4.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silveira KD, Coelho FM, Vieira AT, Sachs D, Barroso LC, Costa VV, et al. Anti-inflammatory effects of the activation of the angiotensin-(1–7) receptor, MAS, in experimental models of arthritis. J Immunol. 2010;185:5569–5576. doi: 10.4049/jimmunol.1000314. [DOI] [PubMed] [Google Scholar]
- Dimitropoulou C, Drakopanagiotakis F, Chatterjee A, Snead C, Catravas JD. Estrogen replacement therapy prevents airway dysfunction in a murine model of allergen-induced asthma. Lung. 2009;187:116–127. doi: 10.1007/s00408-008-9129-z. [DOI] [PubMed] [Google Scholar]
- Du YC, Xu JY, Zhang SJ. Effects of angiotensin II receptor antagonist on expression of collagen III, collagen V, and transforming growth factor beta1 in the airway walls of sensitized rats. Chin Med J (Engl) 2004;117:908–912. [PubMed] [Google Scholar]
- Durrani SR, Viswanathan RK, Busse WW. What effect does asthma treatment have on airway remodeling? Current perspectives. J Allergy Clin Immunol. 2011;128:439–448. doi: 10.1016/j.jaci.2011.06.002. [DOI] [PubMed] [Google Scholar]
- EL-Hashim AZ, Renno WM, Raghupathy R, Abduo HT, Akhtar S, Benter IF. Angiotensin-(1–7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-kappaB-dependent pathways. Br J Pharmacol. 2012;166:1964–1976. doi: 10.1111/j.1476-5381.2012.01905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira AJ, Oliveira TL, Castro MC, Almeida AP, Castro CH, Caliari MV, et al. Isoproterenol-induced impairment of heart function and remodeling are attenuated by the nonpeptide angiotensin-(1–7) analogue AVE 0991. Life Sci. 2007;81:916–923. doi: 10.1016/j.lfs.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Ferreira AJ, Bader M, Santos RA. Therapeutic targeting of the angiotensin-converting enzyme 2/Angiotensin-(1–7)/Mas cascade in the renin-angiotensin system: a patent review. Expert Opin Ther Pat. 2012;22:567–574. doi: 10.1517/13543776.2012.682572. [DOI] [PubMed] [Google Scholar]
- Foster PS, Ming Y, Matthei KI, Young IG, Temelkovski J, Kumar RK. Dissociation of inflammatory and epithelial responses in a murine model of chronic asthma. Lab Invest. 2000;80:655–662. doi: 10.1038/labinvest.3780068. [DOI] [PubMed] [Google Scholar]
- Girodet PO, Ozier A, Bara I, Tunon de Lara JM, Marthan R, Berger P. Airway remodeling in asthma: new mechanisms and potential for pharmacological intervention. Pharmacol Ther. 2011;130:325–337. doi: 10.1016/j.pharmthera.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. 2003;8:223–246. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka S, Tamauchi H, Terashima M, Maruyama H, Habu S, Kitasato H. IL-10 controls Th2-type cytokine production and eosinophil infiltration in a mouse model of allergic airway inflammation. Immunobiology. 2011;216:811–820. doi: 10.1016/j.imbio.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Kumar RK, Herbert C, Yang M, Koskinen AM, McKenzie AN, Foster PS. Role of interleukin-13 in eosinophil accumulation and airway remodelling in a mouse model of chronic asthma. Clin Exp Allergy. 2002;32:1104–1111. doi: 10.1046/j.1365-2222.2002.01420.x. [DOI] [PubMed] [Google Scholar]
- Lampinen M, Carlson M, Hakansson LD, Venge P. Cytokine-regulated accumulation of eosinophils in inflammatory disease. Allergy. 2004;59:793–805. doi: 10.1111/j.1398-9995.2004.00469.x. [DOI] [PubMed] [Google Scholar]
- Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, et al. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res. 2013;112:1104–1111. doi: 10.1161/CIRCRESAHA.113.301077. [DOI] [PubMed] [Google Scholar]
- Lemos VS, Silva DM, Walther T, Alenina N, Bader M, Santos RAS. The endothelium-dependent vasodilator effect of the nonpeptide Ang(1–7) mimic AVE 0991 is abolished in the aorta of mas-knockout mice. J Cardiovasc Pharmacol. 2005;46:274–279. doi: 10.1097/01.fjc.0000175237.41573.63. [DOI] [PubMed] [Google Scholar]
- Lubel JS, Herath CB, Tchongue J, Grace J, Jia Z, Spencer K, et al. Angiotensin-(1–7), an alternative metabolite of the renin-angiotensin system, is up-regulated in human liver disease and has antifibrotic activity in the bile-duct-ligated rat. Clin Sci (Lond) 2009;117:375–386. doi: 10.1042/CS20080647. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay S, de Jongste JC, Saxena PR, Sharma HS. Angiotensin II induces hypertrophy of human airway smooth muscle cells: expression of transcription factors and transforming growth factor-beta1. Am J Respir Cell Mol Biol. 1998;18:823–833. doi: 10.1165/ajrcmb.18.6.2924. [DOI] [PubMed] [Google Scholar]
- Manso L, Reche M, Padial MA, Valbuena T, Pascual C. Diagnostic tools assessing airway remodelling in asthma. Allergol Immunopathol (Madr) 2012;40:108–116. doi: 10.1016/j.aller.2011.11.002. [DOI] [PubMed] [Google Scholar]
- Marques FD, Ferreira AJ, Sinisterra RD, Jacoby BA, Sousa FB, Caliari MV, et al. An oral formulation of angiotensin-(1–7) produces cardioprotective effects in infarcted and isoproterenol-treated rats. Hypertension. 2011;57:477–483. doi: 10.1161/HYPERTENSIONAHA.110.167346. [DOI] [PubMed] [Google Scholar]
- Marques FD, Melo MB, Souza LE, Irigoyen MC, Sinisterra RD, de Sousa FB, et al. Beneficial effects of long-term administration of an oral formulation of angiotensin-(1–7) in infarcted rats. Int J Hypertens. 2012;2012:795452. doi: 10.1155/2012/795452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanese M, Crimi E, Scordamaglia A, Riccio A, Pellegrino R, Canonica GW, et al. On the functional consequences of bronchial basement membrane thickening. J Appl Physiol. 2001;91:1035–1040. doi: 10.1152/jappl.2001.91.3.1035. [DOI] [PubMed] [Google Scholar]
- Palmans E, Kips JC, Pauwels RA. Prolonged allergen exposure induces structural airway changes in sensitized rats. Am J Respir Crit Care Med. 2000;161:627–635. doi: 10.1164/ajrccm.161.2.9902094. [DOI] [PubMed] [Google Scholar]
- Pedersen SE, Hurd SS, Lemanske RF, Jr, Becker A, Zar HJ, Sly PD, et al. Global strategy for the diagnosis and management of asthma in children 5 years and younger. Pediatr Pulmonol. 2011;46:1–17. doi: 10.1002/ppul.21321. [DOI] [PubMed] [Google Scholar]
- Pinheiro SV, Simoes e Silva AC, Sampaio WO, de Paula RD, Mendes EP, Bontempo ED, et al. Nonpeptide AVE 0991 is an angiotensin-(1–7) receptor Mas agonist in the mouse kidney. Hypertension. 2004;44:490–496. doi: 10.1161/01.HYP.0000141438.64887.42. [DOI] [PubMed] [Google Scholar]
- Saldanha JC, Gargiulo DL, Silva SS, Carmo-Pinto FH, Andrade MC, Alvarez-Leite JI, et al. A model of chronic IgE-mediated food allergy in ovalbumin-sensitized mice. Braz J Med Biol Res. 2004;37:809–816. doi: 10.1590/s0100-879x2004000600005. [DOI] [PubMed] [Google Scholar]
- Santos RAS, Ferreira AJ. Pharmacological effects of AVE 0991, a nonpeptide angiotensin-(1–7) receptor agonist. Cardiovasc Drug Rev. 2006;24:239–246. doi: 10.1111/j.1527-3466.2006.00239.x. [DOI] [PubMed] [Google Scholar]
- Santos RAS, Simoes e Silva AC, Maric C, Silva DM, Machado RP, de Buhr I, et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A. 2003;100:8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos RAS, Ferreira AJ, Nadu AP, Braga AN, de Almeida AP, Campagnole-Santos MJ, et al. Expression of an angiotensin-(1–7)-producing fusion protein produces cardioprotective effects in rats. Physiol Genomics. 2004;17:292–299. doi: 10.1152/physiolgenomics.00227.2003. [DOI] [PubMed] [Google Scholar]
- Santos RAS, Ferreira AJ, Pinheiro SV, Sampaio WO, Touyz R, Campagnole-Santos MJ. Angiotensin-(1–7) and its receptor as a potential targets for new cardiovascular drugs. Expert Opin Investig Drugs. 2005;14:1019–1031. doi: 10.1517/13543784.14.8.1019. [DOI] [PubMed] [Google Scholar]
- Tagaya E, Tamaoki J. Mechanisms of airway remodeling in asthma. Allergol Int. 2007;56:331–340. doi: 10.2332/allergolint.R-07-152. [DOI] [PubMed] [Google Scholar]
- Vieira RP, de Andrade VF, Duarte AC, dos Santos AB, Mauad T, Martins MA, et al. Aerobic conditioning and allergic pulmonary inflammation in mice. II. Effects on lung vascular and parenchymal inflammation and remodeling. Am J Physiol Lung Cell Mol Physiol. 2008;295:L670–L679. doi: 10.1152/ajplung.00465.2007. [DOI] [PubMed] [Google Scholar]
- Wang T, Yin KS, Liu KY, Lu GJ, Li YH, Chen JD. Effect of valsartan on the expression of angiotensin II receptors in the lung of chronic antigen exposure rats. Chin Med J (Engl) 2008;121:2312–2319. [PubMed] [Google Scholar]
- Wiemer G, Dobrucki LW, Louka FR, Malinski T, Heitsch H. AVE 0991, a nonpeptide mimic of the effects of angiotensin-(1–7) on the endothelium. Hypertension. 2002;40:847–852. doi: 10.1161/01.hyp.0000037979.53963.8f. [DOI] [PubMed] [Google Scholar]
- Xia YC, Redhu NS, Moir LM, Koziol-White C, Ammit AJ, Al-Alwan L, et al. Pro-inflammatory and immunomodulatory functions of airway smooth muscle: emerging concepts. Pulm Pharmacol Ther. 2012;26:64–74. doi: 10.1016/j.pupt.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Yang YC, Zhang N, Van Crombruggen K, Hu GH, Hong SL, Bachert C. Transforming growth factor-beta1 in inflammatory airway disease: a key for understanding inflammation and remodeling. Allergy. 2012;67:1193–1202. doi: 10.1111/j.1398-9995.2012.02880.x. [DOI] [PubMed] [Google Scholar]
- Zhang F, Hu H, Xu Q, Ye S. Different effects of angiotensin II and angiotensin-(1–7) on vascular smooth muscle cell proliferation and migration. PLoS ONE. 2010;5:e12323. doi: 10.1371/journal.pone.0012323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang WX, Li CC. Airway remodeling: a potential therapeutic target in asthma. World J Pediatr. 2011;7:124–128. doi: 10.1007/s12519-011-0264-x. [DOI] [PubMed] [Google Scholar]