

Abstract
BACKGROUND AND PURPOSE
Clinical studies indicate that statins have a BP-lowering effect in hypercholesterolemic individuals with hypertension. Specifically, statins modulate BP through the up-regulation of endothelial NOS (eNOS) activation in the brain. However, the signalling mechanisms through which statins enhance eNOS activation remain unclear. Therefore, we examined the possible signalling pathways involved in statin-mediated BP regulation in the nucleus tractus solitarii (NTS).
EXPERIMENTAL APPROACH
To investigate the involvement of Ras and other signalling pathways in simvastatin-induced effects on BP, BP and renal sympathetic nerve activity (RSNA) were determined in spontaneously hypertensive rats (SHRs) before and after i.c.v. administration of simvastatin in the absence and presence of a Ras-specific inhibitor (farnesyl thiosalicylic acid, FTS), a geranylgeranyltransferase inhibitor (GGTI-2133), a PI3K inhibitor (LY294002) or a MAPK-ERK kinase (MEK) inhibitor (PD98059).
KEY RESULTS
FTS significantly attenuated the decrease in BP and increased NO evoked by simvastatin and reversed the decrease in basal RSNA induced by simvastatin. Immunoblotting and pharmacological studies showed that inhibition of Ras activity by FTS significantly abolished simvastatin-induced phosphorylation of ERK1/2, ribosomal protein S6 kinase (RSK), Akt and decreased eNOS phosphorylation. Likewise, administration of Akt and ERK1/2 signalling inhibitors, LY294002 and PD98059, attenuated the reduction in BP evoked by simvastatin. Furthermore, i.c.v. simvastatin decreased Rac1 activation and the number of ROS-positive cells in the NTS.
CONCLUSIONS AND IMPLICATIONS
Simvastatin modulates central BP control in the NTS of SHRs by increasing Ras-mediated activation of the PI3K-Akt and ERK1/2-RSK signalling pathways, which then up-regulates eNOS activation.
Keywords: statins, nucleus tractus solitarii, nitric oxide, Ras, hypertension, isoprenylation
Introduction
Statins are 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors and are unambiguously useful for patients with hypercholesterolemia (Halcox and Deanfield, 2004; Kanbay et al., 2005). In addition to their cholesterol-lowering properties, statins exert a number of pleiotropic, cholesterol-independent effects. These effects include improvement in endothelial function, increased NO bioavailability (Kishi et al., 2003) and antioxidant properties (Kishi et al., 2008). Because endothelial dysfunction and reactive oxygen species (ROS) are important pathophysiological determinants of essential hypertension, these effects of statins suggest that statin therapy may be useful for the simultaneous management of clinical hypertension (Koh et al., 2008).
By inhibiting mevalonate synthesis, statins also prevent the synthesis of other important isoprenoid intermediates of the cholesterol biosynthetic pathway, such as farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP) (Liao, 2002). These intermediates serve as important lipid attachments for the post-translational modification of a variety of small GTPase moieties (McTaggart, 2006). Protein isoprenylation provides the lipophilic anchors that facilitate subcellular localization and intracellular trafficking of membrane-associated proteins (Van Aelst and D'Souza-Schorey, 1997).
NO is synthesized by NOS and there are three different NOS isoforms: endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS) (Calabrese et al., 2007). Our previous study revealed that the microinjection of an inhibitor of NOS into the nucleus tractus solitarii (NTS) can increase renal sympathetic nerve activity and induce a pressor effect (Tseng et al., 1996). In addition, we demonstrated that PI3K-Akt and ERK1/2-ribosomal protein S6 kinase (RSK) signallings regulate eNOS to modulate BP in the NTS (Huang et al., 2004; Ho et al., 2008; Cheng et al., 2012a; 2012b). Previously, statins have been reported to increase NO bioavailability in patients with hypercholesterolemia (John et al., 1998; 2001). Moreover, Walsh et al. demonstrated that simvastatin induces Akt-regulated eNOS activation, which leads to NO production and consequently promotes endothelial cell survival (Kureishi et al., 2000). ERK1/2 has an established role in mediating atorvastatin-induced eNOS activation (Merla et al., 2007). However, much remains to be established about the mechanisms through which statins evoke eNOS activation in the NTS.
In this study, we postulated that a central administration of statins could induce depressor effects in the NTS. We clarified which signalling cascades could be modulated by the addition of simvastatin. In addition, we validated which small GTPase was involved in the simvastatin-induced effects observed in the NTS. The results suggest that the Ras-PI3K-Akt-eNOS and Ras-ERK1/2-RSK-eNOS cascades are involved in simvastatin-mediated BP regulation in the NTS.
Methods
Experimental chemicals
All experimental drugs were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise noted.
Farnesyl thiosalicylic acid (FTS) was obtained from Cayman Chemical (Ann Arbor, MI, USA). N(5)-(-iminoethyl)-L-ornithine (L-NIO) was obtained from Calbiochem (Darmstadt, Germany). Simvastatin was obtained from Tocris (Bristol, UK).
Animals
Twenty-week-old male spontaneously hypertensive rats (SHRs) were obtained from the National Science Council Animal Facility and housed in the animal facility of Kaohsiung Veterans General Hospital (Kaohsiung, Taiwan). A total of 108 rats were included in this study. The rats were humanely treated at all times. The rats were housed in individual cages in a room in which lighting was controlled (12 h on/12 h off), and the temperature was maintained at 23 to 24°C. The rats were acclimatized to the housing conditions for 1 week. They were then trained with indirect BP measurements for 1 week. Then, the rats were randomly divided into the four groups, with six rats per group (i) the Con group, in which the SHRs received an i.c.v. injection of artificial CSF (aCSF) as a vehicle; (ii) the Sim group, in which the SHRs received an i.c.v. injection of simvastatin (28.5 nmol); (iii) the FTS group, in which the SHRs received an i.c.v. injection of FTS (0.7 nmol); and (iv) the Sim + FTS group, in which the SHRs received an i.c.v. injection of FTS and then received an i.c.v. infusion of simvastatin. The rats were given normal rat chow (Purina, St. Louis, MO, USA) and tap water ad libitum. All animal research protocols were approved by the Research Animal Facility Committee of Kaohsiung Veterans General Hospital and complied with their guidelines for animal experiments. All results of this study involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
The i.c.v. injection procedure
Surgery was performed under sterile conditions. Rats (n = 90) were anaesthetized with pentobarbital (50 mg kg−1 i.p.) and fixed in a stereotaxic instrument. The depth of anaesthesia was assessed by monitoring palpebral, pedal and corneal reflexes. A stainless steel cannula was positioned in the brain, with the tip placed in the left lateral cerebral ventricle (0.8 mm posterior and 1.5 mm mediolateral, with bregma). Polymerizing dental orthodontic resin was applied to the surface of the skull, and two protective screws were placed around the cannula. The cannula was bent at a 90° angle and fixed again with dental orthodontic resin. The i.c.v. infusion studies were performed after a stabilization period, of at least a 30 min, after insertion of the microinjector into the ventricular-guided cannula. BP was monitored for 3 days after the infusion of the drugs. As a vehicle control, the effect of an i.c.v. injection of aCSF (142 mmol L−1 NaCl, 5 mmol L−1 KCl, 10 mmol L−1 glucose and 10 mmol L−1 HEPES, pH 7.4) was analysed. Simvastatin (28.5 nmol day−1) and L-NIO (48 nmol day−1) were dissolved in aCSF; FTS (0.7 nmol day−1), geranylgeranyltransferase inhibitor (GGTI-2133; 25 pmol day−1), PD98059 (18.7 nmol day−1) and LY294002 (14.5 nmol day−1) were first dissolved in DMSO and then diluted with aCSF to make a 1% final DMSO concentration. The basal BP was examined before injection. The i.c.v. infusions were performed over a 2-min period with drugs and delivered in a volume of 5 μL from day 0 to day 3 by daily injection, single bolus. Simvastatin and inhibitors were injected simultaneously.
BP measurement
Using a tail-cuff method as described previously (Kubota et al., 2006) (Model MK-2000 Storage Pressure Meter, Muromachi Kikai, Tokyo, Japan), 24 rats were randomly divided into four groups for systolic BP (SBP) measurement. The SBP was measured before the start of treatments (day 0). The animals were placed in the fixer for 30 min. With this method, the re-appearance of pulsation on a digital display of the BP cuff was detected by a pressure transducer; this pulsation was amplified and recorded as the SBP. During the measurement, 10 individual readings were obtained in rapid sequence. The highest and the lowest readings were excluded, and the remaining eight readings were averaged.
Renal sympathetic nerve activity (RSNA) recording
The left kidney of the rats studied (n = 24) was exposed using a retroperitoneal dissection. A nerve fascicle to the kidney was isolated. A bipolar electrode was placed under the nerve and covered with silicone gel. The signals were amplified and passed through a band pass filter (10–3 K Hz, DAM50-E, World Precision Instruments Inc., Sarasota, FL, USA) displayed on an oscilloscope. The filtered nerve activity signal was rectified, integrated and collected for displaying and analysis using a PowerLab 35 Series data acquisition system (AD Instruments, Bella Vista, New South Wales, Australia).
Measurement of NO in the NTS
Five groups of rats (six rats per group, n = 30) were enrolled in this experiment. The rats were killed with an overdose of urethane and the brainstems were removed immediately. The NTS was dissected by micropunch (1-mm inner diameter) from a 1-mm thick brainstem slice at the level of the obex under a microscope. Total protein was prepared by homogenizing the NTS tissue in lysis buffer and deproteinized using Microcon YM-30 centrifugal filter units (Millipore, Bedford, MA, USA). The amount of total NO in the samples was determined using a modified procedure based on the purge system of Sievers Nitric Oxide Analyzer (NOA 280i) (Sievers Instruments, Boulder, CO, USA), which involves the use of chemiluminescence (Cheng et al., 2010). Samples (10 μL) were injected into a reflux column containing 0.1 mol L−1 VCl3 in 1 mol L−1 HCl at 90°C to reduce any nitrates and nitrites to NO. NO then combined with the O3 produced by the analyser to form NO2. The resulting emission from the excited NO2 was detected by a photomultiplier tube and recorded digitally (mV). The values were then interpolated using a standard curve of NaNO3 concentrations that was determined concurrently. Measurements were made in triplicate for each sample. The NO levels measured were corrected for the NTS of the rats studied.
In situ detection of ROS in NTS
The endogenous in vivo ROS production of the NTS was determined by dihydroethidium (DHE; Invitrogen, Carlsbad, CA, USA) staining (Cheng et al., 2010). The NTS was dissected from the rats studied (n = 12), quickly frozen, embedded in optimum cutting temperature and then placed in liquid nitrogen. Cryostat slices (30 μm) were stained in the dark for 30 min at 37°C with a 1 μM solution of DHE. The samples were analysed using fluorescence microscopy and the Zeiss LSM Image (Carl Zeiss MicroImaging, Göttingen, Germany) programme.
Immunoblotting analysis
The NTSs of the rats studied (n = 48) were removed after injection of drugs. Total protein was prepared by homogenizing the NTS for 1 h at 4°C in a lysis buffer and proteinase inhibitor cocktail. Protein extracts (20 μg per sample as assessed with the bicinchoninic acid protein assay, Thermo Fisher Scientific Inc., Rockford, IL, USA) were subjected to 6 to 12.5% SDS-Tris glycine gel electrophoresis and transferred to a PVDF membrane (GE Healthcare, Buckingamshire, UK). The membranes were blocked and incubated at 4°C overnight with the appropriate antibody: anti-P-ERK1/2T202/Y204 (1:1000; Cell Signaling Technology, Danvers, MA, USA), anti-ERK1/2 (1:1000; Cell Signaling Technology), anti-P-AktS473 (1:1000; Cell Signaling Technology), anti-Akt (1:1000; Cell Signaling Technology), anti-P-RSKT359/S363 (1:1000; Cell Signaling Technology), anti-RSK (1:1000; Cell Signaling Technology), anti-AMPKT172 (1:1000; Cell Signaling Technology), anti-AMPK (1:1000; Cell Signaling Technology), anti-P-eNOSS1177 (1:1000; BD Biosciences, San Jose, CA, USA) and anti-eNOS (1:1000; BD Biosciences). In addition, anti-actin (1:10000; Millipore), anti-P-nNOSS1416 (1:1000; Abcam, Cambridge, UK), anti-nNOS (1:2000; Millipore) and anti-iNOS (1:1000; Millipore) were also utilized and were diluted in PBS with Tween 20 with 5% BSA. HRP-conjugated anti-mouse (1:10000) or anti-rabbit antibody (1:10000) was used as the secondary antibody, and membranes were incubated with these antibodies at room temperature for 1 h. The membranes were developed with the ECL-Plus detection kit (GE Healthcare).
Ras activation assay
Ras activation was measured using a Ras Activation ELISA Assay Kit (Millipore) following the manufacturer's instructions (n = 24 animals). The assay was initiated by the addition of the recombinant Ras-binding domain of Raf-1 to a glutathione-coated ELISA plate via a GST/glutathione interaction, thus capturing the active Ras and allowing the inactive/GDP-bound Ras to be washed away. The captured active Ras was detected and measured quantitatively through the addition of a monoclonal anti-Ras antibody that detects Ras isoforms from rats. An HRP-conjugated secondary antibody was then added for the detection. Following the addition of the chemiluminescent substrate, signals were measured using a luminometre (Promega, Madison, WI, USA). We also performed a pull-down assay for activated Ras (Millipore). Lysates from the NTS of drug-stimulated rats were precipitated with the Raf-1 Ras-binding domain bound to agarose beads, and the bead-associated proteins were analysed by immunoblotting analysis with an anti-Ras antibody.
Rac1 activation assay
The extent of Rac1 activation was measured with the G-LISA Rac1 Activation Assay Biochem Kit (Cytoskeleton, Denver, CO, USA) according to the manufacturer's protocol (n = 12 rats). This luminescence kit is an ELISA-based assay that measures the GTP-bound form of Rac1. The chemiluminescent signals were obtained using a luminometre (Promega).
Statistical analysis
Student's paired t-test (for comparisons of BP measurements) and one-way anova with Scheffe's post hoc comparison were used to compare group differences. Differences with P < 0.05 were considered significant. All data are expressed as the mean ± SEM.
Results
Central administration of simvastatin induces a systemic vasodepressor effect and NO release through Ras activation in the NTS
In this study, we utilized SHRs as an essential hypertension model to investigate the cardiovascular effects that underlie the i.c.v. infusion of simvastatin into the brain. Figure 1A and Supporting Information Figure S1B show the time course measurements of SBP, taken after the administration of simvastatin beginning at day 0 and continuing to day 3. The maximal decrease in SBP was observed 3 days after simvastatin treatment. DMSO and aCSF (used as solvents) did not influence BP. NO production in the NTS was significantly increased after treatment with simvastatin for 3 days (Figure 1B, lane 1 and lane 2). In addition, we observed a decrease in basal RSNA at day 3 after i.c.v. infusion of simvastatin compared with the control group (Figure 1C). Interestingly, treatment with simvastatin markedly enhanced Ras activation in the NTS (Figure 1D and E, lanes 1 and 2). In addition, we determined whether Ras is involved in the simvastatin-induced depressor response and NO production. Co-administration of simvastatin with a Ras-specific inhibitor, FTS, blocked the simvastatin-induced depressor response in the SHRs, and also the decrease in basal RSNA and NO release in the NTS (Figure 1A–C). The stimulant effect of simvastatin on Ras activation was also reduced after FTS co-treatment (Figure 1D and E, lane 4). These results indicate that Ras activity is required for the simvastatin-induced NO release and depressor response.
Figure 1.
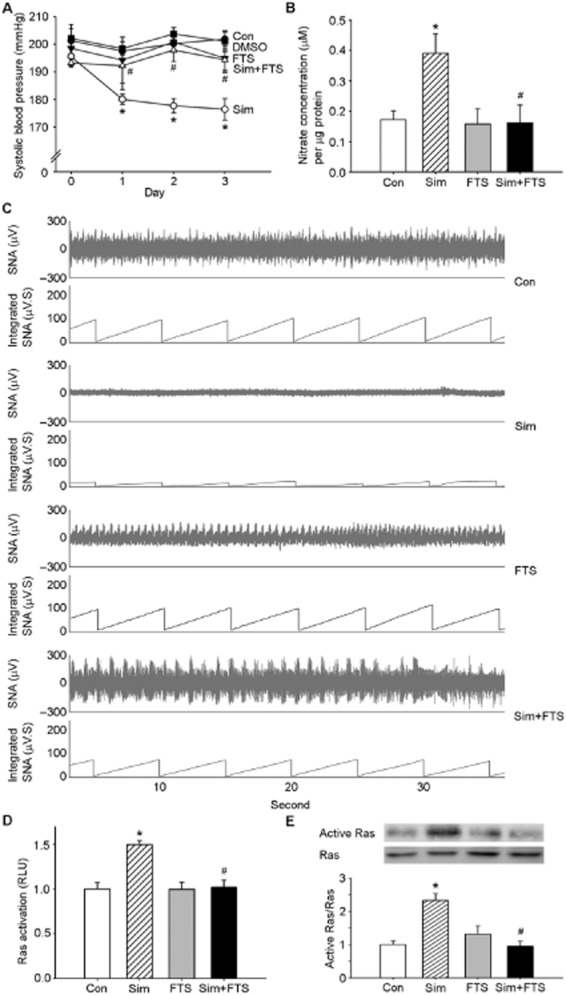
Activation of Ras is required for simvastatin-mediated NO production and BP modulation in the nucleus tractus solitarii (NTS). (A) Time course of SBP after i.c.v. administration of simvastatin for 3 days. SBP was measured on days 0, 1, 2 and 3 after administration. Note that a significant decrease in SBP reached maximal levels at day 3 in the simvastatin-treated group. The depressor effect was reduced by treatment with FTS. (B) Quantification of NO concentrations in the NTS of rats. The bar graph shows the NO concentration. Treatment with simvastatin significantly increased NO levels in the NTS compared with the control group. The elevation of NO production caused by simvastatin was abolished by FTS. (C) Representative tracing show the baseline renal sympathetic nerve activity at day 3 after i.c.v. administration. The i.c.v. infusion of simvastatin produced a decrease in RSNA. Treatment with FTS prevented the sympathoinhibitory effect of simvastatin. (D) Bar diagram showing the activation of Ras after treatment with simvastatin or FTS. (E) Ras activation was determined with a pull-down assay and then detected by immunoblotting analysis using a specific anti-Ras antibody. Note the significant increase in Ras activation after treatment with simvastatin. Simvastatin-mediated Ras activation was attenuated by FTS. Values are shown as the mean ± SEM, n = 6. *P < 0.05 versus control group and #P < 0.05 versus simvastatin group.
Geranylgeranyl pyrophosphate may have a role in central BP modulation
GGPP is a 20-carbon lipid that is necessary for the function of proteins such as Rho and Rac (Maurer-Stroh et al., 2003). We infused GGTI-2133 i.c.v. to explore the effects of GGPP on BP regulation and found that it partially decreased the SBP (Figure 2A). NO production in the NTS increased after treatment with GGTI for 3 days (Figure 2B). From the results shown in Figure 2A and B, GGPP may be involved in central BP regulation in the NTS. Statins can inhibit the synthesis of GGPPs, which help prenylate membrane-bound Rac1 (Goldstein and Brown, 1990). We next determined whether Rac1 was involved in the simvastatin-mediated response. Central administration of simvastatin significantly reduced Rac1 activation in the NTS compared with the control group (Figure 2C). Moreover, previous studies suggested that activation of Rac1 in the NTS contributed to superoxide generation and subsequent hypertension (Nozoe et al., 2007). Figure 2D shows many cells in the NTS of SHRs with high DHE fluorescence activity. Pretreatment with simvastatin significantly decreased the DHE activity.
Figure 2.
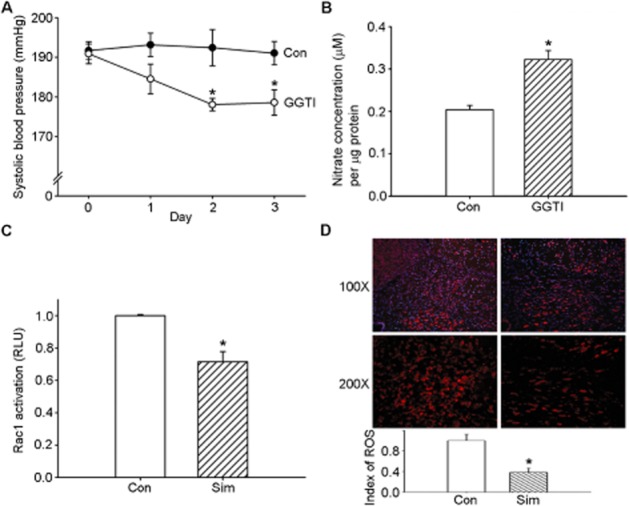
Geranylgeranyl pyrophosphate may be involved in the simvastatin-mediated depressor response. (A) Time course tracing of systolic blood pressure (SBP) after i.c.v. administration of a geranylgeranyl pyrophosphate inhibitor, GGTI, for 3 days. SBP was measured on days 0, 1, 2 and 3 after treatment. (B) Levels ofNO in samples from the nucleus tractus solitarii (NTS) after administration of GGTI. The bar graph shows the NO concentration. (C) Bar graph showing the activation ratio of Rac1 after treatment with simvastatin. Note the significant decrease in Rac1 activation after administration of simvastatin. (D) Representative red fluorescence images depicting reactive oxygen species (ROS)-producing cells in the NTS after the administration of simvastatin. The nuclei in the NTS were counterstained with DAPI and exhibit blue fluorescence. The images were photographed at 100 and 200 × magnification. Bar graph presentation of the ROS index in the NTS of the simvastatin-treated and control groups. The ROS index is the relative mean intensity of fluorescence of DHE. Values are shown as the mean ± SEM, n = 6. *P < 0.05 versus control group.
Simvastatin may activate the PI3K-Akt-eNOS and ERK1/2-RSK-eNOS cascades through Ras signalling to modulate BP control in the NTS
We assessed the role of Ras in simvastatin-mediated NO production within the NTS. Central administration of simvastatin markedly increased the level of eNOS phosphorylation at Ser1177; the addition of FTS reversed simvastatin-induced eNOS phosphorylation (Figure 3A). Similarly, there were increases in the phosphorylation of Akt, ERK1/2 and RSK in the NTS following central administration of simvastatin. In addition, Figure 3E shows that the simvastatin-induced depressor effect was attenuated by the NOS inhibitor, L-NIO. Conversely, immunoblotting analyses of proteins extracted from the NTS demonstrated that i.c.v. treatment with simvastatin did not influence nNOS phosphorylation at Ser1416 or iNOS protein expression compared with the control group (Figure 3F). An i.c.v. infusion of FTS with simvastatin abolished the simvastatin-mediated phosphorylation of Akt, ERK1/2 and RSK in the NTS, suggesting that simvastatin-induced activation of eNOS is dependent on the Ras-mediated up-regulation of the PI3K-Akt-eNOS and ERK1/2-RSK-eNOS cascades.
Figure 3.
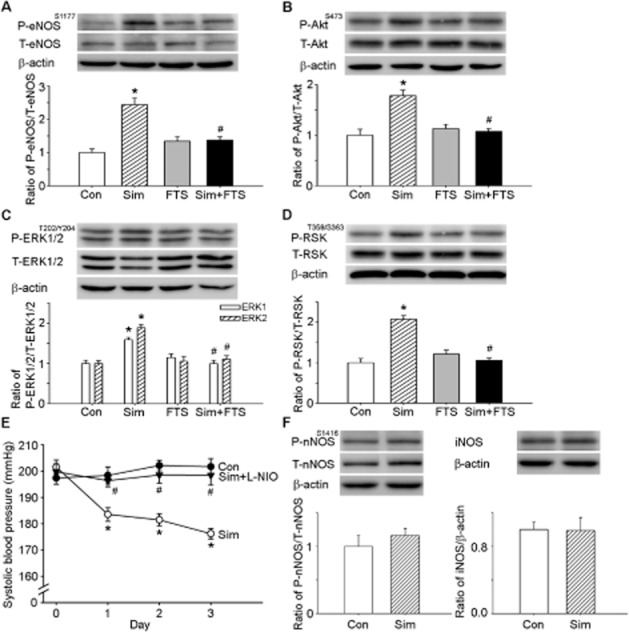
Ras is a critical determinant of simvastatin-enhanced Akt, ERK1/2, ribosomal protein S6 kinase (RSK) and endothelial NOS (eNOS) phosphorylation. (A) The quantitative immunoblotting analysis demonstrates that simvastatin treatment increased the level of phosphorylation of eNOS at Ser1177(P-eNOSS1177) in the nucleus tractus solitarii (NTS). The elevated eNOS phosphorylation ratio observed with simvastatin treatment was reduced by farnesyl thiosalicylic acid (FTS). Densitometric analysis of P-eNOSS1177 protein levels after administration of simvastatin or FTS. (B) The quantitative immunoblotting analysis demonstrates that simvastatin treatment increased the level of P-AktS473 protein in the NTS. The elevated Akt phosphorylation ratio observed with simvastatin treatment was reduced by FTS. Densitometric analysis of P-AktS473 protein levels after administration of simvastatin or FTS. (C) The quantitative immunoblotting analysis demonstrates that simvastatin treatment increased the level of P-ERK1/2T202/Y204 protein in the NTS. The elevated ERK1/2 phosphorylation ratio observed with simvastatin treatment was reduced by FTS. Densitometric analysis of P-ERK1/2T202/Y204 protein levels after administration of simvastatin or FTS. (D) The quantitative immunoblotting analysis demonstrates that simvastatin treatment increased the level of P-RSKT359/S363 protein in the NTS. The elevated RSK phosphorylation ratio observed with simvastatin treatment was reduced by FTS. Densitometric analysis of P-RSKT359/S363 protein levels after administration of simvastatin. (E) Time course of systolic blood pressure (SBP) after i.c.v. administration of simvastatin for 3 days; L-NIO, N(5)-(-iminoethyl)-L-ornithine group. SBP was measured on days 0, 1, 2 and 3 after administration. Note that a significant decrease in SBP reached maximal levels at day 3 in the simvastatin-treated group. The depressor effect was reduced by treatment with L-NIO. (F) Immunoblots depicting the levels of P-nNOSS1416 and iNOS protein in the NTS of the control group and the simvastatin-treated group. Densitometric analysis of P-nNOSS1416 and iNOS protein levels after the administration of simvastatin. Note that there are no significant differences in the nNOSS1416 phosphorylation levels and iNOS expression levels in the NTS between SHR controls and the simvastatin-treated groups. Values are shown as the mean ± SEM, n = 6. *P < 0.05 versus control group and #P < 0.05 versus simvastatin group.
A PI3K inhibitor and a MAPK-ERK kinase (MEK) inhibitor attenuated simvastatin-induced PI3K-Akt-eNOS and ERK1/2-RSK-eNOS pathway activity in the NTS
Next, we performed pharmacological studies to further elucidate the involvement of the PI3K-Akt and MEK-ERK1/2-RSK cascades in simvastatin-induced eNOS phosphorylation. As depicted in Figure 4A, immunoblotting analyses showed that i.c.v. infusion of LY294002 with simvastatin attenuated the simvastatin-induced phosphorylation of Akt at Ser473 and eNOS at Ser1177 in the NTS. In addition, i.c.v. infusion of PD98059 with simvastatin attenuated the simvastatin-induced phsphorylation of ERK1/2 at Thr202/Tyr204, RSK at Thr359/Ser363 and eNOS at Ser1177 in the NTS (Figure 4B). Supporting Information Figure S3 shows that bilateral microinjection of LY294002 (6 pmol·60 nL–1) or PD98059 (6 pmol·60 nL–1) into the NTS of simvastatin-treated SHRs also attenuated the simvastatin-induced activation of PI3K-Akt-eNOS and MEK-ERK1/2-RSK signalling. These results suggest that the effects of simvastatin are due to activation of the PI3K-Akt and MEK-ERK1/2-RSK pathways.
Figure 4.
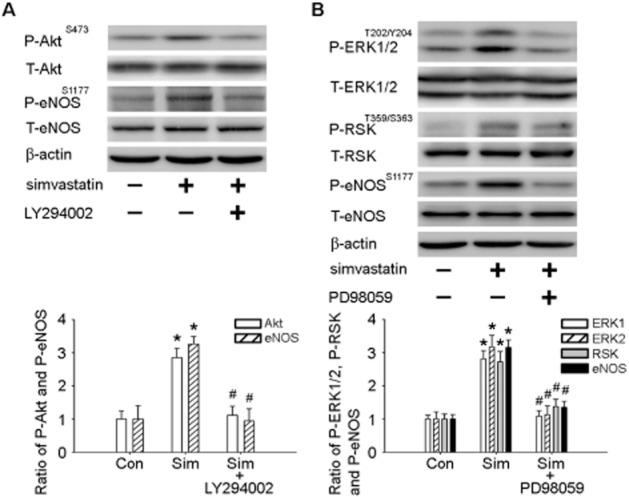
PI3K inhibitor and MAP kinase-ERK kinase (MEK) inhibitor attenuated simvastatin-induced PI3K-Akt-endothelial NOS (eNOS) and ERK1/2-ribosomal protein S6 kinase (RSK)-eNOS pathway activity. (A) Immunoblot showing P-AktS473 and P-eNOSS1177 protein levels after simvastatin treatment with the PI3K inhibitor LY294002. The elevated Akt and eNOS phosphorylation ratios observed with simvastatin treatment were reduced by LY294002 in the nucleus tractus solitarii (NTS). (B) Immunoblot showing P-ERK1/2T202/Y204, P-RSKT359/S363 and P-eNOSS1177 protein levels after simvastatin treatment with the MEK inhibitor PD98059. The elevated ERK1/2, RSK and eNOS phosphorylation ratios observed with simvastatin were reduced by PD98059 in the NTS. Values are shown as the mean ± SEM, n = 6. *P < 0.05 versus control group and #P < 0.05 versus simvastatin group.
Discussion and conclusions
Previously, Lorkowska et al. (2006) demonstrated that there are no significant differences in total cholesterol, low-density lipoprotein (LDL) and high-density lipoprotein levels between hypertensive SHRs and normotensive Wistar-Kyoto rats. Additionally, several clinical studies indicated that statins do not result in reduced BP in moderate hypercholesterolemic, normotensive patients (Kanbay et al., 2005; Terzoli et al., 2005). However, atorvastatin decreases BP and the plasma concentrations of triglycerides and cholesterol in normocholesterolemic SHRs (Wassmann et al., 2001). Also Ferrier et al. (2002) suggested that atorvastatin treatment reduces BP, LDL and triglyceride levels in normolipidaemic patients with isolated systolic hypertension. Therefore, we used SHRs as an animal model in our current study to investigate the molecular signalling mechanisms involved in simvastatin-mediated BP control in the NTS. Moreover, our findings in SHRs support those obtained in the clinical trial with normocholesterolemic subjects, which found that statin treatment is beneficial in hypertensive patients (Ferrier et al., 2002). However, we could not exclude the effects of cholesterol in the present study and the relationship between cholesterol and BP control after simvastatin treatment needs to be clarified.
Farnesyltransferase and geranylgeranyltransferase I have the same CAAX motif near the cysteine residue in their substrates. Many small GTPase proteins belong to the family of CAAX proteins (Maurer-Stroh et al., 2003; McTaggart, 2006). Modification of the Ras family of proteins by FPP is essential for their proper localization and Rac1 and RhoA are also isoprenylated by geranylgeranyltransferase I. However, farnesyltransferase and geranylgeranyltransferase I have cross-activity. K-Ras and N-Ras can be farnesylated by farnesyltransferase; but when farnesyltransferase activity is abolished by FTI, K-Ras and N-Ras can become substrates for geranylgeranylation by geranylgeranyltransferase I (Whyte et al., 1997). Therefore, we utilized FTS to specifically inhibit Ras activity in this study. Interestingly, inhibition of Ras completely inhibited the simvastatin-induced depressor response and NO production in the NTS (Figure 1A and B). Our previous study showed that Ras plays a critical role in the renin-mediated modulation of central BP (Cheng et al., 2012b). However, statins not only inhibit HMG-CoA reductase and inhibit cholesterol synthesis, but they also block the synthesis of FPP and GGPP, which are substrates for isoprenylation. In addition, simvastatin decreases the activation of Ras in the brain (Ghosh et al., 2009; Takayama et al., 2011). In the present study, we determined the pharmacological effects of a Ras inhibitor on cardiovascular parameters and showed that Ras is activated in the NTS after administration of simvastatin, which suggests that cardiovascular regulation by simvastatin in the NTS occurs predominantly through Ras activation.
In this study, we also showed that simvastatin attenuated Rac1 activity and abolished ROS generation in the NTS (Figure 2C and D). ROS have already been found to contribute to neurogenic hypertension in the brain by enhancing the activity of the sympathetic nervous system (Kishi et al., 2004; Hirooka, 2008; Cheng et al., 2010). Furthermore, Nozoe et al. (2007) demonstrated that activation of Rac1 in the NTS can elevate BP and this is associated with increased NAD(P)H oxidase and subsequent generation of intracellular ROS. Our data for Rac1 activity and in situ expression of ROS in the NTS of SHRs provide further support for these observations. Furthermore, we tested the effect of administration of the geranylgeranyltransferase I inhibitor GGTI-2133 into the NTS to provide further evidence that Rac1 and RhoA are involved in central BP regulation and subsequent increased NO release (Figure 2C and D). Statins have been shown to down-regulate Rac1 activity and lower BP through the suppression of oxidative stress (Kishi et al., 2010). The RhoA signalling pathway has also been shown to play a critical role in the simvastatin-mediated BP-lowering effect (Liu et al., 2010; Takayama et al., 2011). In the present study, we observed differences between the effects of central administration of GGTI and simvastatin on BP and NO production. Rac1 and RhoA are isoprenylated by geranylgeranyltransferase I. We utilized GGTI-2133 to directly inhibit the geranylgeranylation of Rac1 and RhoA, whereas simvastatin prevents the synthesis of FPP and GGPP. It has been shown that Rho GTPases may active independently from their membrane anchoring (Samuel and Hynds, 2010). These results indicate that a non-GGPP-dependent mechanism may exist. Neurotrophin-3 induces Ras activation and ultimately activates Rac1 and Schwann cell migration (Yamauchi et al., 2005). Further studies are needed to validate the possible BP-regulating mechanisms that connect simvastatin and the small GTPase signalling cascade in the NTS.
As previously indicated, NO and NOS have an essential role in the modulation of sympathetic nerve activity by BP (Tseng et al., 1996). Many studies have also demonstrated that eNOS gene delivery into the NTS induces a depressor response (Sakai et al., 2000; Tai et al., 2004). Additionally, statins exert beneficial effects on the activation and expression of eNOS in SHRs (Kishi et al., 2008; Ohkawara et al., 2010). However, it has also been demonstrated that administration of statin decreases sympathetic nerve activity by up-regulating NOS expression and NO bioavailability (Kishi et al., 2003; Gao et al., 2008). In addition, atorvastatin has been found to reduce sympathetic nerve activity through reduction of oxidative stress in the CNS (Kishi et al., 2008; 2010). Oxidative stress in the brain contributes to the neuropathogenesis of hypertension (Paravicini and Touyz, 2006). Our results also demonstrated that simvastatin acts as an antioxidant agent; it reduced Rac1 activity and the expression of superoxide in the NTS (Figure 2C and 2D). We also found that simvastatin increased eNOS phosphorylation at Ser1177 in the NTS (Figure 3A and 3F); however, it did not affect the activity or expression of nNOS or iNOS. The inhibition of Ras activity by FTS in the presence of simvastatin decreased eNOS phosphorylation at Ser1177 in the NTS. This suggests that eNOS might be one of the Ras signalling downstream targets of simvastatin that is involved in the sympathetic NO release and regulates BP function in the NTS of SHRs. It has been reported that statins can induce eNOS phosphorylation at Ser1177 and increase NO bioavailability by activation of AMPK (Sun et al., 2006). Previous studies demonstrated that AMPK-Rac1-Akt signalling is involved in the activation of eNOS by simvastatin (Levine et al., 2007; Kou et al., 2009). Laufs et al. revealed that treatment with simvastatin decreases Rho GTP-binding activity, which then induces an up-regulation of eNOS (Laufs et al., 2000a; 2000b). Van Linthout et al. (2007) also demonstrated that treatment with atorvastatin increases eNOS levels by inhibiting RhoA and Rac1 activity in diabetic cardiomyopathy. Atorvastatin increases activation of eNOS via inhibition of Rho activity, but not Ras (Sabbatini et al., 2004). We previously reported that renin modulates BP control in the NTS by affecting the Ras-PI3K-Akt signal-regulated phosphorylation of eNOS (Cheng et al., 2012b). Simvastatin enhances Akt-eNOS signalling in normocholesterolemic rabbits (Kureishi et al., 2000). Furthermore, eNOS activity is increased by activation of the ERK1/2-RSK signalling pathway in rats (Cheng et al., 2012a). In our experiments, i.c.v. infusion of simvastatin into the NTS of SHRs induced phosphorylation of Akt at Ser473, ERK1/2 at Thr202/Tyr204 and RSK at Thr359/Ser363, and these phosphorylation events were reduced by the central administration of FTS (Figure 3). Inhibition of PI3K and MEK blocked the activation of eNOS induced by simvastatin (Figure 4 and Supporting Information Figure S3). However, no significant differences in AMPK phosphorylation or protein expression levels were detected in the NTS after treatment with simvastatin or inhibition of Ras (Supporting Information Figure S2). Thus, we propose that activation of the PI3K-Akt and ERK1/2-RSK pathways may result in the Ras-mediated increase in eNOS by simvastatin in the NTS of SHRs.
In previous studies it was demonstrated that the effect of simvastatin on BP in SHRs varies (Bezerra and Mandarim-de-Lacerda, 2005; Takayama et al., 2011). In clinical investigations, simvastatin was also observed to either lower BP or have no effect (Borghi et al., 2004; Lewandowski et al., 2010). Statins are classified into hydrophilic and lipophilic groups. Simvastatin is highly lipophilic at physiological pH and is smaller in size (Serajuddin et al., 1991; Ramirez et al., 2011; Sierra et al., 2011). Simvastatin is the most effective statin at penetrating the blood-brain barrier (Sierra et al., 2011). We observed slight differences between effects in the pravastatin-treated group (hydrophilic) and the simvastatin-treated group (lipophilic) (Supporting Information Figure S1A). This may suggest that lipophilic statins are more potent at modulating BP through CNS control than hydrophilic statins. Numerous cardioregulatory regions are located in the CNS [e.g. hypothalamus paraventricular nucleus, rostral ventrolateral medulla (RVLM) and NTS] (Guyenet, 2006) and it has been suggested that statins reduce central sympathetic outflow by the up-regulation of NOS in the RVLM (Kishi et al., 2003; Gao et al., 2008). The NTS plays an important role in regulating the set point of BP. In addition, the NTS is also the first integrative site for baroreflex afferent signals in the CNS. Previously, we observed that microinjection of L-arginine into the NTS and RVLM induces depressor effects and reduces SNA (Tseng et al., 1996). Our present data also showed that i.c.v. infusion of simvastatin increased NO production in the NTS and attenuated SNA (Figure 1B and C). Although in this study direct observations were made only in the NTS, we assume that the BP response that we measured after the i.c.v. infusion of simvastatin was mediated by its actions on all of these cardiovascular regulatory centres.
In conclusion, we suggest that simvastatin can modulate central BP control in the NTS and that this control is accomplished by activation of the Ras-PI3K-Akt and Ras-ERK1/2-RSK signalling-regulated phosphorylation of eNOS. We have shown for the first time that Ras contributes to the simvastatin-induced depressor effect and NO release in the NTS. Furthermore, our results indicate that simvastatin may regulate central BP through the blockade of Rac1 activation and down-regulation of superoxide production in the NTS. The current data demonstrate that simvastatin modulates central BP control via activation of the Ras-PI3K-Akt and Ras-ERK1/2-RSK signalling cascades and downstream eNOS.
Acknowledgments
The authors gratefully acknowledge Ms. Yi-Shan Wu for technical assistance, Mr. Chia-Yu Lee for language editing and Mr. Bo-Zone Chen for his invaluable input to this manuscript.
Glossary
- ACSF
artificial CSF
- DAB
diaminobenzidine
- eNOS
endothelial NOS
- FPP
farnesyl pyrophosphate
- FTI
farnesyltransferase inhibitor
- FTS
farnesyl thiosalicylic acid
- GGPP
geranylgeranyl pyrophosphate
- GGTI
geranylgeranyltransferase inhibitor
- iNOS
inducible NOS
- LDL
low-density lipoprotein
- MEK
MAP kinase-ERK kinase
- L-NIO
N(5)-(-iminoethyl)-L-ornithine
- nNOS
neuronal NOS
- NTS
nucleus tractus solitarii
- PBST
PBS with Tween 20
- ROS
reactive oxygen species
- RSK
ribosomal protein S6 kinase
- RSNA
renal sympathetic nerve activity
- RVLM
rostral ventrolateral medulla
- SBP
systolic BP
- SHR
spontaneously hypertensive rats
Sources of funding
This work was supported by funding from the National Science Council NSC100-2321-B-075B-002 and NSC 101-2320-B-075B-002-MY3 (to C-J T), Kaohsiung Veterans General Hospital VGHKS101-024 (to C-J T) and Kaohsiung Medical University Hospital, Kaohsiung Medical University (to W-Y H).
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12317
Figure S1 The blood pressure effects in SHRs after treatment with pravastatin or simvastatin. (A) Time-course tracing of SBP in SHRs after administration of pravastatin (20 mg kg−1 day−1) or simvastatin (20 mg kg−1 day−1) by gavage for 14 days. Filled circles represent SHR control, open circles represent SHR + pravastatin, and inverted filled triangles represent SHR + simvastatin. The rats receiving pravastatin or simvastatin had a decreased SBP as compared to the control group. Values are shown as the mean ± SEM, n = 6. *,#P < 0.05 versus control group. (B) Time course of SBP before and after recovery from simvastatin infused i.c.v. Filled circles represent the contol group, open circles represent the simvastatin group. Note that a significant decrease in SBP was reached with maximal levels at day 3 in the simvastatin-treated SHRs. At Day 4, the administration of the drugs was stopped and the rats began to recover from simvastatin treatment. At Day 7, 3 days after recovery from simvastatin treatment, the SBP of the treated rats had risen to the same level as observed in control rats. Values are shown as the mean ± SEM, n = 6. *P < 0.05 versus control group.
Figure S2 Quantitative immunoblotting analysis of AMPK phosphorylation and expression in the NTS following treatment with simvastatin or FTS. Densitometric analysis of P-AMPKT172 protein levels after the administration of simvastatin or FTS. Note that there are no significant differences in the AMPKT172 phosphorylation levels in the NTS between SHR controls and the simvastatin-treated or simvastatin + FTS-treated groups. Values are shown as the mean ± SEM, n = 6.
Figure S3 Microinjection of a PI3K inhibitor or MAP kinase-ERK kinase (MEK) inhibitor attenuated simvastatin-induced PI3K-Akt-endothelial NOS (eNOS) and ERK1/2-ribosomal protein S6 kinase (RSK)-eNOS pathway activity. (A) Immunoblot showing P-AktS473 and P-eNOSS1177 protein levels after simvastatin treatment with the PI3K inhibitor LY294002. The elevated Akt and eNOS phosphorylation ratios observed with simvastatin treatment were reduced by microinjection of LY294002 in the nucleus tractus solitarii (NTS). (B) Immunoblot showing P-ERK1/2T202/Y204, P-RSKT359/S363 and P-eNOSS1177 protein levels after simvastatin treatment with the MEK inhibitor PD98059. The elevated ERK1/2, RSK and eNOS phosphorylation ratios observed with simvastatin were reduced by microinjection of PD98059 in the NTS. Values are shown as the mean ± SEM, n = 6. *P < 0.05 versus control group and #P < 0.05 versus simvastatin group.
References
- Bezerra DG, Mandarim-de-Lacerda CA. Beneficial effect of simvastatin and pravastatin treatment on adverse cardiac remodelling and glomeruli loss in spontaneously hypertensive rats. Clin Sci (Lond) 2005;108:349–355. doi: 10.1042/CS20040292. [DOI] [PubMed] [Google Scholar]
- Borghi C, Dormi A, Veronesi M, Sangiorgi Z, Gaddi A Brisighella Heart Study Working P. Association between different lipid-lowering treatment strategies and blood pressure control in the Brisighella Heart Study. Am Heart J. 2004;148:285–292. doi: 10.1016/j.ahj.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- Cheng PW, Wu AT, Lu PJ, Yang YC, Ho WY, Lin HC, et al. Central hypotensive effects of neuropeptide Y are modulated by endothelial nitric oxide synthase after activation by ribosomal protein S6 kinase. Br J Pharmacol. 2012a;167:1148–1160. doi: 10.1111/j.1476-5381.2012.02077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WH, Lu PJ, Ho WY, Tung CS, Cheng PW, Hsiao M, et al. Angiotensin II inhibits neuronal nitric oxide synthase activation through the ERK1/2-RSK signaling pathway to modulate central control of blood pressure. Circ Res. 2010;106:788–795. doi: 10.1161/CIRCRESAHA.109.208439. [DOI] [PubMed] [Google Scholar]
- Cheng WH, Lu PJ, Hsiao M, Hsiao CH, Ho WY, Cheng PW, et al. Renin activates PI3K-Akt-eNOS signalling through the angiotensin AT(1) and Mas receptors to modulate central blood pressure control in the nucleus tractus solitarii. Br J Pharmacol. 2012b;166:2024–2035. doi: 10.1111/j.1476-5381.2012.01832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier KE, Muhlmann MH, Baguet JP, Cameron JD, Jennings GL, Dart AM, et al. Intensive cholesterol reduction lowers blood pressure and large artery stiffness in isolated systolic hypertension. J Am Coll Cardiol. 2002;39:1020–1025. doi: 10.1016/s0735-1097(02)01717-5. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang W, Zucker IH. Simvastatin inhibits central sympathetic outflow in heart failure by a nitric-oxide synthase mechanism. J Pharmacol Exp Ther. 2008;326:278–285. doi: 10.1124/jpet.107.136028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Roy A, Matras J, Brahmachari S, Gendelman HE, Pahan K. Simvastatin inhibits the activation of p21ras and prevents the loss of dopaminergic neurons in a mouse model of Parkinson's disease. J Neurosci. 2009;29:13543–13556. doi: 10.1523/JNEUROSCI.4144-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7:335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- Halcox JP, Deanfield JE. Beyond the laboratory: clinical implications for statin pleiotropy. Circulation. 2004;109:II42–II48. doi: 10.1161/01.CIR.0000129500.29229.92. [DOI] [PubMed] [Google Scholar]
- Hirooka Y. Role of reactive oxygen species in brainstem in neural mechanisms of hypertension. Auton Neurosci. 2008;142:20–24. doi: 10.1016/j.autneu.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Ho WY, Lu PJ, Hsiao M, Hwang HR, Tseng YC, Yen MH, et al. Adenosine modulates cardiovascular functions through activation of extracellular signal-regulated kinases 1 and 2 and endothelial nitric oxide synthase in the nucleus tractus solitarii of rats. Circulation. 2008;117:773–780. doi: 10.1161/CIRCULATIONAHA.107.746032. [DOI] [PubMed] [Google Scholar]
- Huang HN, Lu PJ, Lo WC, Lin CH, Hsiao M, Tseng CJ. In situ Akt phosphorylation in the nucleus tractus solitarii is involved in central control of blood pressure and heart rate. Circulation. 2004;110:2476–2483. doi: 10.1161/01.CIR.0000145116.75657.2D. [DOI] [PubMed] [Google Scholar]
- John S, Schlaich M, Langenfeld M, Weihprecht H, Schmitz G, Weidinger G, et al. Increased bioavailability of nitric oxide after lipid-lowering therapy in hypercholesterolemic patients: a randomized, placebo-controlled, double-blind study. Circulation. 1998;98:211–216. doi: 10.1161/01.cir.98.3.211. [DOI] [PubMed] [Google Scholar]
- John S, Delles C, Jacobi J, Schlaich MP, Schneider M, Schmitz G, et al. Rapid improvement of nitric oxide bioavailability after lipid-lowering therapy with cerivastatin within two weeks. J Am Coll Cardiol. 2001;37:1351–1358. doi: 10.1016/s0735-1097(01)01128-7. [DOI] [PubMed] [Google Scholar]
- Kanbay M, Yildirir A, Bozbas H, Ulus T, Bilgi M, Muderrisoglu H, et al. Statin therapy helps to control blood pressure levels in hypertensive dyslipidemic patients. Ren Fail. 2005;27:297–303. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG Group NCRRGW. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi T, Hirooka Y, Mukai Y, Shimokawa H, Takeshita A. Atorvastatin causes depressor and sympatho-inhibitory effects with upregulation of nitric oxide synthases in stroke-prone spontaneously hypertensive rats. J Hypertens. 2003;21:379–386. doi: 10.1097/00004872-200302000-00030. [DOI] [PubMed] [Google Scholar]
- Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contribute to neural mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109:2357–2362. doi: 10.1161/01.CIR.0000128695.49900.12. [DOI] [PubMed] [Google Scholar]
- Kishi T, Hirooka Y, Shimokawa H, Takeshita A, Sunagawa K. Atorvastatin reduces oxidative stress in the rostral ventrolateral medulla of stroke-prone spontaneously hypertensive rats. Clin Exp Hypertens. 2008;30:3–11. doi: 10.1080/10641960701813429. [DOI] [PubMed] [Google Scholar]
- Kishi T, Hirooka Y, Konno S, Sunagawa K. Sympathoinhibition induced by centrally administered atorvastatin is associated with alteration of NAD(P)H and Mn superoxide dismutase activity in rostral ventrolateral medulla of stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol. 2010;55:184–190. doi: 10.1097/FJC.0b013e3181ce9681. [DOI] [PubMed] [Google Scholar]
- Koh KK, Quon MJ, Waclawiw MA. Are statins effective for simultaneously treating dyslipidemias and hypertension? Atherosclerosis. 2008;196:1–8. doi: 10.1016/j.atherosclerosis.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou R, Sartoretto J, Michel T. Regulation of Rac1 by simvastatin in endothelial cells: differential roles of AMP-activated protein kinase and calmodulin-dependent kinase kinase-β. J Biol Chem. 2009;284:14734–14743. doi: 10.1074/jbc.M808664200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota Y, Umegaki K, Kagota S, Tanaka N, Nakamura K, Kunitomo M, et al. Evaluation of blood pressure measured by tail-cuff methods (without heating) in spontaneously hypertensive rats. Biol Pharm Bull. 2006;29:1756–1758. doi: 10.1248/bpb.29.1756. [DOI] [PubMed] [Google Scholar]
- Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufs U, Endres M, Custodis F, Gertz K, Nickenig G, Liao JK, et al. Suppression of endothelial nitric oxide production after withdrawal of statin treatment is mediated by negative feedback regulation of rho GTPase gene transcription. Circulation. 2000a;102:3104–3110. doi: 10.1161/01.cir.102.25.3104. [DOI] [PubMed] [Google Scholar]
- Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, et al. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest. 2000b;106:15–24. doi: 10.1172/JCI9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine YC, Li GK, Michel T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells: evidence for an AMPK → Rac1 → Akt → endothelial nitric-oxide synthase pathway. J Biol Chem. 2007;282:20351–20364. doi: 10.1074/jbc.M702182200. [DOI] [PubMed] [Google Scholar]
- Lewandowski J, Sinski M, Bidiuk J, Abramczyk P, Dobosiewicz A, Ciarka A, et al. Simvastatin reduces sympathetic activity in men with hypertension and hypercholesterolemia. Hypertens Res. 2010;33:1038–1043. doi: 10.1038/hr.2010.137. [DOI] [PubMed] [Google Scholar]
- Liao JK. Isoprenoids as mediators of the biological effects of statins. J Clin Invest. 2002;110:285–288. doi: 10.1172/JCI16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ, Wang XG, Tang YB, Chen JH, Lv XF, Zhou JG, et al. Simvastatin ameliorates rat cerebrovascular remodeling during hypertension via inhibition of volume-regulated chloride channel. Hypertension. 2010;56:445–452. doi: 10.1161/HYPERTENSIONAHA.110.150102. [DOI] [PubMed] [Google Scholar]
- Lorkowska B, Bartus M, Franczyk M, Kostogrys RB, Jawien J, Pisulewski PM, et al. Hypercholesterolemia does not alter endothelial function in spontaneously hypertensive rats. J Pharmacol Exp Ther. 2006;317:1019–1026. doi: 10.1124/jpet.105.098798. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTaggart SJ. Isoprenylated proteins. Cell Mol Life Sci. 2006;63:255–267. doi: 10.1007/s00018-005-5298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer-Stroh S, Washietl S, Eisenhaber F. Protein prenyltransferases. Genome Biol. 2003;4:212–220. doi: 10.1186/gb-2003-4-4-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merla R, Ye Y, Lin Y, Manickavasagam S, Huang MH, Perez-Polo RJ, et al. The central role of adenosine in statin-induced ERK1/2, Akt, and eNOS phosphorylation. Am J Physiol Heart Circ Physiol. 2007;293:H1918–H1928. doi: 10.1152/ajpheart.00416.2007. [DOI] [PubMed] [Google Scholar]
- Nozoe M, Hirooka Y, Koga Y, Sagara Y, Kishi T, Engelhardt JF, et al. Inhibition of Rac1-derived reactive oxygen species in nucleus tractus solitarius decreases blood pressure and heart rate in stroke-prone spontaneously hypertensive rats. Hypertension. 2007;50:62–68. doi: 10.1161/HYPERTENSIONAHA.107.087981. [DOI] [PubMed] [Google Scholar]
- Ohkawara H, Ishibashi T, Saitoh S, Inoue N, Sugimoto K, Kamioka M, et al. Preventive effects of pravastatin on thrombin-triggered vascular responses via Akt/eNOS and RhoA/Rac1 pathways in vivo. Cardiovasc Res. 2010;88:492–501. doi: 10.1093/cvr/cvq221. [DOI] [PubMed] [Google Scholar]
- Paravicini TM, Touyz RM. Redox signaling in hypertension. Cardiovasc Res. 2006;71:247–258. doi: 10.1016/j.cardiores.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Ramirez C, Tercero I, Pineda A, Burgos JS. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J Alzheimers Dis. 2011;24:161–174. doi: 10.3233/JAD-2010-101653. [DOI] [PubMed] [Google Scholar]
- Sabbatini M, Pisani A, Uccello F, Serio V, Seru R, Paterno R, et al. Atorvastatin improves the course of ischemic acute renal failure in aging rats. J Am Soc Nephrol. 2004;15:901–909. doi: 10.1097/01.asn.0000119573.01290.ae. [DOI] [PubMed] [Google Scholar]
- Sakai K, Hirooka Y, Matsuo I, Eshima K, Shigematsu H, Shimokawa H, et al. Overexpression of eNOS in NTS causes hypotension and bradycardia in vivo. Hypertension. 2000;36:1023–1028. doi: 10.1161/01.hyp.36.6.1023. [DOI] [PubMed] [Google Scholar]
- Samuel F, Hynds DL. RHO GTPase signaling for axon extension: is prenylation important? Mol Neurobiol. 2010;42:133–142. doi: 10.1007/s12035-010-8144-2. [DOI] [PubMed] [Google Scholar]
- Serajuddin AT, Ranadive SA, Mahoney EM. Relative lipophilicities, solubilities, and structure-pharmacological considerations of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitors pravastatin, lovastatin, mevastatin, and simvastatin. J Pharm Sci. 1991;80:830–834. doi: 10.1002/jps.2600800905. [DOI] [PubMed] [Google Scholar]
- Sierra S, Ramos MC, Molina P, Esteo C, Vazquez JA, Burgos JS. Statins as neuroprotectants: a comparative in vitro study of lipophilicity, blood-brain-barrier penetration, lowering of brain cholesterol, and decrease of neuron cell death. J Alzheimers Dis. 2011;23:307–318. doi: 10.3233/JAD-2010-101179. [DOI] [PubMed] [Google Scholar]
- Sun W, Lee T-S, Zhu M, Gu C, Wang Y, Zhu Y, et al. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation. 2006;114:2655–2662. doi: 10.1161/CIRCULATIONAHA.106.630194. [DOI] [PubMed] [Google Scholar]
- Tai MH, Hsiao M, Chan JY, Lo WC, Wang FS, Liu GS, et al. Gene delivery of endothelial nitric oxide synthase into nucleus tractus solitarii induces biphasic response in cardiovascular functions of hypertensive rats. Am J Hypertens. 2004;17:63–70. doi: 10.1016/j.amjhyper.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Takayama N, Kai H, Kudo H, Yasuoka S, Mori T, Anegawa T, et al. Simvastatin prevents large blood pressure variability induced aggravation of cardiac hypertrophy in hypertensive rats by inhibiting RhoA/Ras-ERK pathways. Hypertens Res. 2011;34:341–347. doi: 10.1038/hr.2010.229. [DOI] [PubMed] [Google Scholar]
- Terzoli L, Mircoli L, Raco R, Ferrari AU. Lowering of elevated ambulatory blood pressure by HMG-CoA reductase inhibitors. J Cardiovasc Pharmacol. 2005;46:310–315. doi: 10.1097/01.fjc.0000175432.56789.e6. [DOI] [PubMed] [Google Scholar]
- Tseng CJ, Liu HY, Lin HC, Ger LP, Tung CS, Yen MH. Cardiovascular effects of nitric oxide in the brain stem nuclei of rats. Hypertension. 1996;27:36–42. doi: 10.1161/01.hyp.27.1.36. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Van Linthout S, Riad A, Dhayat N, Spillmann F, Du J, Dhayat S, et al. Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia. 2007;50:1977–1986. doi: 10.1007/s00125-007-0719-8. [DOI] [PubMed] [Google Scholar]
- Wassmann S, Laufs U, Baumer AT, Muller K, Ahlbory K, Linz W, et al. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension. 2001;37:1450–1457. doi: 10.1161/01.hyp.37.6.1450. [DOI] [PubMed] [Google Scholar]
- Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272:14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- Yamauchi J, Miyamoto Y, Tanoue A, Shooter EM, Chan JR. Ras activation of a Rac1 exchange factor, Tiam1, mediates neurotrophin-3-induced Schwann cell migration. Proc Natl Acad Sci U S A. 2005;102:14889–14894. doi: 10.1073/pnas.0507125102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.