

Abstract
BACKGROUND AND PURPOSE
The cannabinoid receptor type 1 (CB1) has an allosteric binding site. The drugs ORG27569 {5-chloro-3-ethyl-N-[2-[4-(1-piperidinyl)phenyl]ethyl]-1H-indole-2-carboxamide} and PSNCBAM-1 {1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl)phenyl]urea} have been extensively characterized with regard to their effects on signalling of the orthosteric ligand CP55,940 {(−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol}, and studies have suggested that these allosteric modulators increase binding affinity but act as non-competitive antagonists in functional assays. To gain a deeper understanding of allosteric modulation of CB1, we examined real-time signalling and trafficking responses of the receptor in the presence of allosteric modulators.
EXPERIMENTAL APPROACH
Studies of CB1 signalling were carried out in HEK 293 and AtT20 cells expressing haemagglutinin-tagged human and rat CB1. We measured real-time accumulation of cAMP, activation and desensitization of potassium channel-mediated cellular hyperpolarization and CB1 internalization.
KEY RESULTS
ORG27569 and PSNCBAM-1 produce a complex, concentration and time-dependent modulation of agonist-mediated regulation of cAMP levels, as well as an increased rate of desensitization of CB1-mediated cellular hyperpolarization and a decrease in agonist-induced receptor internalization.
CONCLUSIONS AND IMPLICATIONS
Contrary to previous studies characterizing allosteric modulators at CB1, this study suggests that the mechanism of action is not non-competitive antagonism of signalling, but rather that enhanced binding results in an increased rate of receptor desensitization and reduced internalization, which results in time-dependent modulation of cAMP signalling. The observed effect of the allosteric modulators is therefore dependent on the time frame over which the signalling response occurs. This finding may have important consequences for the potential therapeutic application of these compounds.
Keywords: cannabinoid receptor, allosteric modulator, desensitization, internalization, receptor signalling
Introduction
The cannabinoid type 1 receptor (CB1) is a GPCR present at high levels throughout the CNS (Glass et al., 1997). The signal transduction pathways described for CB1 include pertussis toxin (PTX)-sensitive Gαi-mediated inhibition of AC as well as activation of AC after inhibition of the Gαi-mediated pathway by PTX treatment (Glass and Felder, 1997). CB1 has also been shown to be linked to Gαq and produce elevations in intracellular Ca2+ in a cell type and ligand-specific manner (Sugiura et al., 1996; Lauckner et al., 2005). Additionally, stimulation of CB1 leads to Gβγ-mediated activation of G protein-coupled inwardly rectifying potassium channels (GIRKs) (Mackie et al., 1995) and inhibition of voltage-gated calcium channels as well as activation of the MAPK pathways leading to ERK1/2 phosphorylation (Howlett, 2005).
Allosteric modulation of GPCRs is an emerging therapeutic strategy that can potentially provide improved selectivity and safety, along with maintenance of spatial and temporal regulation associated with native receptor signalling (May et al., 2004). The binding of an allosteric modulator may cause a conformational change in the receptor protein that is transmitted to the orthosteric site, essentially creating a GPCR with novel binding and functional properties (Kenakin and Miller, 2010; Luttrell and Kenakin, 2011; Valant et al., 2012; Wootten et al., 2012).
CB1 is of considerable therapeutic interest for the treatment of neurodegenerative disease (Scotter et al., 2010; Gowran et al., 2011), pain (Sagar et al., 2009) and multiple sclerosis (Correa et al., 2007; Docagne et al., 2007). Interest is now turning to improved drug therapies for these receptors through development of biased ligands and allosteric modulators. Several CB1 allosteric modulators have been described. Price et al. (2005) reported a series of structurally related small molecules (ORG27569, ORG27759 and ORG29647) that were shown to be allosteric enhancers of the CP55,940 {(−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol} binding, but which produced insurmountable antagonism (allosteric inhibition) in guanine 5′-3-O-(thio)triphosphate (GTPγS) assays of both CP55,940 and WIN55,212-2 {R(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo [1,2,3,-de]-1,4-benzoxacin-6-yl]-1-naphtalenylmethanone mesylate} (Price et al., 2005). PSNCBAM-1 {1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl)phenyl]urea}, initially described by Horswill et al. (2007), likewise enhanced agonist binding while antagonizing GTPγS activation and the ability of CB1 to reduce mIPSC frequency in cerebellar neurons (Horswill et al., 2007; Wang et al., 2011). PSNCBAM-1 showed more pronounced inhibition of CP55,940 efficacy than that of WIN55,212-2, suggesting ligand-dependent allosteric modulation. ORG27569 {5-chloro-3-ethyl-N-[2-[4-(1-piperidinyl)phenyl]ethyl]-1H-indole-2-carboxamide} has been shown to act as an allosteric agonist in relation to ERK phosphorylation (Ahn et al., 2012; Baillie et al., 2013), although there are conflicting results on whether this is G-protein dependent or independent (Ahn et al., 2012; Baillie et al., 2013). Additionally, ORG27569 has been shown to act as an allosteric enhancer of ERK phosphorylation in the presence of CP55,940 (Baillie et al., 2013).
Almost all of the existing work has utilized signalling assays that examine the cumulative signalling response at a given time point following receptor activation. In this study, we have examined receptor signalling and regulation using real-time kinetic assays, and in doing so have revealed that the allosteric modulator ORG27569 and PSNCBAM-1 show divergent modulation of cAMP, GIRK-mediated hyperpolarization and desensitization and agonist-induced CB1 internalization.
Methods
Drugs
Drug stocks were made up in ethanol (CP55,940 and WIN55,212-2) or DMSO (forskolin, ORG27569 and PSNCBAM-1) and diluted to give final solvent concentrations of 0.05–0.2%. Anandamide, CP55,940, WIN55,212-2, forskolin and ORG27569 were purchased from Tocris Bioscience (Bristol, UK), Cayman Chemical Company (Ann Arbor, MI, USA) or Ascent Scientific (Bristol, UK); SR141716A [N-(piperidin-1-yl)-5-(4-cholrophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide] was a gift from National Institute of Drug Abuse (Rockville, MD, USA) and PSNCBAM-1 was purchased from Axon Medchem (Groningen, the Netherlands). Our drug/molecular target nomenclature conforms to the British Journal of Pharmacology's Guide to receptors and channels (Alexander et al., 2011).
Stable cell lines and cell maintenance
Tissue culture media and reagents were from Life Technologies (Grand Island, NY, USA) or Sigma (Castle Hill, Australia); tissue culture plasticware was from Corning (Corning, NY, USA) or Becton Dickinson (North Ryde, Australia). Cell lines were cultivated in DMEM supplemented with 10% FBS and appropriate selection antibiotics, and incubated in 5% CO2 at 37°C in a humidified atmosphere.
Human embryonic kidney 293 (HEK) cells (ATCC #CRL-1573) were transfected with human CB1 (hCB1) chimerized with three haemagglutinin (HA) tags at the amino terminus (HEK 3HA-hCB1). The hCB1 cDNA construct (Missouri S&T cDNA Resource Center, http://www.cdna.org, #CNR01LTN00) had been sub-cloned via KpnI/PmeI restriction sites from pcDNA3.1(+) to pEF4A (Life Technologies) and sequence verified. Linearized plasmid DNA was transfected into cells with Lipofectamine 2000. A clonal population stably expressing the receptor was isolated and maintained with 250 μg Zeocin·mL−1.
The Mus musculus brain neuroblastoma cell line Neuro-2a (ATCC #CRL-131) was maintained in DMEM supplemented with 10% FBS and 25 mM HEPES (pH 7.4). AtT-20 cells stably transfected with single HA-tagged rat CB1 (AtT-20 HA-rCB1) were a gift from Professor Ken Mackie (Indiana University, Bloomington, IN, USA) (Mackie et al., 1995) and were maintained under selection with 400 μg G418·mL−1.
Assay for cAMP measurement
Cellular cAMP was measured utilizing the pcDNA3L-His-CAMYEL plasmid (ATCC, Manassas, VA, USA), which encodes the cAMP sensor YFP-Epac-RLuc (CAMYEL), a BRET sensor (Jiang et al., 2007). HEK 3HA-hCB1 cells were plated in 10 cm dishes, 1 day prior to transfection. pcDNA3L-His-CAMYEL (3–5 μg) was transfected into cells utilizing linear polyethyleneimine (mw 25 kDa; Polysciences, Warrington, PA, USA) (Verzijl et al., 2008). Twenty-four hours after transfection cells were re-plated in poly-L-lysine (0.2 mg·mL−1 in PBS; Sigma-Aldrich, St. Louis, MO, USA) coated white CulturPlate™-96 plates (PerkinElmer, Waltham, MA, USA) at a density of 55 000 cells per well. Another 24 hours later, cells were serum-starved in HBSS (pH 7.4) containing 1 mg·mL−1 BSA (ICPBio, Auckland, New Zealand) for 30 min prior to assay. Cells were treated with 5 μM Coelenterazine-h (Promega, Madison, WI, USA) for 5 min prior to addition of drugs/vehicle in HBSS plus 1 mg·mL−1 BSA. PTX (Sigma-Aldrich) treatment of HEK 3HA-hCB1 was carried out by incubating cells with PTX (100 ng·mL−1) for 16 h prior to assay.
Emission signals were detected simultaneously at 460/25 nm (Renilla luciferase, RLuc) and 535/25 nm (yellow fluorescent protein, YFP), immediately following drug addition with a Victor-Lite (PerkinElmer) at 37°C. Raw data are presented as an inverse BRET ratio of emission at 460/535 such that an increase in ratio correlates with an increase in cAMP production. AUC analysis was performed in GraphPad Prism (Version 5.02, GraphPad Software, Inc., La Jolla, CA, USA) with data normalized to individual assay basal and forskolin values. pEC50 values were then calculated for individual experiments by fitting sigmoidal dose–response curves.
Data were also modelled by subtracting agonist plus forskolin from the comparative treatments at each time point to normalize the data such that agonist plus forskolin equalled zero and fitting ‘plateau followed by one phase association’ curves. Parameters of interest were ‘X0’, the length (in minutes) of the initial plateau phase that represented cAMP levels consistent with agonist plus forskolin, and the ‘top plateau’, which indicated the maximum measured cAMP level relative to that for forskolin alone. Curves were fitted either with weighting of 1/x2 (so as to give precedence to earlier time points in the traces when raw data were most consistent between intra-experiment replicates) or without weighting as best fit the data for the individual trace (r squared values). Top plateau data for individual experiments were subsequently normalized to forskolin alone (100%), plotted, and pEC50 values calculated by fitting sigmoidal dose–response curves in GraphPad Prism.
Membrane potential assay
AtT20 HA-rCB1 cells grown to optimum confluence (approximately 90%), trypsinized and transferred into clear-bottomed black walled 96-well plates in L15 medium supplemented with 1% FBS. The cells were plated in a volume of 100 μL and were incubated in humidified room air at 37°C overnight. Changes in membrane potential were measured with the membrane potential blue dye kit from Molecular Devices (Sunnyvale, CA, USA) using a FLEX Station 3 Microplate Reader (Molecular Devices), as outlined in Knapman et al. (2013). Briefly, membrane potential blue dye was dissolved in low potassium HEPES-buffered saline (HBS) of composition (mM): NaCl 140, CaCl2 1.3, MgCl2 0.5, HEPES 22, Na2HPO4 0.338, NaHCO3 4.17, KH2PO4 0.44, MgSO4 0.4, glucose 10 (pH to 7.3, osmolarity = 315 ± 15 mosmol) and 100 μL was loaded into each well of the plate 1 h prior to testing in the FLEX station at 37°C. Fluorescence was measured every 2 s (λexcitation = 530 nm, λemission = 565 nm) for the duration of the experiment (Knapman et al., 2013). Drugs were added after at least 2 min of baseline recording. In experiments where one drug addition was made, 50 μL of drug dissolved in HBS was added, or for two drug additions, 25 μL was added each time. Concentration–response curves were fitted to individual experiments utilizing sigmoidal dose–response curve (constrained to bottom = 0% change in RFU) to produce pEC50 values.
Internalization assay
Surface hCB1 expression and the degree of internalization was determined utilizing a live cell antibody feeding technique and quantified via the Discovery-1 automated fluorescent microscope (Molecular Devices) as previously described (Cao et al., 1999; Grimsey et al., 2008). In brief, HEK 3HA-hCB1 cells were seeded at 30 000 cells per well in poly-L-lysine treated 96-well, flat bottom clear plates (Nunc, Roskilde, Denmark). Approximately 24 h later, cells were serum-starved in DMEM with 5 mg·mL−1 BSA (DMEM-BSA) for 30 min at 37°C and subsequently incubated with anti-mouse Monoclonal HA11 antibody (MMS-101P, Covance, Princeton, NJ, USA) diluted 1:500 in DMEM-BSA at 37°C for 30 min. After two washes with DMEM-BSA, drugs were applied to cells. For concentration–response experiments, all drugs were applied for 60 min, whereas for time course experiments, all conditions were incubated in equivalent vehicle for the duration of the experiment and drugs were added in a time series at twice the final desired concentration. Following drug incubation, plates were placed on ice to prevent any further receptor trafficking after which they were incubated with Alexa Fluor® 488-conjugated goat anti-mouse antibody (Life Technologies) diluted 1:300 in DMEM-BSA at room temperature for 30 min. Cells were then washed twice in DMEM-BSA, fixed with 4% paraformaldehyde, and stained with Hoechst 33258 (Life Technologies) diluted 1:500 in PBS with 0.2% Triton-X. Images of the cells were acquired with a Discovery-1 microscope (10× objective, four images per well) and experimental effects quantified using MetaMorph software (v.6.2r6, Molecular Devices) by calculating the intensity of fluorescent labelling per cell (Grimsey et al., 2008). Sigmoidal concentration–response curves were fitted utilizing GraphPad Prism (constrained to top plateau equal or below 100%) to produce pEC50 values for independent experiments, and time course experiments were fitted with a one phase exponential decay curve with no constraints.
Statistical analysis
Statistical tests were carried out with SigmaPlot (v.11.0, Systat Software, Chicago, IL, USA). t-Tests were utilized when comparing two data points, one-way anova for more than two data points with one independent variable and two-way anova for datasets with two independent variables. Normality and equality of variance assumptions were tested with Shapiro–Wilk and Levene's tests respectively. When these assumptions were not satisfied, data were either transformed appropriately or an equivalent non-parametric test was utilized. The Holm–Šídák post-test was used to assess multiple comparisons in parametric anova tests. The P-values reported have been adjusted for multiplicity from the test output to allow simultaneous inference to a critical level of 0.05 using (1 – pu)m, where pu is the unadjusted P-value and m is the rank position of pu when ordered largest to smallest (Wright, 1992). In graphical representations, * indicates a P-value of 0.01–0.05.
Results
Real-time cAMP BRET measurement from hCB1 stimulated with CP55,940 and allosteric modulators ORG27569 and PSNCBAM-1
Using a real-time kinetic BRET CAMYEL assay (Jiang et al., 2007) in the absence of PDE inhibitor, we found that the HEK 3HA-hCB1 cells produced a rapid increase in cAMP in response to forskolin (10 μM) that plateaued within approximately 5 min and was maintained for the entire time course of the assay (approximately 30 min) (Figure 1A). Treatment with the CB1 agonist CP55,940 produced an immediate concentration-dependent inhibition of forskolin-mediated cAMP production. AUC analysis for CP55,940 with forskolin demonstrated a pEC50 of 9.40 ± 0.14 (n = 3) (Figure 1B).
Figure 1.
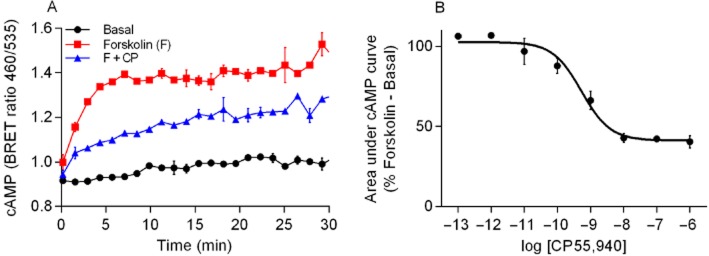
(A) Representative experiment for real-time cAMP BRET assay. HEK 3HA-hCB1 cells were stimulated in the presence of vehicle, forskolin (F) and forskolin plus CP55,940 (1 μM) (F + CP), and emission data for RLuc and YFP were collected over time. Values plotted as raw ratio (SEM) of emissions 460/535 over time (min). (B) Area under the curve analysis for CP55,940 in the presence of 10 μM forskolin. The area under the curve was calculated for the different CP55,940 concentrations used as well as forskolin and basal. This is a representative CP55,940 concentration curve. Data were normalized to forskolin (100%) and basal (0%).
We assessed the kinetics of the CP55,940-induced inhibition of forskolin-mediated cAMP production in the presence of ORG27569 and PSNCBAM-1 (1 μM of either; Figure 2). PSNCBAM-1 completely prevented the CP55,940 (1 μM)-induced inhibition of cAMP at all time points. Interestingly, ORG27569 did not affect the initial inhibition of cAMP accumulation by CP55,940, but approximately 5 min following drug addition, ORG27569 produced an apparent signalling switch that resulted in antagonism of CP55,940-mediated inhibition of AC and subsequently enhanced cAMP levels above those produced by forskolin alone. To ensure that the apparent time lag in producing a response was not an issue with drug solubility or access to the receptor site, assays were repeated with the allosteric modulator pre-incubated for 20 min prior to exposure to forskolin and the orthosteric ligand with no observable difference (data not shown).
Figure 2.
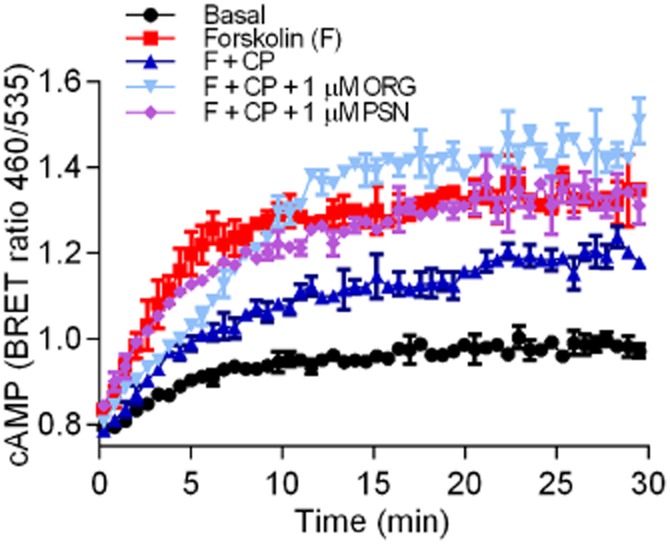
An individual representative real-time cAMP BRET assay for HEK 3HA-hCB1, CP55,940 (CP) and allosteric modulators ORG27569 (ORG) and PSNCBAM-1 (PSN). HEK 3HA-hCB1 cells were stimulated with forskolin (F) and 1 μM CP55,940 in the presence and absence of 1 μM of either ORG or PSN. Values plotted as raw ratio (± SEM) of emissions 460/535 over time (min).
Concentration dependence and detailed analysis of allosteric modulator effects
Levels of cAMP were measured over time with a high concentration of CP55,940 (1 μM) in the presence of varying concentrations of ORG27569 (Figure 3A) and PSNCBAM-1 (Figure 3B). Concentration-dependent effects were apparent with both allosteric modulators, with higher concentrations producing inhibition of the CP55,940 response at earlier time points. Data analysis (as described in the Methods section) allowed for analysis of two parameters: the time of the initial plateau ‘X0’ indicating the time prior to reversal of the inhibition during which there is no antagonism of CP55,940 signalling apparent and the top plateau representing the maximum measured cAMP level in proportion to orthosteric agonist plus forskolin (0%) and forskolin alone (100%). An example of data analysed by this method is provided in Figure 4A. It is apparent that the maximum levels of cAMP production in the presence of CP55,940 and allosteric modulator is concentration dependent (Figure 4B), with ORG27569 and PSNCBAM-1 exhibiting similar potencies (pEC50 = 6.75 ± 0.06 and 6.44 ± 0.14, respectively; P = 0.128). At high concentrations of both allosteric modulators in the presence of CP55,940, the maximum cAMP levels were significantly greater than produced by forskolin alone (≥0.3 μM ORG27569 P = 0.001–0.008; ≥1 μM PSNCBAM-1 P = 0.004–0.012), with extent of the effect being similar for the drugs at the highest concentration (30 μM; P = 0.924). In addition, the concentrations of allosteric modulator that produced a statistically significant inhibition of the CP55,940 response did so with a concentration-dependent lag prior to the initiation of antagonism. As illustrated in Figure 4C, high concentrations of ORG27569 and PSNCBAM-1 antagonized the CP55,940 effect immediately and therefore had an X0 plateau time with CP55,940 plus forskolin that was close to zero. In contrast, intermediate concentrations exhibited a period of uninhibited CP55,940 signalling prior to the onset of antagonism, represented by an X0 plateau time greater than zero. This effect was present with both allosteric modulators but was particularly pronounced with ORG27569. For example, while 1 μM PSNCBAM-1 and 0.3 μM ORG27569 ultimately produced a similar change in CP55,940 plus forskolin signalling, the time to onset of antagonism was 1.6 ± 0.5 min for PSNCBAM-1 and 10.4 ± 2.1 min for ORG27569. We also compared the antagonism produced by ORG27569 and PSNCBAM-1 with that of the orthosteric inverse agonist SR141716A in the presence of CP55,940. As expected, SR141716A produced an immediate antagonism of CP55,940 at each active concentration, with no lag detected at any concentration (Figure 4C). At a high concentration of SR141716A (30 μM), apparent blockade of CB1 constitutive activity was observed (Bouaboula et al., 1997; Landsman et al., 1997), resulting in an increase in cAMP levels above those produced by forskolin alone (P < 0.006; Figure 4B). The elevation of cAMP by 30 μM SR141716A was significantly greater than that produced by ORG27569 and PSNCBAM-1 in the presence of CP55,940 (P = 0.004 and 0.003 respectively).
Figure 3.
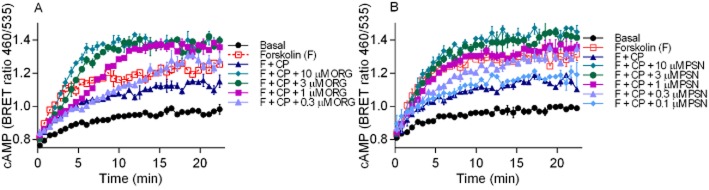
An individual representative cAMP BRET assay for HEK 3HA-hCB1 cells with 10 μM forskolin (F) and CP55,940 (1 μM) in the presence of (A) 0.1–10 μM ORG27569 (ORG) and (B) 0.1–10 μM PSNCBAM-1 (PSN). Emission data for RLuc and YFP were collected over time and values plotted as raw ratio (± SEM) of emissions 460/535 over time (min).
Figure 4.

(A) An individual representative real-time cAMP BRET assay for HEK 3HA-hCB1 cells normalized such that F + CP equalled zero at each point, while forskolin alone equalled 100%. This normalization facilitated fitting of a ‘plateau then exponential association’ curve that allowed measurement of the ‘X0’ plateau, indicating the length of time the allosteric modulator trace tracked with F + CP prior to the initiation of antagonism, while the top plateau of the exponential association allowed measurement of the maximum cAMP level reached (arrows). (B) Summary data for the maximum cAMP level reached (as measured by the top plateau of ‘plateau then exponential association curves’) for HEK 3HA-hCB1 stimulated with 10 μM forskolin and 1 μM CP55,940 in the presence of 1 nM – 30 μM ORG27569 (ORG), PSNCBAM-1 (PSN) or SR141716A (SR). (C) Summary data for the time prior to detection of inhibition (as measured by the ‘X0’ time of ‘plateau then exponential association curves’) of HEK 3HA-hCB1 signalling with 10 μM forskolin and 1 μM CP55,940 (F + CP) in the presence of 0.1–30 μM ORG27569 (ORG), PSNCBAM-1 (PSN) or SR141716A (SR). Only concentrations of allosteric modulator that ultimately produced maximum cAMP levels statistically different from F + CP are represented. For (B) and (C), raw data were normalized to F + CP (0%) and forskolin alone (100%), and plotted as the mean ± SEM of four to five independent experiments.
We then tested whether the allosteric modulators produced any modification of cAMP levels in the absence of orthosteric ligand. ORG27569 or PSNCBAM-1 had no impact on cAMP levels in the absence of forskolin (data not shown) but high concentrations of ORG27569 or PSNCBAM-1 applied with forskolin significantly enhanced cAMP levels above those produced by forskolin alone (ORG27569 10 μM & 30 μM P < 0.05; PSNCBAM-1 30 μM P = 0.007; Figure 5A). SR141716A co-applied with forskolin also enhanced cAMP levels relative to forskolin alone, however, to a significantly greater extent than the allosteric modulators (30 μM P = 0.016–0.024). Interestingly, although 30 μM ORG27569 in the presence and absence of CP55,940 ultimately produced an indistinguishable elevation of forskolin-stimulated cAMP (P = 0.963), qualitative assessment of these experiments indicated that ORG27569 exerted its effects at a slower rate in the absence of CP55,940 (Figure 5B). While the enhancement of forskolin-stimulated cAMP by SR141716A alone was also not significantly different from that observed when applied with CP55,940 (P = 0.147), SR141716A potency appeared to be markedly greater when it was applied alone, consistent with a competitive orthosteric interaction. This is a qualitative observation, as the SR141716A pEC50 could not be accurately defined in the presence of CP55,940. In contrast, both ORG27569 and PSNCBAM-1 exhibited reduced potency (P < 0.001, P = 0.034, respectively) when applied in the absence of CP55,940.
Figure 5.
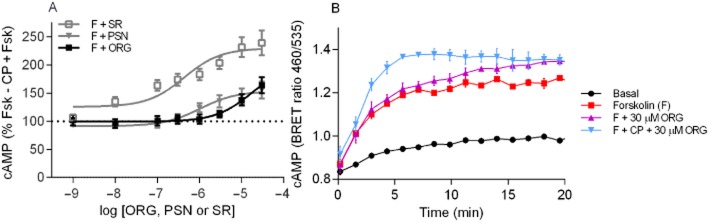
(A) Summary data for the maximum cAMP level reached (as measured by the top plateau of ‘plateau then exponential association curves’) for HEK 3HA-hCB1 cells stimulated with 10 μM forskolin (F) plus 1 nM – 10 μM ORG27569 (ORG), PSNCBAM-1 (PSN) or SR141716A (SR). Raw data were normalized to 1 μM CP55,940 plus forskolin (0%) and forskolin alone (100%), and plotted as the mean ± SEM of four to six independent experiments. (B) An individual representative cAMP BRET assay for hCB1 with 5 μM forskolin (F) and 30 μM ORG27569 (ORG), in the presence and absence of CP55,940 (CP). Emission data for RLuc and YFP were collected over time and values plotted as raw ratio (SEM) of emissions 460/535 over time (min).
PTX treatment unmasks CB1 coupling to Gαs which is blocked by allosteric modulators and prevents the increase in cAMP produced by allosteric modulators in the absence of agonist
We hypothesized that the allosteric modulators may stabilize a conformation of hCB1 that allowed for a time-dependent switch from Gαi-dependent cAMP inhibition to Gαs-dependent stimulation of cAMP. We investigated this theory by pre-incubating the HEK 3HA-hCB1 cells overnight with PTX (100 ng·mL−1). As previously reported (Glass and Felder, 1997), CP55,940 (1 μM) treatment resulted in enhancement of cAMP production above that produced by forskolin alone in PTX-treated cells. Intriguingly, from the raw data traces for these experiments, it is clear that the stimulation of cAMP by hCB1 in the presence of forskolin and PTX is not immediate, but rather only begins to be detected after about 3–5 min. The CP55,940-mediated increase in cAMP was completely abrogated by either 1 μM ORG27569 or PSNCBAM-1 (Figure 6A,B). As described above, in non-PTX-treated cells both ORG27569 and PSNCBAM-1 in the presence of forskolin increased the maximum level of cAMP produced above that produced by forskolin alone; following PTX treatment neither 30 μM ORG27569 nor 30 μM PSNCBAM-1 produced a level of cAMP that was significantly different from forskolin alone (ORG27569 P = 0.677, PSNCBAM-1 P = 0.353).
Figure 6.
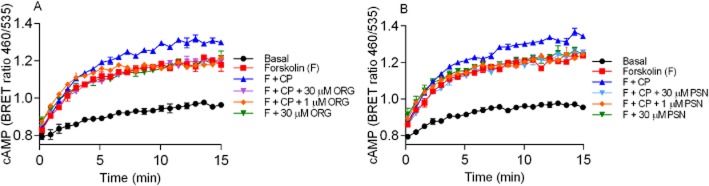
Representative cAMP BRET assay for PTX treated HEK 3HA-hCB1 cells with 10 μM forskolin (F) and forskolin plus 1 μM CP55,940 (F + CP) with (A) 30 μM ORG27569 and 1 μM ORG27569 (ORG) and (B) 30 μM PSNCBAM-1 and 1 μM PSNCBAM-1 (PSN). Additionally forskolin plus 30 μM ORG (A) and 30 μM PSN (B) were assayed in the absence of CP55,940. Emission data for RLuc and YFP were collected over time and values plotted as raw ratio (± SEM) of emissions 460/535 over time (min). Data are a representative of three individual experiments.
Allosteric effects on cAMP BRET measurement in cells endogenously expressing CB1
To verify that the allosteric drug-induced delay in cAMP signalling observed in HEK 3HA-hCB1 cells was not due to receptor overexpression, we investigated the effects of ORG27569 and PSNCBAM-1 in Neuro-2A cells that endogenously express mCB1. We assessed the kinetics of the CP55,940-induced inhibition of forskolin-mediated cAMP production in the presence of either 1 μM ORG27569 or 0.1 μM PSNCBAM-1 (Figure 7A,B respectively). As in HEK 3HA-hCB1 cells, ORG27569 (1 μM) and PSNCBAM-1 (0.1 μM) did not affect the initial inhibition of cAMP accumulation by CP55,940, but minutes following drug addition, ORG27569 and PSNCBAM-1 produced antagonism of CP55,940-mediated inhibition of AC back to the levels produced by forskolin alone. Interestingly, and unlike the recombinant cells, in the presence of CP55,940 neither ORG27569 (1 μM) nor PSNCBAM-1 (0.1 μM) produced enhanced cAMP levels above forskolin alone. To investigate the level of constitutive CB1 activity detectable in these cells, we examined the effect of the inverse agonist SR141716A (1 μM) on forskolin-mediated cAMP. As can be seen in Figure 7A and B, SR141716A did not alter cAMP level from that produced by forskolin alone.
Figure 7.
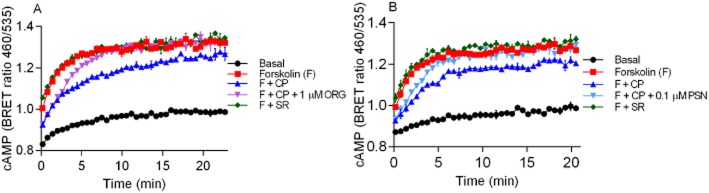
Representative cAMP BRET assay for Neuro-2A cells endogenously expressing mCB1 with 10 μM forskolin (F), forskolin plus 1 μM CP55,940 (F + CP) and, forskolin plus 1 μM SR141716A (F + SR), and (A) 1 μM ORG27569 (ORG) or (B) 0.1 μM PSNCBAM-1 (PSN) in the presence and absence of 1 μM CP55,940. Emission data for RLuc and YFP were collected over time and values plotted as raw ratio (± SEM) of emissions 460/535 over time (min). Data are a representative of three individual experiments.
Activation of inwardly rectifying potassium channels
Activation of G protein-gated inwardly rectifying potassium channels (GIRKs) by G protein βγ subunits represents a relatively direct measure of GPCR activation (Logothetis et al., 1987; Connor et al., 2004). CB1 couples to endogenous GIRKs in AtT-20 cells (Mackie et al., 1995) and we have recently reported that prolonged activation of GIRKs in AtT-20 cells can be studied using a membrane potential sensitive dye (Knapman et al., 2013). In this assay, a decrease in observed fluorescence represents a hyperpolarization of the cell. CP55,940 hyperpolarized AtT-20 cells expressing rCB1 (AtT-20 HA-rCB1) in a concentration-dependent manner (pEC50 8.7 ± 0.1, maximum decrease in fluorescence 36 ± 2%, n = 5; Figure 8A). Pre-incubation of AtT-20 cells with ORG27569 (10 μM, 5 min) did not affect the potency or maximal effect of CP55,940 subsequently applied in the continued presence of the allosteric modulator (CP55,940 + ORG27569 pEC50 8.7 ± 0.1, max 36 ± 2%, n = 5, P = 0.784; Figure 8A). By contrast, in the presence of PSNCBAM-1 (10 μM), the hyperpolarization produced by CP55,940 (300 nM) was decreased to 24 ± 4% (P < 0.005) (Figure 8B). Lower concentrations of PSNCBAM (100 nM, 1 μM) did not affect the peak response (P = 0.23, 0.18 respectively) (Figure 8B).
Figure 8.
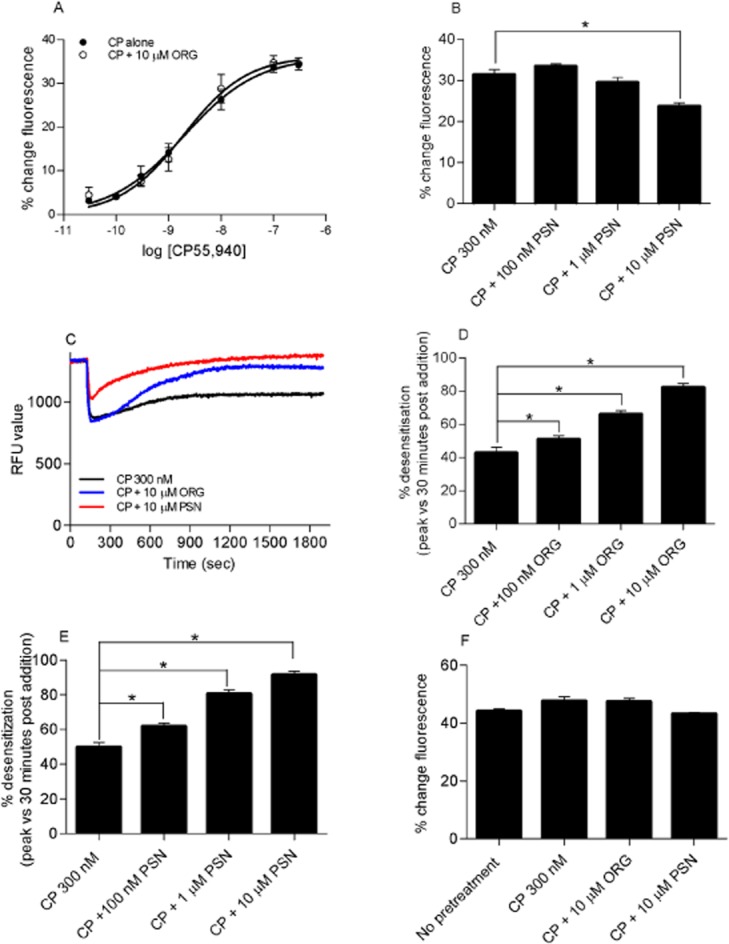
(A) CP55,940 concentration–response curve of hyperpolarization of AtT-20 HA-rCB1 cells in the presence of 10 μM ORG27569. Cells were pre-incubated with either vehicle or 10 μM of ORG27569 (ORG) for 5 min prior to subsequent application of CP55,940 in the continued presence of either vehicle or ORG. In this assay, change in the observed fluorescence represents a hyperpolarization of the cell (n = 5). (B) Hyperpolarization of AtT-20 HA-rCB1 cells stimulated with 300 nM of CP55,940 (CP) in the presence of 10 μM, 1 μM and 100 nM PSNCBAM-1 (PSN). In this assay, change in the observed fluorescence represents a hyperpolarization of the cell (n = 6). (C) Desensitization of AtT-20 HA-rCB1 cells after stimulation with 300 nM of CP in the presence or 10 μM ORG or PSN. This figure shows a representative trace for 300 nM CP, 300 nM CP and 10 μM ORG and 300 nM CP and 10 μM PSN. Drug treatments were added 2 min into the experiment. (D) Desensitization of AtT-20 HA-rCB1 cells after stimulation with CP in the presence of 10 μM, 1 μM and 100 nM ORG. This graph shows the percentage desensitization comparing peak fluorescence after the addition of drug and 30 min post-drug addition (n = 6). (E) Desensitization of AtT-20 HA-rCB1 cells after stimulation with CP in the presence of 10 μM, 1 μM and 100 nM PSN. This graph shows the percentage desensitization comparing peak fluorescence after the addition of drug and 30 min post-drug addition (n = 6). (F) Somatostatin challenge of AtT-20 HA-rCB1 cells after no pretreatment, pretreatment in the presence of CP or CP with either 10 μM ORG or PSN. After 30 min pre-incubation, cells were stimulated with 100 nM somatostatin and the hyperpolarization or % change in fluorescence was measured (n = 5). * indicates a P-value of 0.01–0.05.
Prolonged application of CP55,940 (300 nM) produced a hyperpolarization that slowly reversed over time (Figure 8C). In cells pre-incubated with ORG27569 or PSNCBAM-1 (10 μM, 5 min), the hyperpolarization produced by CP55,940 reversed more rapidly when applied in the continued presence of either ORG27569 or PSNCBAM-1 (Figure 8C). Because of the relatively slow reversal of hyperpolarization produced by CP55,940 alone, we measured the extent of desensitization 30 min after agonist application. The effects of ORG27569 and PSNCBAM-1 were concentration-dependent, with a greater tachyphylaxis of the CP55,940 response at higher concentrations of allosteric modulators (Figure 8D,E). Both ORG27569 (Figure 8D) and PSNCBAM-1 (Figure 8E) significantly enhanced desensitization of CP55,940 at concentrations of 10 μM, 1 μM and 100 nM as compared to CP55,940 alone (ORG27569, P = 0.002–0.036, and PSNCBAM-1, P = 0.002–0.006). The reversal of the cannabinoid-induced hyperpolarization of AtT-20 cells could reflect desensitization of CB1 or their downstream effectors. To investigate this, cells were challenged with somatostatin, which couples to endogenous somatostatin receptors to hyperpolarize AtT-20 cells via GIRKs (Knapman et al., 2013). Application of 100 nM somatostatin after 30 min of CP55,940 (300 nM) with or without ORG27569 (10 μM) or PSNCBAM (10 μM) produced a hyperpolarization that was not significantly different to that produced by somatostatin alone (P = 0.69–0.75 respectively; Figure 8F). This suggests that that desensitization was mediated at the level of CB1.
Receptor internalization
As previously reported (Hsieh et al., 1999), CP55,940 produced concentration-dependent internalization of hCB1 (pEC50 at 60 min = 10.18 ± 0.25). An approximate EC90 concentration of CP55,940 (1 nM) was then used to investigate the effect of ORG27569 and PSNCBAM-1 on orthosteric ligand-induced internalization. Both allosteric modulators produced a concentration-dependent inhibition of CP55,940-induced receptor internalization with pEC50 5.32 ± 0.24 for ORG27659 (n = 3) and pEC50 5.81 ± 0.21 for PSNCBAM-1 (n = 3) (Figure 9A; pEC50s not significantly different P = 0.199). To further understand the mechanism by which this blockade of internalization occurs, the time course of internalization was examined for 1 μM CP55,940 in the presence of 10 μM ORG27569 or PSNCBAM-1 (n = 4) (Figure 9B). CP55,940 induced internalization with a t1/2 of 4.2 ± 0.2 min. In the presence of ORG27569, this trended towards an increased half time (7.5 ± 2.0 min) but this did not reach statistical significance (P > 0.05). PSNCBAM-1 produced a significant increase in half time (10.5 ± 1.5 min) (P < 0.05). As predicted from the concentration–response data, both allosteric modulators significantly inhibited the extent of internalization produced by CP55,940 alone. While CP55,940 produced essentially complete internalization of receptors (0.8 ± 0.7% remaining on cell surface), this was decreased to 43 ± 8% in the presence of ORG27569 or 45 ± 5% of receptors remaining on the cell surface in the presence of PSNCBAM-1. ORG27569 and PSNCBAM-1 at concentrations up to 30 μM produced no change in cell surface expression in the absence of orthosteric ligand (data not shown).
Figure 9.
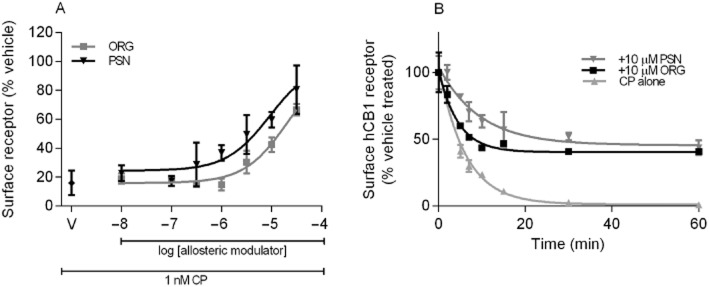
(A) Concentration-dependent inhibition of CP55,940-induced in HEK 3HA-hCB1 internalization with allosteric modulators ORG27569 (ORG) and PSNCBAM-1 (PSN). A representative graph that depicts the fluorescently tagged HEK 3HA-hCB1 surface receptor for each concentration of allosteric modulator normalized to 3HA surface receptor in vehicle-treated cells. CP with allosteric modulator vehicle is shown as V on the graph. (B) Time course of internalization of hCB1 with CP (1 μM) in the presence and absence of either 10 μM ORG or 10 μM PSN. This is a representative time course of internalization. The graph depicts the fluorescently tagged HA-hCB1 surface normalized to vehicle/no treatment.
Orthosteric agonist-specific effects with ORG27569 and PSNCBAM-1
As there has previously been a suggestion of agonist specificity in the response of the allosteric modulators (Wang et al., 2011; Baillie et al., 2013), we compared the effects of the allosteric modulators in the presence of CP55,940 and WIN55,212-2 in all of the reported assays, and on the endogenous agonist anandamide in key assays.
In cAMP assays, both ORG27569 (Figure 10A) and PSNCBAM-1 (Figure 10B) produced a time- and concentration-dependent antagonism of WIN55,212-2-mediated inhibition of forskolin-stimulated cAMP. AUC analysis (GraphPad Prism) showed that WIN55,212-2 inhibited the forskolin-stimulated cAMP accumulation with a pEC50 of 7.90 ± 0.074 (n = 4). ORG27569 and PSNCBAM-1 antagonized 1 μM WIN55,212-2-induced inhibition of forskolin-stimulated cAMP accumulation (pEC50s: ORG27569 = 5.49 ± 0.08; PSNCBAM-1 = 5.98 ± 0.11). ORG27569 (30 μM, P < 0.004), but not PSNCBAM-1 (30 μM, P = 0.131), applied together with WIN55,212-2 resulted in an increase in cAMP levels greater than that produced by forskolin alone (Figure 10C). This is in contrast to the results with CP55,940, where both orthosteric ligands potentiated cAMP accumulation in the presence of forskolin and agonist. ORG27569 was less potent (P < 0.001) but similarly efficacious (30 μM P = 0.600) at antagonizing CB1 signalling when WIN55,212-2 rather than CP55,940 was the agonist. PSNCBAM-1 was equally potent (P = 0.055) but less efficacious (30 μM P = 0.028) at antagonizing WIN55,212-2 rather than CP55,940. Within the concentrations of allosteric modulator that produced a statistically significant inhibition of the WIN55,212-2 response, concentration-dependent lags prior to the initiation of antagonism were again observed (Figure 10D). As seen for CP55,940, ORG27569 produces a more pronounced lag phase than PSNCBAM-1 with WIN55,212-2.
Figure 10.

A representative cAMP BRET assay for HEK 3HA-hCB1 cells with 10 μM forskolin (F) and WIN55,212-2 (1 μM) in the presence of (A) 0.1–10 μM ORG27569 (ORG) and (B) 0.1–10 μM PSNCBAM-1 (PSN). Emission data for RLuc and YFP were collected over time and values plotted as raw ratio (± SEM) of emissions 460/535 over time (min). (C) Summary data for the maximum cAMP level reached (as measured by the top plateau of ‘plateau then exponential association curves’) for hCB1 stimulated with 10 μM forskolin plus 1 μM WIN55,212-2 (F + WIN) and 1 nM–30 μM ORG27569 (ORG) or PSNCBAM-1 (PSN). Raw data were normalized to F + WIN (0%) and forskolin alone (100%), and plotted as the mean ± SEM of three independent experiments. (D) Summary data for the time prior to detection of inhibition (as measured by the ‘X0’ time of ‘plateau then exponential association curves’) of hCB1 signalling with 10 μM forskolin plus 1 μM WIN55,212-2 (F + WIN) and 0.1 μM – 30 μM ORG or PSN. Only concentrations of allosteric modulator that ultimately produced maximum cAMP levels statistically different from F + WIN are represented. Raw data were normalized to forskolin plus WIN55,212-2 (0%) and forskolin alone (100%), and plotted as the mean ± SEM of three independent experiments. (E) An individual representative real-time cAMP BRET assay for hCB1, anandamide (AEA) and allosteric modulators. HEK 3HA-hCB1 cells were stimulated with vehicle, 5 μM forskolin (F), 10 μM AEA as well as either 1 μM of ORG or PSN. Values plotted as raw ratio (SEM) of emissions 460/535 over time (min). (F) Desensitization of AtT-20 HA-rCB1 cells after stimulation with 10 μM WIN in the presence of 10 μM ORG or 10 μM PSN. This graph shows the percentage desensitization comparing peak fluorescence after the addition of drug and 30 min post-drug addition (n = 5). (G) Desensitization of AtT-20 HA-rCB1 cells after stimulation with AEA in the presence of 10 μM ORG or 10 μM PSN. This graph shows percentage desensitization comparing peak fluorescence after the addition of drug and 30 min post-drug addition (n = 5). (H) Concentration-dependent inhibition of 400 nM WIN55,212-2 (WIN)-induced hCB1 internalization with allosteric modulators ORG27569 (ORG) and PSNCBAM-1 (PSN). A representative graph that depicts the fluorescently tagged HA-hCB1 surface receptor for each concentration of allosteric modulator normalized to surface receptor in vehicle-treated cells. WIN with allosteric modulator vehicle is shown as V on the graph. * indicates a P-value of 0.01–0.05.
The endogenous cannabinoid anandamide (10 μM) inhibited forskolin-stimulated cAMP accumulation in HEK 3HA-hCB1 cells; this effect was antagonized by ORG27569 and PSNCBAM-1 (1 μM each) with a lag time prior to antagonism followed by an increase in cAMP over and above that produced by forskolin alone (Figure 10E).
WIN55,212-2 hyperpolarized AtT-20 HA-rCB1 cells with a pEC50 6.5 ± 0.1 and a maximum decrease in fluorescence of 33 ± 3% (n = 5). The desensitization of the WIN55,212-2 (10 μM) hyperpolarization was potentiated by either ORG27569 (10 μM P < 0.007) or PSNCBAM-1 (10 μM, P < 0.008) (Figure 10F). Similarly, 10 μM anandamide-induced hyperpolarization of AtT-20 HA-rCB1 cells was significantly potentiated by ORG27569 (10 μM) or PSNCBAM-1 (10 μM) (P = 0.013 and 0.002 respectively) (Figure 10G).
In receptor internalization assays, WIN55,212-2 produced a concentration-dependent reduction in cell surface receptor expression (pEC50 at 60 min = 7.47 ± 0.10 n = 3) (Figure 10H). Both ORG27569 and PSNCBAM-1 prevented the internalization produced by an approximate EC90 concentration of WIN55,212-2 (400 nM) in a concentration-dependent manner (ORG27569 pEC50 at 60 min = 4.64 ± 0.22; PSNCBAM-1 pEC50 5.15 ± 0.05), these potencies were equivalent to those measured in the presence of CP55,940 (P = 0.159–0.213).
Discussion and conclusions
In this study, we have used real-time kinetic assays to provide novel insights into the interactions between CB1 agonists and allosteric modulators. Using the CAMYEL BRET assay (Jiang et al., 2007), we were able to characterize in detail the real-time HEK 3HA-hCB1 cAMP signalling responses of allosteric modulators ORG27569 and PSNCBAM-1 in the presence and absence of orthosteric ligands. We found that the allosteric modulators exhibited a concentration-dependent time delay in their ability to antagonize the agonist-mediated response, in contrast to the classical inverse agonist SR141716A that produced immediate antagonism of the agonist response (Figure 4C). Thus, at EC50 concentrations of ORG27569 and PSNCBAM-1, there was no immediate effect on the ability of CP55,940 to inhibit cAMP accumulation, but after approximately 9 and 3 min, respectively, the agonist-mediated inhibition of cAMP accumulation was antagonized (Figure 4C). Such complex behaviour would not be apparent in classical cAMP accumulation assays that include a PDE inhibitor and are usually carried out for 15–30 min. Our findings highlight the advantage of using kinetic systems for analysis of signalling interactions.
In addition to showing the delay in action of allosteric modulators, we also demonstrate that they do not simply reverse the agonist-mediated inhibition of forskolin-stimulated cAMP accumulation, but their application together with agonists results in cAMP levels significantly above that produced by forskolin alone. While CB1 dominantly couples to Gαi signalling, it can, under some circumstances, couple to the stimulation of cAMP through a putative Gαs pathway (Bonhaus et al., 1998; Glass and Felder, 1997). We tested whether the allosteric modulators might stabilize a Gαs preferring conformation of the receptor by pre-treating the cells with PTX, which disrupts Gαi-mediated signalling (Katada et al., 1983). In PTX-treated cells, CP55,940 increased cAMP in the presence of forskolin (Glass and Felder, 1997), and interestingly, the CP55,940-induced cAMP increase was completely inhibited by PSNCBAM-1 and ORG27569. If the increase in cAMP above forskolin levels was due to Gαs pathway, it would be expected that PTX treatment would result in the allosteric modulators either not affecting or enhancing the agonist-induced cAMP production observed when competing Gαi pathways were inhibited. Instead, our results suggest that the cAMP increase seen with the allosteric modulators in the presence of agonist is produced downstream of Gαi. Our data also demonstrate that allosteric modulators completely abolish the putative Gαs coupling of CB1 observed in PTX-treated cells. Furthermore, and in contrast to Baillie et al. (2013), our data show that the increase in cAMP above that of forskolin in the presence of high concentrations of allosteric modulators alone is abolished after PTX treatment. Treatment with inverse agonists such as SR141716A also produces an increase in cAMP above that produced by forskolin alone (Bouaboula et al., 1997). This has been suggested to be due to the inverse agonists reducing Gαi-linked constitutive activity of the receptor and is blocked by PTX (Bouaboula et al., 1997; Landsman et al., 1997; MacLennan et al., 1998; Glass and Northup, 1999). Thus, the increase in cAMP produced by the allosteric modulators could also represent inverse agonism by these compounds. Consistent with this, in Neuro-2a cells that endogenously express CB1 at low levels, the allosteric modulators displayed an equivalent delay in antagonizing orthosteric agonist signalling, but no subsequent increase in cAMP above forskolin levels. As seen in Figure 7, no constitutive activity was detected by SR141716A in the Neuro-2a cells either, which is consistent with constitutively active receptors being required for the allosteric modulator response to be detected.
As the delay in antagonism of cAMP signalling did not appear to be due to either drug solubility or a switch in signalling pathways from Gαi to Gαs, we hypothesized that the receptor may be desensitizing more rapidly in the presence of the allosteric modulators, and adopting a constitutively inactive conformation. GIRK activation by CB1 and subsequent channel closing provides a more direct pathway than cAMP for investigating desensitization (Mackie et al., 1995); therefore, we investigated the effect of the allosteric modulators on recovery of membrane potential following agonist-mediated hyperpolarization. Interestingly, ORG27569 at concentrations up to 10 μM did not produce any decrease in maximum hyperpolarization produced by CP55,940. A small but significant reduction in maximal hyperpolarization was observed at 10 μM of PSNCBAM-1 but not 1 μM. Despite this, at concentrations as low as 100 nM, both ORG27569 and PSNCBAM-1 produced a significantly greater repolarization of the cell 30 min following agonist addition, strongly suggesting that the receptors desensitize more rapidly in the presence of the allosteric modulators. We verified that this likely represents desensitization of the receptor and not the ion channel by demonstrating normal hyperpolarization in response to subsequent activation of endogenous somatostatin receptors.
Classical GPCR regulation paradigms suggest that following desensitization by phosphorylation of the receptor, arrestins are recruited, and receptors are subsequently internalized (Magalhaes et al., 2012). Baillie et al. (2013) measured arrestin recruitment by CB1 in the presence of CP55,940 and found that it was antagonized in the presence of ORG27569. Consistent with this observation, our internalization studies showed that both ORG27569 and PSNCBAM-1 produced a concentration-dependent decrease in the extent and rate of agonist-induced CB1 internalization. While it may initially appear counter-intuitive that desensitization is increased while arrestin recruitment and internalization are decreased, our study is not the first to find an apparent divergence of these pathways. For example, previous studies have identified that a non-desensitizing mutant of CB1 recruits β-arrestin-2 (Daigle et al., 2008) and internalizes normally (Jin et al., 1999). Our data, combined with that of Baillie et al. (2013), might therefore suggest that the desensitized conformation of CB1 adopted in the presence of the allosteric modulators is different from that induced in the presence of agonist alone and is less favourable for arrestin recruitment and subsequent internalization.
The effect of ORG27569 on agonist-induced internalization has previously been studied by Ahn et al. (2012), but in contrast with a number of prior studies (Rinaldi-Carmona et al., 1998; Hsieh et al., 1999; Grimsey et al., 2010), they were unable to detect ligand induced wild-type hCB1 internalization due to very low surface expression of the wild-type receptor. Ahn et al. (2012) therefore investigated the effect of ORG27569 on a constitutively inactive form of hCB1-GFP (T210A) and suggested more rapid internalization with co-treatment of ORG27569 and CP55,940 as compared with agonist alone. Subsequently, they have shown that this is β-arrestin-2 dependent (Ahn et al., 2013). While we cannot fully explain this discrepancy, the inactive mutant form of CB1 presumably adopts a suite of receptor conformations quite distinct from those of the wild-type receptor.
CB1 normally degrades following internalization and the cell is re-sensitized by the production and delivery of newly synthesized receptors (Martini et al., 2007; Rozenfeld and Devi, 2008; Grimsey et al., 2010). The longer term consequences of holding desensitized CB1 at the cell surface are difficult to predict and require further study. Cell surface trapping of desensitized receptors may result in an increase in total receptor expression; furthermore, there is also precedent for potential re-sensitization without the requirement for internalization/recycling (Murphy et al., 2011; Doll et al., 2012). Experiments to investigate such possibilities are particularly challenging for cannabinoids due to the inability to washout these highly lipophilic compounds (Grimsey et al., 2010). Uncoupling of the receptor from its protean Gαi coupling in the absence of subsequent internalization could potentially result in the increased cAMP accumulation seen at later time points in our assays via receptor coupling to alternative signalling pathways, such has been observed with the β-adrenergic receptor that switches from Gαs to Gαi coupling after phosphorylation (Lefkowitz et al., 2002). Our findings, however, point more strongly to the allosteric modulators stabilizing a constitutively inactive form of the receptor on the cell surface. Surprisingly, the maximal extent of cAMP produced by the allosteric modulators was significantly lower than that produced by the inverse agonist SR141716A, suggesting an intermediate conformation of the receptor leading to only partial blockade of constitutive activity. Interestingly, Fay and Farrens (2012) suggested that ORG27569 may stabilize an intermediate structure, ‘one that is on the pathway that flows from agonist binding to full receptor activation’, our data rather suggest it is an intermediate on the pathway to full receptor inactivation. As would be expected for an orthosteric ligand, the potency at which SR141716A enhances cAMP production was reduced in the presence of CP55,940; in contrast, the allosteric modulators both exhibited greater potency in the presence of CP55,940, suggesting that the presence of the orthosteric ligand enhances the ability of the allosteric ligands to alter receptor conformation. Consistent with this suggestion, the cAMP data suggested that the rate at which the receptors were ‘turned off’ was more rapid in the presence of the orthosteric ligand (Figure 5B).
Ligand selectivity has been described previously in relation to CB1 and allosteric modulators ORG27569 and PSNCBAM-1 (Wang et al., 2011; Baillie et al., 2013). Our cAMP signalling results suggested that ORG27569 had a slightly lower potency against WIN55,212-2 than CP55,940, whereas PSNCBAM-1 had equivalent potency. Perhaps more surprising however is that the maximal extent of cAMP production reached by PSNCBAM-1 in the presence of WIN55,212-2 was not significantly greater than that produced by forskolin alone, suggesting that WIN55,212-2 prevented the receptor from adopting an inactive conformation. This difference, and those described (Wang et al., 2011; Baillie et al., 2013) likely reflects the differences in proposed binding sites of WIN55,212-2 and CP55,940 to CB1 (Song and Bonner, 1996). Importantly, our studies have also indicated that the allosteric modulators appear to produce a similar time and concentration-dependent modulation of anandamide-induced cAMP accumulation, and an enhanced extent of receptor desensitization to anandamide-induced hyperpolarization, indicating that this mechanism is not constrained to synthetic orthosteric agonists.
Considering our observations, we hypothesize that the allosteric-induced enhancement of orthosteric agonist binding initially results in minimal alteration to Gαi-mediated pathways including cAMP signalling and GIRK-mediated hyperpolarization; however, as a consequence of an enhanced rate of CB1 desensitization, cytoplasmic cAMP levels and hyperpolarization states return to baseline more rapidly than for orthosteric agonist alone, and additionally constitutive receptor activity is further reduced. It is therefore critically important to note that contrary to prior assumptions, these ligands do not act strictly as allosteric antagonists towards Gαi-mediated AC inhibition and GIRK channel activation, but rather reduce the temporal window within which classical signalling may occur. Thus, rather than preventing all signalling, signalling bias may be produced as early signalling events may remain unaffected, while later signalling events may be antagonized. The downstream metabolic and network level effects of this complex paradigm remain to be seen but must be understood if the potential therapeutic application of these compounds is to be realized.
Acknowledgments
The authors thank Dr Meritxell Canals (Monash University) for helpful advice on establishing the CAMYEL assay and Ken Mackie (University of Indiana) for the AtT-20 rCB1. Funding was received for EC from the Neurological Foundation of New Zealand Repatriation Fellowship. C. B. was supported by the PJ Smith Travelling Fellowship and University of Auckland Doctoral Scholarship. M. C. was supported by NHMRC Grant 1002680, W. J. R. by an International Scholarship from Macquarie University.
Glossary
- AEA
arachidonoyl ethanolamide (anandamide)
- CAMYEL
cAMP sensor using YFP-Epac-RLuc
- CB1
cannabinoid receptor type 1
- CP55,940
(−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol
- GIRK
G protein-coupled inwardly-rectifying potassium channel
- GTPγS
guanine 5′-3-O-(thio)triphosphate
- HA
haemagglutinin
- ORG27569
5-chloro-3-ethyl-N-[2-[4-(1-piperidinyl)phenyl]ethyl]-1H-indole-2-carboxamide
- PSNCBAM-1
1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl)phenyl]urea
- PTX
pertussis toxin
- Rluc
Renilla luciferase
- SR141716A
N-(piperidin-1-yl)-5-(4-cholrophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- WIN55,212-2
R(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo [1,2,3,-de]-1,4-benzoxacin-6-yl]-1-naphtalenylmethanone mesylate
- YFP
yellow fluorescent protein
Conflict of interest
The authors declare that they have no conflict of interest.
References
- Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem. 2012;287:12070–12082. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn KH, Mahmoud MM, Shim JY, Kendall DA. Distinct roles of beta-arrestin 1 and beta-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor one (CB1) J Biol Chem. 2013;288:9790–9800. doi: 10.1074/jbc.M112.438804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl 1):S1–324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie GL, Horswill JG, Anavi-Goffer S, Reggio PH, Bolognini D, Abood ME, et al. CB(1) receptor allosteric modulators display both agonist and signaling pathway specificity. Mol Pharmacol. 2013;83:322–338. doi: 10.1124/mol.112.080879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhaus DW, Chang LK, Kwan J, Martin GR. Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist-specific trafficking of intracellular responses. J Pharmacol Exp Ther. 1998;287:884–888. [PubMed] [Google Scholar]
- Bouaboula M, Perrachon S, Milligan L, Canat X, Rinaldi-Carmona M, Portier M, et al. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J Biol Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. A kinase-regulated PDZ-domain interaction controls endocytic sorting of the beta2-adrenergic receptor. Nature. 1999;401:286–290. doi: 10.1038/45816. [DOI] [PubMed] [Google Scholar]
- Connor M, Osborne PB, Christie MJ. Mu-opioid receptor desensitization: is morphine different? Br J Pharmacol. 2004;143:685–696. doi: 10.1038/sj.bjp.0705938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa F, Docagne F, Mestre L, Loria F, Hernangomez M, Borrell J, et al. Cannabinoid system and neuroinflammation: implications for multiple sclerosis. Neuroimmunomodulation. 2007;14:182–187. doi: 10.1159/000110644. [DOI] [PubMed] [Google Scholar]
- Daigle TL, Kearn CS, Mackie K. Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology. 2008;54:36–44. doi: 10.1016/j.neuropharm.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docagne F, Muneton V, Clemente D, Ali C, Loria F, Correa F, et al. Excitotoxicity in a chronic model of multiple sclerosis: neuroprotective effects of cannabinoids through CB1 and CB2 receptor activation. Mol Cell Neurosci. 2007;34:551–561. doi: 10.1016/j.mcn.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Doll C, Poll F, Peuker K, Loktev A, Gluck L, Schulz S. Deciphering micro-opioid receptor phosphorylation and dephosphorylation in HEK293 cells. Br J Pharmacol. 2012;167:1259–1270. doi: 10.1111/j.1476-5381.2012.02080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay JF, Farrens DL. A key agonist-induced conformational change in the cannabinoid receptor CB1 Is blocked by the allosteric ligand Org 27569. J Biol Chem. 2012;287:33873–33882. doi: 10.1074/jbc.M112.352328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Felder CC. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci. 1997;17:5327–5333. doi: 10.1523/JNEUROSCI.17-14-05327.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Northup JK. Agonist selective regulation of G proteins by cannabinoid CB(1) and CB(2) receptors. Mol Pharmacol. 1999;56:1362–1369. doi: 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RL. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77:299–318. doi: 10.1016/s0306-4522(96)00428-9. [DOI] [PubMed] [Google Scholar]
- Gowran A, Noonan J, Campbell VA. The multiplicity of action of cannabinoids: implications for treating neurodegeneration. CNS Neurosci Ther. 2011;17:637–644. doi: 10.1111/j.1755-5949.2010.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsey NL, Narayan PJ, Dragunow M, Glass M. A novel high-throughput assay for the quantitative assessment of receptor trafficking. Clin Exp Pharmacol Physiol. 2008;35:1377–1382. doi: 10.1111/j.1440-1681.2008.04991.x. [DOI] [PubMed] [Google Scholar]
- Grimsey NL, Graham ES, Dragunow M, Glass M. Cannabinoid Receptor 1 trafficking and the role of the intracellular pool: implications for therapeutics. Biochem Pharmacol. 2010;80:1050–1062. doi: 10.1016/j.bcp.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Horswill JG, Bali U, Shaaban S, Keily JF, Jeevaratnam P, Babbs AJ, et al. PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br J Pharmacol. 2007;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett AC. Cannabinoid receptor signaling. Handb Exp Pharmacol. 2005;168:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- Hsieh C, Brown S, Derleth C, Mackie K. Internalization and recycling of the CB1 cannabinoid receptor. J Neurochem. 1999;73:493–501. doi: 10.1046/j.1471-4159.1999.0730493.x. [DOI] [PubMed] [Google Scholar]
- Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T, et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J Biol Chem. 2007;282:10576–10584. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Brown S, Roche JP, Hsieh C, Celver JP, Kovoor A, et al. Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J Neurosci. 1999;19:3773–3780. doi: 10.1523/JNEUROSCI.19-10-03773.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katada T, Tamura M, Ui M. The A protomer of islet-activating protein, pertussis toxin, as an active peptide catalyzing ADP-ribosylation of a membrane protein. Arch Biochem Biophys. 1983;224:290–298. doi: 10.1016/0003-9861(83)90212-6. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev. 2010;62:265–304. doi: 10.1124/pr.108.000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapman A, Santiago M, Du YP, Bennallack PR, Christie MJ, Connor M. A continuous, fluorescence-based assay of mu-opioid receptor activation in AtT-20 cells. J Biomol Screen. 2013;18:269–276. doi: 10.1177/1087057112461376. [DOI] [PubMed] [Google Scholar]
- Landsman RS, Burkey TH, Consroe P, Roeske WR, Yamamura HI. SR141716A is an inverse agonist at the human cannabinoid CB1 receptor. Eur J Pharmacol. 1997;334:R1–R2. doi: 10.1016/s0014-2999(97)01160-6. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, Mackie K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A. 2005;102:19144–19149. doi: 10.1073/pnas.0509588102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, Pierce KL, Luttrell LM. Dancing with different partners: protein kinase a phosphorylation of seven membrane-spanning receptors regulates their G protein-coupling specificity. Mol Pharmacol. 2002;62:971–974. doi: 10.1124/mol.62.5.971. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Kurachi Y, Galper J, Neer EJ, Clapham DE. The beta gamma subunits of GTP-binding proteins activate the muscarinic K+ channel in heart. Nature. 1987;325:321–326. doi: 10.1038/325321a0. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Kenakin TP. Refining efficacy: allosterism and bias in G protein-coupled receptor signaling. Methods Mol Biol. 2011;756:3–35. doi: 10.1007/978-1-61779-160-4_1. [DOI] [PubMed] [Google Scholar]
- Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan SJ, Reynen PH, Kwan J, Bonhaus DW. Evidence for inverse agonism of SR141716A at human recombinant cannabinoid CB1 and CB2 receptors. Br J Pharmacol. 1998;124:619–622. doi: 10.1038/sj.bjp.0701915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes AC, Dunn H, Ferguson SS. Regulation of GPCR activity, trafficking and localization by GPCR-interacting proteins. Br J Pharmacol. 2012;165:1717–1736. doi: 10.1111/j.1476-5381.2011.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini L, Waldhoer M, Pusch M, Kharazia V, Fong J, Lee JH, et al. Ligand-induced down-regulation of the cannabinoid 1 receptor is mediated by the G-protein-coupled receptor-associated sorting protein GASP1. FASEB J. 2007;21:802–811. doi: 10.1096/fj.06-7132com. [DOI] [PubMed] [Google Scholar]
- May LT, Avlani VA, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Curr Pharm Des. 2004;10:2003–2013. doi: 10.2174/1381612043384303. [DOI] [PubMed] [Google Scholar]
- Murphy JE, Roosterman D, Cottrell GS, Padilla BE, Feld M, Brand E, et al. Protein phosphatase 2A mediates resensitization of the neurokinin 1 receptor. Am J Physiol Cell Physiol. 2011;301:C780–C791. doi: 10.1152/ajpcell.00096.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MR, Baillie GL, Thomas A, Stevenson LA, Easson M, Goodwin R, et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Le Duigou A, Oustric D, Barth F, Bouaboula M, Carayon P, et al. Modulation of CB1 cannabinoid receptor functions after a long-term exposure to agonist or inverse agonist in the Chinese hamster ovary cell expression system. J Pharmacol Exp Ther. 1998;287:1038–1047. [PubMed] [Google Scholar]
- Rozenfeld R, Devi LA. Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3. FASEB J. 2008;22:2311–2322. doi: 10.1096/fj.07-102731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar DR, Gaw AG, Okine BN, Woodhams SG, Wong A, Kendall DA, et al. Dynamic regulation of the endocannabinoid system: implications for analgesia. Mol Pain. 2009;5:59. doi: 10.1186/1744-8069-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotter EL, Goodfellow CE, Graham ES, Dragunow M, Glass M. Neuroprotective potential of CB1 receptor agonists in an in vitro model of Huntington's disease. Br J Pharmacol. 2010;160:747–761. doi: 10.1111/j.1476-5381.2010.00773.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song ZH, Bonner TI. A lysine residue of the cannabinoid receptor is critical for receptor recognition by several agonists but not WIN55212-2. Mol Pharmacol. 1996;49:891–896. [PubMed] [Google Scholar]
- Sugiura T, Kodaka T, Kondo S, Tonegawa T, Nakane S, Kishimoto S, et al. 2-Arachidonoylglycerol, a putative endogenous cannabinoid receptor ligand, induces rapid, transient elevation of intracellular free Ca2+ in neuroblastoma x glioma hybrid NG108-15 cells. Biochem Biophys Res Commun. 1996;229:58–64. doi: 10.1006/bbrc.1996.1757. [DOI] [PubMed] [Google Scholar]
- Valant C, Robert Lane J, Sexton PM, Christopoulos A. The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2012;52:153–178. doi: 10.1146/annurev-pharmtox-010611-134514. [DOI] [PubMed] [Google Scholar]
- Verzijl D, Storelli S, Scholten DJ, Bosch L, Reinhart TA, Streblow DN, et al. Noncompetitive antagonism and inverse agonism as mechanism of action of nonpeptidergic antagonists at primate and rodent CXCR3 chemokine receptors. J Pharmacol Exp Ther. 2008;325:544–555. doi: 10.1124/jpet.107.134783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Horswill JG, Whalley BJ, Stephens GJ. Effects of the allosteric antagonist 1-(4-chlorophenyl)-3-[3-(6-pyrrolidin-1-ylpyridin-2-yl)phenyl]urea (PSNCBAM-1) on CB1 receptor modulation in the cerebellum. Mol Pharmacol. 2011;79:758–767. doi: 10.1124/mol.110.068197. [DOI] [PubMed] [Google Scholar]
- Wootten D, Savage EE, Valant C, May LT, Sloop KW, Ficorilli J, et al. Allosteric modulation of endogenous metabolites as an avenue for drug discovery. Mol Pharmacol. 2012;82:281–290. doi: 10.1124/mol.112.079319. [DOI] [PubMed] [Google Scholar]
- Wright P. Adjusted P-values for simultaneous inference. Biometrics. 1992;48:1005–1013. [Google Scholar]