

Abstract
Mutations in superoxide dismutase-1 (SOD1) cause a form of the fatal paralytic disorder amyotrophic lateral sclerosis (ALS), presumably by a combination of cell-autonomous and non–cell-autonomous processes. Here, we show that expression of mutated human SOD1 in primary mouse spinal motor neurons does not provoke motor neuron degeneration. Conversely, rodent astrocytes expressing mutated SOD1 kill spinal primary and embryonic mouse stem cell–derived motor neurons. This is triggered by soluble toxic factor(s) through a Bax-dependent mechanism. However, mutant astrocytes do not cause the death of spinal GABAergic or dorsal root ganglion neurons or of embryonic stem cell–derived interneurons. In contrast to astrocytes, fibroblasts, microglia, cortical neurons and myocytes expressing mutated SOD1 do not cause overt neurotoxicity. These findings indicate that astrocytes may play a role in the specific degeneration of spinal motor neurons in ALS. Identification of the astrocyte-derived soluble factor(s) may have far-reaching implications for ALS from both a pathogenic and therapeutic standpoint.
ALS is an adult-onset motor neuron disease that can be induced by dominantly inherited mutations in the gene encoding the enzyme SOD1 (refs. 1,2). Mice that are transgenic for the human SOD1 mutations recapitulate the paralytic phenotype of ALS3–5, but neither the mechanism of SOD1 toxicity nor its cellular site of action have been elucidated. Expression of mutated SOD1 in both motor neurons and non-neuronal cells has been suggested as contributing to the disease process in vivo6–8. Studies in chimeric mice composed of cells expressing either wild-type or mutated SOD1 (ref. 6) have shown that wild-type motor neurons surrounded by non-neuronal cells expressing mutated SOD1 can acquire ubiquitin-positive protein aggregates6, a sign of neuronal damage in this ALS model9. However, the precise identity of the mutant non-neuronal cells that mediate toxicity to motor neurons has remained an open question.
Astrocytes are the most abundant non-neuronal cells in the central nervous system, and their role in neurodegenerative processes is becoming increasingly appreciated5,10,11. To determine the contribution of astrocytes to neurodegeneration in the mutant SOD1 ALS mouse model, we used cocultures composed of an astrocyte monolayer (AML) and primary spinal (PMN) or embryonic stem cell–derived (ESMN) motor neurons. This culture system showed that whereas expression of mutated SOD1 in PMNs caused neuronal morphometric alterations, its expression in astrocytes affected both morphometry and survival of PMNs and ESMNs. We also found that these deleterious effects (i) were mediated by soluble factors; (ii) recapitulated the selectivity of ALS neurodegeneration, as mutant astrocytes were toxic to PMNs and ESMNs but not to dorsal root ganglion (DRG) neurons or spinal primary GABAergic or embryonic stem cell–derived interneurons; (iii) were specific to astrocytes, as other cell types, including microglia, expressing mutated SOD1 did not induce overt motor neuron degeneration; and (iv) were abrogated by a soluble inhibitor of the pro–cell death protein Bax. Our data provide evidence that astrocytes are specific contributors to spinal motor neuron degeneration in SOD1-linked ALS.
RESULTS
Primary and stem cell-based cultures are complementary
We used cultured motor neurons derived from mouse embryonic spinal cord, as PMNs have been shown to be suitable for probing the molecular basis of selective motor neuron degeneration caused by mutated SOD1 (ref. 12), and from mouse embryonic stem cells, as ESMNs not only show many of the molecular markers and functional properties of spinal motor neurons13,14 but also have the unique characteristic of being readily expandable.
We obtained PMNs from embryonic day (E) 12.5 mouse spinal cords. They yielded multipolar motor neurons immunopositive for microtubule-associated protein 2 (MAP2), for the motor neuron–specific transcription factor HB9 and for unphosphorylated neurofilament with the SMI32 antibody (Fig. 1a,b). PMNs were produced from transgenic mouse embryos expressing either SOD1G93A, SOD1G85R or SOD1G37R (G93A, G85R or G37RPMNs, respectively)—the three best characterized mouse models of ALS3–5—as well as from their nontransgenic littermates (NTgPMN). We also prepared PMNs from transgenic embryos expressing human wild-type SOD1 (WTPMNs). To facilitate the identification of motor neurons, we also used transgenic Hlxb9-GFP1Tmj mouse embryos expressing enhanced green fluorescent protein under the control of the HB9 promoter (eGFPPMNs)13. eGFP fluorescence was observed in both cell bodies and processes of large MAP2+ neurons, and it colocalized with the cholinergic transmitter synthetic enzyme choline acetyltransferase (ChAT) (Fig. 1c,d).
Figure 1.

Our two culture systems show well defined spinal motor neurons. (a,b) Immunostaining of primary neuronal cultures showing MAP2+HB9+ (a) and SMI32+HB9+ (b) large multipolar PMNs derived from nontransgenic mouse embryos. All were plated on NTgAML. (c,d) Double immunostaining of primary neuronal cultures showing MAP2+eGFP+ (c) and ChAT+eGFP+ (d) PMNs derived from a transgenic Hlxb9-GFP1Tmj embryo. (e–h) ESMNs expressing eGFP under the HB9 promoter cultured for 7 d on spinal cord astrocyte monolayers. Confirming their motor neuron phenotype, eGFP+HB9+ ESMNs are immunopositive for MAP2 (e,f), ChAT (g) and Islet (Isl) 1/2 (h). Scale bars, 50 μm (a–d,f–h) and 100 μm (e).
We generated ESMNs from stem cells derived from the transgenic Hlxb9-GFP1Tmj embryos13. This culture system typically contained ~30% eGFP+ neurons (Fig. 1e). All of these neurons expressed MAP2 (Fig. 1e,f) and were immunoreactive with both an antibody recognizing ChAT and one recognizing the LIM homeodomain proteins Islet 1 and Islet 2 (Islet 1/2) (Fig. 1g,h), confirming their motor neuron phenotype.
Mutant SOD1 motor neurons show morphometric alterations
To first examine whether expression of mutated SOD1 promoted the degeneration of spinal motor neuron, we plated G93A, G37R, G85R, WTand NTgPMNs on NTgAMLs. All transgenic and nontransgenic cultures were plated at the same density of neurons (see Methods) and resulted in the same number of PMNs 1 d after plating (Fig. 2a). Thereafter, the numbers of surviving G37R, WT and NTgPMNs declined by ~25% over 14 d without any genotypic difference (Fig. 2a). The absence of genotypic effect could not be attributed to a loss of expression of mutated SOD1 in transgenic neurons15, as its level did not change during the course of the experiments (Supplementary Fig. 1 online).
Figure 2.
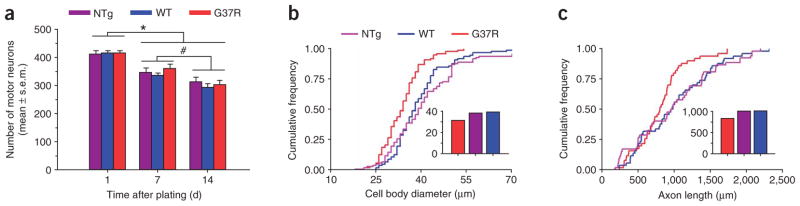
Mutated SOD1 expressed in primary spinal-cord motor neurons provokes a mild cell-autonomous phenotype. (a) Quantification of NTg, WT and G37RPMNs at 1, 7 and 14 d after plating, showing progressive decreases (P < 0.01) in PMN numbers, but no difference among the various genotypes (P > 0.05). *Lower (P < 0.01) than day 1; different from each other (P < 0.05). Values represent means ± s.e.m. from at least three independent experiments performed at least in triplicate, analyzed by two-way ANOVA followed by a Newman-Keuls post hoc test. (b,c) At 14 d after plating, G37RPMNs show (Kolmogorov-Smirnov test; P < 0.001) smaller cell body diameters (b) and shorter axon lengths (c) than WT and NTgPMNs. Insets represent the respective medians. For these analyses, we used data from 50–100 motor neurons per genotype.
Contrasting with the lack of difference in PMN numbers among the genotypes (F2,10 = 0.47, P = 0.64), at both 1 d (data not shown) and 14 d in culture, G37RPMN cell body diameters were 19% smaller (Kolmogorov-Smirnov test; P < 0.001) than those of WT and NTgPMNs (Fig. 2b). Likewise, G37RPMNs had a much lower frequency of axons ≥700 μm long compared with WTand NTgPMNs (Fig. 2c). We obtained similar results with G93A and G85RPMNs (data not shown). Thus, these findings demonstrate that, under the present experimental conditions, expression of mutated SOD1 in motor neuron caused a mild cell-autonomous abnormal phenotype.
Mutant SOD1 astrocytes kill motor neurons
We then asked whether the above neuronal morphometric abnormalities caused by mutated SOD1 result solely from a cell-autonomous mechanism. We thus plated NTgPMNs and ESMNs on G93A and NTgAMLs. At 14 d after plating, NTgPMNs grown on G93AAMLs showed the same morphometric alterations as G37R, G85R and G93APMNs grown on NTgAMLs (data not shown). Similarly, at 3 and 7 d after plating, ESMNs grown on G93AAMLs showed reduced axonal lengths and cell body diameters compared with ESMNs grown on NTgAMLs (Fig. 3a,b).
Figure 3.

Marked toxicity of mutated SOD1–expressing astrocytes to motor neurons. (a,b) ESMNs show shorter axonal lengths (a) and smaller cell body diameters (b) when plated on AMLs expressing SOD1G93A, compared with their counterparts plated on NTgAMLs (Kolmogorov-Smirnov test; P < 0.005). (c) The decay in numbers of eGFPPMNs plated on G93AAMLs is greater (F3,37 = 6.3, P = 0.0015) than that of eGFPPMNs plated on NTgAMLs. (d) At 7 d after plating, there are consistently fewer eGFPPMNs (P < 0.01) in G93A, G37R and G85RAML cocultures than in NTgAML cocultures. However, WT and NTgAML cocultures have similar (P > 0.05) numbers of NTgPMNs. (e,f) ESMNs plated on either NTg or TgAMLs behave similarly to PMNs. (g) There are comparable numbers (P > 0.05) of NTg and G37RPMNs surviving at all time points after plating on NTgAMLs. There are fewer (*P < 0.01) NTg and G37RPMNs surviving on G93AAMLs than on NTgAMLs. The loss of G37RPMNs plated on G93AAMLs is comparable (P > 0.05) to that of NTgPMNs. Values represent means ± s.e.m. from at least three independent experiments performed at least in triplicate, analyzed by two-way ANOVA followed by a Newman-Keuls post hoc test. Tg, transgenic.
Because the neuronal morphometric alterations could be recapitulated through a non–cell-autonomous process, we then asked whether the expression of mutated SOD1 in astrocytes could also produce more profound neuronal damage (Fig. 3c–f). Initially in this experiment, we plated eGFPPMNs and ESMNs on G93A and NTgAMLs from rats16 to increase the yield of glial monolayers. The numbers of eGFPPMNs plated on NTgAMLs decreased by ~25% over 14 d (Fig. 3c). Under the same conditions, 70–80% of the 1,076 ± 19 ESMNs counted at day 1 were lost over the next 4 d; thereafter, the loss of ESMNs slowed, decaying at a rate reminiscent of that of PMNs. Because of the pronounced death of ESMNs in cocultures with astrocytes of either genotype, we normalized the numbers of ESMNs plated on G93AAMLs to the numbers of those plated on NTgAMLs (Fig. 3e). Following this data transformation, PMN and ESMN values could be compared (Fig. 3c,e). This analysis showed that the losses of both eGFPPMNs (F3,37 = 6.3, P = 0.0015) and ESMNs (F2,24 = 6.0, P = 0.003) were more profound when grown on G93AAMLs than when grown on NTgAMLs (Fig. 3c–f).
We obtained similar results if, instead of rat, we used mouse G93AAMLs (data not shown) or if, instead of eGFPPMNs, we used NTgPMNs at seeding densities varying from 1,000 to 10,000 per cm2 (Supplementary Fig. 2 online). Furthermore, G37R or G85RAMLs caused comparable toxicity to eGFPPMNs or ESMNs as did G93AAMLs (Fig. 3d,f). In contrast, the effect of WTAMLs on the survival of eGFPPMNs or ESMNs did not differ from that of NTgAMLs (Fig. 3d,f), ruling out the possibility that the toxicity of mutant AMLs to PMNs and ESMNs was merely due to SOD1 overexpression. These results show that expression of mutated SOD1 in astrocytes represents a toxic pathway, which is in keeping with a non–cell-autonomous mechanism in ALS pathogenesis.
Mutant astrocytes affect wild-type and mutant motor neurons
We then asked whether the combination of mutant motor neurons grown on mutant astrocytes would give rise to a more severe neurodegenerative phenotype than any other coculture combination. Although at day 1 the numbers of PMNs were identical among the different coculture combinations (Fig. 3g), at days 7 and 14 there were significantly fewer surviving PMNs of either genotype when they were cultured on G93AAMLs than when they were cultured on NTgAMLs (Fig. 3g). However, none of these time points showed a loss of G37RPMNs different (Newman-Keuls, P > 0.3) from that of NTgPMNs grown on G93AAMLs (Fig. 3g). Likewise, the morphometric alterations in G37RPMNs did not differ from those of NTgPMNs grown on G93AAMLs (Supplementary Fig. 3 online). In addition, at day 7, transgenic PMNs expressing various SOD1 mutations and grown on G93AAMLs were identically affected (Supplementary Fig. 2). Conversely, high expression of human SOD1WT in PMNs seemed to attenuate G93AAML-mediated toxicity (Supplementary Fig. 2). Thus, these data indicate that expression of mutated SOD1 in both astrocytes and motor neurons did not exacerbate the death or the morphometric changes of PMNs caused by its expression in astrocytes alone.
Mutant astrocytes release soluble neurotoxic factor(s)
To determine whether the effect of mutant astrocytes on PMNs and ESMNs was caused by soluble factor(s), we plated eGFPPMNs onto coverslips coated with poly-D-lysine and laminin. These preparations were then cultured for 7 d with media preconditioned by either G93A or NTgAMLs (Fig. 4). Astrocyte conditioned media were frozen until assay and resupplemented before use with glucose and trophic factors (see Methods). The numbers of eGFPPMNs remaining after exposure to G93AAML conditioned medium were significantly lower (Newman-Keuls, P < 0.01) than the numbers remaining after exposure to NTg or WTAML conditioned medium (Fig. 4a). These observations support the hypothesis that mutant astrocytes exert toxicity on motor neurons through the release of soluble factor(s). This putative toxic mediator(s) seems specific to astrocytes, as media conditioned in the same manner by primary skeletal myocytes, spinal cord microglia, cerebral cortical neurons or skin fibroblasts expressing comparable levels of SOD1G93A did not cause comparable reductions in eGFPPMN or ESMN counts (Fig. 4b,c).
Figure 4.
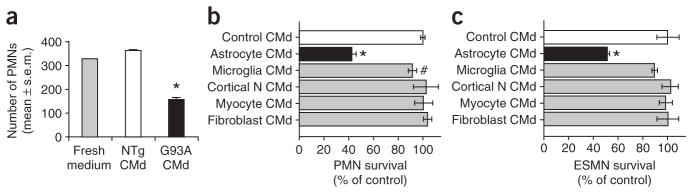
Media conditioned specifically by astrocytes expressing mutated SOD1 kill primary spinal cord and embryonic stem cell–derived motor neurons. (a) There are more eGFPPMNs surviving after 7 d of culture in fresh medium or NTgAML conditioned medium (CMd) than in G93AAML conditioned medium (*P < 0.01). (b,c) In contrast to the medium conditioned with G93AAML (*P < 0.01), media conditioned with SOD1G93A cerebral cortical neurons, skeletal myotubes or skin fibroblasts have no effect (P > 0.05) on either PMN or ESMN survival compared with controls (ESMNs cultured with medium conditioned with nontransgenic cells). Medium conditioned with spinal SOD1G93A microglia plated at a density twice that of astrocytes has only mild toxic effects on PMN (#P < 0.05) or ESMN (P > 0.05) survival. Values represent means ± s.e.m. from at least three independent experiments performed at least in triplicate, analyzed by two-way ANOVA followed by a Newman-Keuls post hoc test.
Mutant astrocyte toxic effect is specific to motor neurons
To determine whether the observed astrocyte toxicity is specific to motor neurons, we evaluated the fate of primary spinal GABAergic or DRG neurons (Fig. 5a–d) and non-motor neuron embryonic stem cell–derived neurons such as eGFP−MAP2+ neurons or Lim1+ and Lim2+ (Lim1/2+) D3 and LH2+ D1 interneurons (Fig. 5e–h).
Figure 5.

Neither mutant SOD1 AMLs nor conditioned media impair survival of neurons other than motor neurons. (a) eGFPPMNs on an AML, immunostained for eGFP and GABA at 7 d. (b) The percentage of surviving eGFPPMNs on G93AAMLs at 7 d is lower (*P < 0.004) than that at 1 d, whereas that of surviving GABAergic neurons on G93AAMLs is identical at 1 and 7 d (P > 0.5). (c,d) The percentages of surviving MAP2+ DRG neurons cultured with G93AAML conditioned medium for 2 d or for 7 d are also identical (P > 0.05). (e) Within the same culture, among the embryonic stem cell–derived MAP2+ neurons, some are eGFP+HB9+ (ESMNs) and others are eGFP−HB9−; among the latter, some are Lim1/2+. (f) Five days after embryonic stem cell–derived neurons were plated, the percentage of surviving ESMNs on G93AAMLs is lower (*P < 0.01) than that on NTgAMLs. In contrast, the percentages of neither MAP2+eGFP−HB9−(MAP2+GFP−) (F3,24 = 0.4, P = 0.8) nor MAP2+eGFP−HB9−Lim1/2+ (Lim1/2+) neurons (F3,16 = 0.1, P = 0.9) differs between the two AML cocultures. (g) Embryonic stem cell–derived neurons differentiated into posterior interneurons expressing the LH2 marker. (h) The percentages of surviving LH2+ or MAP2+ neurons are not different on NTg or G93AAMLs at 2 or 7 d (P > 0.05). Values represent means ± s.e.m. from at least three independent experiments performed at least in triplicate, analyzed by two-way ANOVA followed by a Newman-Keuls post hoc test. Scale bars, 100 μm (a), 20 μm (c) and 50 μm (e,g).
By day 7, unlike eGFPPMNs, GABAergic (Fig. 5a,b) and DRG neurons (Fig. 5c,d) respectively plated on G93AAMLs or exposed to G93AAML conditioned medium were similar in numbers to ones grown on NTgAMLs or in NTgAML conditioned medium. Similarly, neither soma diameter nor axonal length of GABAergic interneurons plated on G93AAMLs differed from that of their counterparts plated on NTgAMLs (Supplementary Fig. 4 online).
One day after plating, our embryonic stem cell–astrocyte cocultures contained, in addition to ESMNs, 54% (that is, 1,094 ± 36) eGFP−MAP2+ neurons, of which 15% (that is, 312 ± 26) were Lim1/2+ D3 interneurons (Fig. 5e,f). In contrast with the numbers of ESMNs, those of neither the eGFP−HB9−MAP2+ nor the eGFP−Lim1/2+ neurons differed between G93AAMLs and NTgAMLs cocultures over 14 d (Fig. 5f). In a variation on the experiment, we subjected embryoid bodies to a modified protocol of differentiation giving rise to 70% D1 interneurons expressing the transcription factors Lhx2 or Lhx9 (recognized by the LH2 antibody)17, as a percentage of the total MAP2+ neuronal population (Fig. 5g). Confirming further the selectivity of mutant astrocyte toxicity for the motor neuron phenotype, the numbers of eGFP−LH2+ interneurons were similar between cocultures with G93AAMLs and NTgAMLs, at both 2 and 7 d post plating (Fig. 5h). Thus, motor neuron identity may confer susceptibility to mutant astrocyte–mediated toxicity.
Astrocyte-induced motor neuron death depends on Bax
To confirm that the differences in the numbers of ESMNs reflected differences in cell survival, we compared the proportion of dying neurons between the two coculture genotypes using the DNA dye ethidium homodimer (EthD), which selectively permeates the broken membranes of dying cells. The percentage of EthD-labeled embryonic stem cell–derived MAP2+ neurons at 7 d was 1.34-fold higher upon culture with G93AAMLs than upon culture with NTgAMLs (Fig. 6a). To ask whether the depleted survival of ESMNs reflected the activation of the apoptotic pathway, we stained with both the membrane-permeant DNA dye Hoechst 33342 (Fig. 6b,c) and an antibody against fractin (Fig. 6b), which is a 32-kDa fragment of β-actin generated by activated caspase-3 (ref. 18). This showed that by 7 d, the percentage of fractin-immunostained cells with condensed nuclei was 1.51-fold higher in cocultures with G93AAMLs than in those with NTgAMLs (Fig. 6d). All cell nuclei with condensed chromatin evidenced by Hoechst 33342 were also positive for apoptotic DNA fragmentation evidenced by terminal dUTP nick-end labeling (TUNEL; Fig. 6c).
Figure 6.
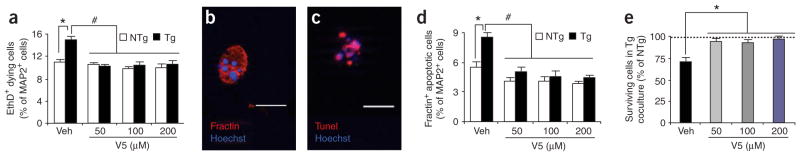
ESMNs die in response to mutant AMLs through a Bax-dependent mechanism. (a–e) Death of ESMNs assessed by immunostaining for fractin and by EthD uptake. At 7 d after plating, the Bax inhibitor V5 decreases (P < 0.01) the percentages of ESMNs labeled with EthD (a) and embryonic stem cell–derived MAP2+fractin+ neurons (d) and increases (*P < 0.01) the percentages of surviving ESMNs (e). Values are expressed as percentage of NTgAML-coculture values and represent means ± s.e.m. from at least three independent experiments performed at least in triplicate, analyzed by two-way ANOVA followed by a Newman-Keuls post hoc test. *Higher (P < 0.01) than NTgAML cocultures; #lower (P < 0.001) than TgAML cocultures with vehicle (veh). (b,c) All ESMNs immunopositive for fractin show DNA condensations, as evidenced by Hoechst 33342 (b), and all ESMNs with Hoechst 33342–labeled chromatin clumps show DNA fragmentation, as evidenced by TUNEL (c). Scale bar, 20 μm.
To characterize the biochemical pathway activated in ESMNs by mutant astrocytes, we incubated cultures with the membrane-permeant pentapeptide VPMLK (V5), which inhibits the death agonist Bax (ref. 19). This treatment reduced specifically the numbers of fractin-positive cells in cocultures with G93AAMLs (Fig. 6d). In addition, V5 diminished the numbers of EthD-labeled cells specifically in cocultures with G93AAMLs (Fig. 6a) and augmented the numbers of surviving ESMNs plated on G93AAMLs (Fig. 6e). Thus, the soluble factors produced by mutant astrocytes kill ESMNs through the activation of a Bax-dependent cell death pathway.
DISCUSSION
A role for non-neuronal cells in the demise of neighboring motor neurons in familial ALS caused by SOD1 mutations is increasingly recognized, and the underpinning of this non–cell-autonomous pathogenic element is just beginning to be uncovered. Our use of a neuronalglial coculture system provides evidence that astrocytes expressing either mutated catalytically active or inactive SOD1 cause death of wild-type PMNs and ESMNs (Fig. 3). This toxic effect is mediated through the release of soluble factor(s) from astrocytes and culminates in the recruitment of a Bax-dependent death machinery within motor neurons. The present findings indicate that this neurotoxicity is produced by a specific interaction between astrocytes and motor neurons, in that, among a variety of non-neuronal cell types, astrocytes are the only ones found to be endowed with a potent toxic property, and, among a variety of neuronal subtypes, motor neurons are the only ones found to succumb to this astrocyte-induced toxicity. Indeed, contrasting with mutant astrocytes, mutant microglia, cortical neurons, fibroblasts or myocytes exert minimal or no effect on motor neuron survival (Fig. 4), and whereas mutant astrocytes kill wild-type motor neurons, they do not affect the survival of DRG neurons or spinal interneurons (Fig. 5). Thus, our data suggest that astrocytes play a specific role in spinal motor neuron degeneration in ALS.
Our results are in agreement with those in the companion paper20 in that both studies show that primary astrocytes expressing SOD1G93A induce the death of wild-type mouse ESMNs. Given these data, we propose that, in spinal cords of chimeric mice6 mutant astrocytes may contribute to the development of the pathological features described in the neighboring wild-type motor neurons. However, transgenic mice expressing mutated SOD1 driven by the astrocyte-specific glial acidic fibrillary protein (GFAP) promoter have a more subtle phenotype characterized by gliosis but no overt motor neuron degeneration21. Without knowing the proportion of astrocytes expressing mutated SOD1 or the level of expression of this toxic protein per astrocyte, one cannot exclude the possibility that the amount of astrocytic mutated SOD1 might not have been sufficient to cause motor neuron degeneration in these transgenic mice. Furthermore, precedent exists for the ability of astrocytes expressing mutated proteins to induce neurodegeneration in vivo. Indeed, it has been shown that expression of mutated ataxin-7 in astrocytes causes degeneration of wild-type Purkinje cells in a mouse model of spinocerebellar atrophy10.
Although our data implicate astrocytes in mutated SOD1–induced neurotoxicity, they do not preclude a role for other cell types in the disease process. The emergence of the ALS phenotype can be retarded by decreasing the expression of mutated SOD1 selectively in motor neurons of transgenic mutant SOD1 mice8,15. Also relevant to this idea is the demonstration that a transgenic mouse line engineered to express the highest levels of mutated SOD1 in both neurons and astrocytes does develop an ALS phenotype22. Although our data showed that mutated SOD1 expressed in PMNs did not kill spinal PMNs by 14 d in culture, the companion study20 did find that survival of ESMNs expressing SOD1G93A was reduced compared with their wild-type counterparts after culture for more than 14 d. Thus, mutated SOD1 may act in concert in astrocytes and, apparently in a more protracted manner, in motor neurons to kill spinal motor neurons. Deletion of mutated SOD1 in microglia8 or the absence of microglia expressing mutated SOD17 also prolongs survival in transgenic mutant SOD1 mice, but without delaying the age at onset of the disease phenotype. Furthermore, mutant SOD1 microglia do not induce the death of wild-type motor neurons either in vitro (present work) or in vivo7. These results indicate that microglial expression of mutated SOD1 alters disease duration, but does not induce neurodegeneration in this ALS model. They also indicate that among glial cells, both microglia and astrocytes may contribute to the ALS phenotype by playing complementary roles in the disease process.
Although this work focuses on neuronal death, we also examined morphometric parameters and found that mutant SOD1 PMNs had smaller cell bodies and shorter axons than their wild-type counterparts (Fig. 2). We showed, however, that these mild morphological alterations could be reproduced non–cell autonomously (Supplementary Fig. 3) and they were seemingly not exacerbated by having mutated SOD1 expressed in both PMNs and astrocytes (Supplementary Fig. 3). Although the molecular basis of these morphometric changes remains to be determined, they may reflect abnormalities specific to motor neurons, as under similar experimental conditions we did not observe them in GABAergic interneurons (Supplementary Fig. 4). Under the various coculture combinations, the morphological alterations occurred before or in the absence of detectable PMN death, and in the absence of neuromuscular junctions, implying that they represent early abnormalities initiated within the motor neurons. Such early neuromuscular junction–independent change is reminiscent of the aberrant hyperexcitability23 and Na+-channel dysfunction seen in mutant SOD1 motor neurons24. One interesting possibility is that these morphological and electrophysiological perturbations may represent different manifestations of a common pathogenic mechanism in motor neurons.
As for the molecular basis of this astrocytic non–cell-autonomous toxicity to spinal motor neurons, the inhibitor of soluble Fas ligand Fas:Fc (Supplementary Fig. 5 online), the pan-caspase inhibitor zVAD-fmk (Supplementary Fig. 6 online) and neutralizing antibodies against nerve growth factor (data not shown)—which have all been shown to attenuate apoptosis in cultured motor neurons12,25—failed to improve survival of ESMNs. In contrast, Bax inhibition provided strong protection specifically against astrocyte-mediated motor neuron death (Fig. 6). This result is consistent with the in vivo demonstration that Bax ablation completely prevents the loss of spinal motor neurons in transgenic mutant SOD1 mice26. However, Bax deletion did little for the lifespan of transgenic mutant SOD1 mice26, indicating that pathways other than the Bax pathway may also contribute to the motor neuron damage in vivo and, possibly, in our cell culture system. Our experiments with conditioned media (Fig. 4) indicate that mutant astrocytes produce soluble molecule(s) which impair motor neuron survival. Studies of Huntington disease27 and spinocerebellar atrophy10 have implicated neuronal excitotoxicity due to decreased glutamate uptake by mutant astrocytes as the basis of their non–cell-autonomous properties. In ALS a similar mechanism may operate, as expression of the astrocytic glutamate transporter EAAT2 is reduced by 90% in the ventral horn of paralyzed transgenic SOD1G93A rats28. However, extracellular glutamate concentrations were not increased in our cocultures from mutant SOD1 animals, glutamate uptake was not impaired in mutant astrocyte cultures (Supplementary Fig. 5) and a potent antagonist of both AMPA and kainate receptors failed to prevent mutant astrocyte toxicity (Supplementary Fig. 5). Furthermore, key toxic chemokines such as interleukin-1β, interleukin-6, interferon-γ and tumor necrosis factor-α were either undetectable or produced similarly by wild-type and mutant astrocytes (Supplementary Fig. 5).
ESMNs have emerged as a potential repair reagent for the treatment of spinal cord diseases such as ALS. However, our study and the companion work20 show that mutant astrocytes impair the survival of wild-type ESMNs. This fact implies that wild-type ESMNs, upon engraftment into ALS spinal cords, may be subjected to a hostile cellular environment challenging their ability to survive and to grow processes. Alternatively, embryonic stem cells may provide attractive prospects of therapies for ALS by avenues distinct from the mere replacement of motor neurons. Our demonstration that ESMNs respond to mutant astrocyte–mediated toxicity in the same manner as PMNs suggests that ESMNs offer an invaluable, readily expandable cellular tool for the high-throughput screening of small neuroprotective molecules in ALS. In addition, embryonic stem cells may also be differentiated into astrocytes29. In light of our data and those in the companion paper20, it is possible that grafting wild-type embryonic stem cell–derived astrocytes into ALS-affected spinal cords may be useful for diluting the non–cell-autonomous toxic phenotype, thereby attenuating the degeneration of neighboring motor neurons.
The identification of the toxic factor(s) responsible for the effects of mutant astrocytes on motor neuron survival represents an important challenge that may greatly benefit from new technologies such as the informatics-assisted protein profiling that has been used in transgenic mutant SOD1 mice30. Once known, the toxic factors may provide new insights into the mechanism by which motor neurons die. This work may be relevant, not only to the rare familial form of ALS linked to mutated SOD1, but also to the more common sporadic form of this incurable disease. Early diagnosis of ALS is difficult and often delayed by the insidious onset of symptoms that mimic other conditions, and clinical trials are slow in determining whether a treatment is efficacious. The discovery of astrocyte toxic mediators may thus lead to their use as biomarkers for the early diagnosis of ALS, to measure the progression of the disease, and to assess the effects of treatment as well as to develop new therapies aimed at mitigating motor neuron degeneration in ALS.
METHODS
Animals
Procedures using laboratory animals were in accordance with the US National Institutes of Health guidelines for the use of live animals and were approved by the Institutional Animal Care and Use Committee of Columbia University. In this study, PMNs and ESMNs expressing eGFP were derived from the transgenic HB9::eGFP mouse line13, which is now designated as Hlxb9-GFP1Tmj (Jackson Laboratory).
Primary astrocyte culture
We prepared glial monolayers from spinal cords of transgenic SODG93A, SODG37R, SODG85R and SODWT newborn pups and of their nontransgenic littermates as previously described31. We plated cell suspensions in glial medium: DMEM (Invitrogen) containing 10% FBS (Invitrogen), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin (penicillin/streptomycin, Invitrogen). After 2 weeks, glial cultures contained 95% GFAP+ astrocytes, 5% CD11b+ microglia and no neurons or oligodendrocytes as indicated by the lack respectively of MAP2 or 2′-3′-cyclic nucleotide phosphohydrolase immunoreactivity (data not shown). To eliminate residual microglia, 2-week-old flasks were agitated (200 r.p.m. for 6 h) and astrocytes were detached by 0.25% trypsin (Invitrogen) and plated onto coverslips at a density of 20,000 cells per cm2.
Embryonic stem cell–derived neuron cultures
Cells were derived from Hlxb9-GFP1Tmj transgenic mice13 and differentiated into ESMNs as described previously13. To form embryoid bodies, we grew cells for 2 d in 1:1 (vol/vol) DMEM/Ham’s F-12 medium (Invitrogen) containing B27 supplement (Invitrogen), penicillin/streptomycin and 0.1 mM 2-mercaptoethanol (Sigma). We treated them with 1 μM retinoic acid (Sigma) and 400 nM sonic hedgehog agonist (Hh-Ag1.3, Curis Inc.) for 5 d and then dissociated them with papain (Worthington). We then plated them at 1,600 eGFP+ cells per cm2 in motor neuron medium: neurobasal medium (Invitrogen) containing 2% horse serum (heat inactivated; Invitrogen), B27 supplement, 0.5 mM glutamine (Invitrogen), 25 μM 2-mercaptoethanol, and penicillin/streptomycin. To obtain LH2+ neurons, once embryoid bodies formed we treated them with 0.1 μM retinoic acid and 15 ng ml−1 bone morphogenetic protein 4 (R&D Systems) for 5 d before papain dissociation. Then we plated 1,500 cells per cm2 onto an AML in motor-neuron medium.
Primary neuronal cultures
We performed spinal neuronal cultures from E12.5 Hlxb9-GFP1Tmj transgenic, wild-type or transgenic SODG93A, SODG37R, SODG85R or SODWT rodents as previously described12. Cells were plated at 1,500 eGFP+ cells per cm2 for Hlxb9::eGFP and at 5,000 cells per cm2 for the other cultures, either on coverslips coated with 0.01% poly-D-lysine and 10 μg ml−1 laminin (poly-D-lysine/laminin) or on astrocyte monolayers. The culture medium was either motor neuron medium supplemented with a cocktail of trophic factors composed of 0.5 ng ml−1 glia-derived neurotrophic factor, 1 ng ml−1 brain-derived neurotrophic factor and 10 ng ml−1 ciliary neurotrophic factor (trophic factor cocktail, R&D Systems); or astrocyte-conditioned motor neuron medium (see section below).
We prepared DRG cultures as previously described32,33. Cell suspensions were plated at 1,500 cells per cm2 onto poly-D-lysine/laminin coated coverslips in motor-neuron medium conditioned for 1 week by the various astrocyte monolayers (see below) and supplemented with 10 ng ml−1 of neurotrophin-3 and nerve growth factor (R&D Systems).
We prepared cortical neuron cultures as previously described34 from E17.5 mouse brains. Neurons were plated in neurobasal medium supplemented with B27, 0.5 mM glutamine and penicillin/streptomycin onto poly-D-lysine/laminin coated dishes.
Primary non-neuronal cultures
We collected microglia from glial monolayers as described above and centrifuged them (500g, 5 min). We resuspended the cells in fresh glial medium and plated them at 40,000 cells per cm2. We obtained fibroblasts as described previously35 and plated them onto dishes in minimum essential medium (Invitrogen) containing 10% FBS and penicillin/streptomycin. Myoblasts we obtained from rat pup skin after digestion with dispase/collagenase IV (37 °C, 25 min; Worthington). Cells were suspended in growth medium (Hams F-10 medium (Invitrogen) supplemented with 15% horse serum, penicillin/streptomycin and 5 ng ml−1 b-fibroblast growth factor (R&D Systems) and plated on collagen-coated dishes. At 90% of confluency, cells were differentiated with 6 μg ml−1 insulin and cultured in Hams F-10 medium supplemented with 1.5% horse serum and penicillin/streptomycin. Myotubes formed in 2–3 d.
Conditioned medium preparation
Cultures of astrocytes, microglia, fibroblasts and muscle were prepared from both wild-type and transgenic SOD1G93A rodents. Once they reached confluency or differentiation, we replaced their culture media with either motor neuron or DRG medium. After 7 d, we collected conditioned media, centrifuged them (500g for 10 min) to eliminate floating cells, and collected and froze the supernatants. Before use, conditioned media were supplemented with 4.5 mg ml−1 D-glucose (final concentration), penicillin/streptomycin and the cocktail of trophic factors and filtered.
Immunocytochemistry and cell labeling
For EthD (Molecular Probes) estimation of death, we incubated cells with 2 μM EthD (diluted in Dulbecco’s PBS; 45 min; 25°C) as we previously reported36. For immunocytochemistry and other cell labelings, we processed cells as we previously described36. Primary antibodies were rabbit polyclonal antibodies to eGFP (1:2,000; Molecular Probes), fractin (1:5,000; BD Pharmingen), GABA (1:2,000; Chemicon), protein gene product 9.5 (PG-P 9.5; 1:2,000; Chemicon), Lim2 (1:150; ref. 13) and HB9 (1:1,000; ref. 13); goat polyclonal antibody to ChAT (1:100; Chemicon); sheep polyclonal antibody to SOD1 (1:500; Calbiochem) and mouse monoclonal antibodies to MAP-2 (1:1,000; Chemicon), GFAP (1:1,000; Sigma), Islet 1/2 (1:100; ref. 37), LH2 (1:2; ref. 38) and SMI-32 for unphosphorylated neurofilament heavy chain (1:1,000, Sternberger Monoclonals). We performed TUNEL (in situ cell death detection kit, Roche Diagnostics) following the manufacturer’s recommendations.
Pharmacological treatments
zVAD-fmk and V5 (Sigma) were dissolved in DMSO and added to cultures to final concentrations ranging from 10–20 μM for the former and 50–200 μM for the latter. We added fresh drugs daily. We evaluated cell survival and death at 7 d by counting eGFP+HB9+ neurons and EthD- and fractin-labeled cells as described below.
Morphometric analysis
For estimation of motor neuron morphometric parameters, we captured images at ×50 under fluorescence examination using Axiovision LE Rel. 4.2 software (Zeiss) and measured soma diameter and length distance between two points for axon length.
Cell counting and statistics
Results are expressed as mean ± s.e.m. for 3–6 independent experiments. Each experiment corresponds to ≥3 transgenic and nontransgenic cultures from the same litter and three coverslips per time point and per condition. We counted each coverslip in its entirety at ×100 under fluorescence examination. To determine the proportion of dying or apoptotic neurons, we counted ≥750 MAP2+ neurons per culture. Differences between means were analyzed by a two-tailed Student’s t-test, whereas differences among means were analyzed by one- or two-way ANOVA with the different types of mice, treatment doses or time as the independent factors. When ANOVA showed significant differences, we performed pair-wise comparisons between means by Newman-Keuls post hoc testing. We used SigmaStat for Windows (version 3.1; Jandel) for all of these statistical analyses. For morphometric studies, we analyzed differences by the Kolmogorov-Smirnov test (http://www.physics.csbsju.edu/stats/KS-test.n.plot_form.html). In all analyses, the null hypothesis was rejected at the 0.05 level.
Supplementary Material
Acknowledgments
The authors wish to thank H. Mitsumoto for his support, C. Henderson, W. Dauer, E. Schon and J. Krakauer for their comments and advice, and J. Jeon for assistance in preparing this manuscript. We thank T. Maniatis and K. Eggan for communicating their results before publication and for insightful discussions about the study. This study is supported by Muscular Dystrophy Association/Wings-over-Wall Street, the ALS Association, Project-ALS, US National Institutes of Health NS42269, NS38370, NS11766, AG 21617, ES013177 and DK58056, US Department of Defense Grant DAMD 17-03-1, the Parkinson’s Disease Foundation and the Bernard and Anne Spitzer Fund. M.N. is the recipient of the Gardner’s fellowship from the Muscular Dystrophy Association. T.M.J. is an investigator of the Howard Hughes Medical Institute. D.B.R. is the recipient of a Philippe Foundation grant for exchange programs between France and the United States.
Footnotes
Note: Supplementary information is available on the Nature Neuroscience website.
AUTHOR CONTRIBUTIONS
M.N., D.B.R. and T.N. conducted all experiments and participated in designing them and writing the manuscript; H.W. and A.C. assisted in the experiments and in writing the manuscript; T.M.J. and S.P. conducted the data analyses and wrote the manuscript; and S.P. supervised the project. H.W. and T.M.J. also provided critical reagents.
COMPETING INTERESTS STATEMENT
The authors declare no competing financial interests.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions
References
- 1.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 2.Deng HX, et al. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- 3.Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 4.Wong PC, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- 5.Bruijn LI, et al. ALS-linked SOD1 mutant G85R mediated damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- 6.Clement AM, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 7.Beers DR, et al. Wild-type microglia extend survival in PU. 1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boillee S, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 9.Kostic V, Jackson-Lewis V, De Bilbao F, Dubois-Dauphin M, Przedborski S. Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science. 1997;277:559–562. doi: 10.1126/science.277.5325.559. [DOI] [PubMed] [Google Scholar]
- 10.Custer SK, et al. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- 11.Das S, Potter H. Expression of the Alzheimer amyloid-promoting factor antichymotrypsin is induced in human astrocytes by IL-1. Neuron. 1995;14:447–456. doi: 10.1016/0896-6273(95)90300-3. [DOI] [PubMed] [Google Scholar]
- 12.Raoul C, et al. Motoneuron death triggered by a specific pathway downstream of Fas. Potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–1083. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- 13.Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 14.Miles GB, et al. Functional properties of motoneurons derived from mouse embryonic stem cells. J Neurosci. 2004;24:7848–7858. doi: 10.1523/JNEUROSCI.1972-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raoul C, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
- 16.Nagai M, et al. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee KJ, Mendelsohn M, Jessell TM. Neuronal patterning by BMPs: a requirement for GDF7 in the generation of a discrete class of commissural interneurons in the mouse spinal cord. Genes Dev. 1998;12:3394–3407. doi: 10.1101/gad.12.21.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suurmeijer AJ, van der Wijk J, van Veldhuisen DJ, Yang F, Cole GM. Fractin immunostaining for the detection of apoptotic cells and apoptotic bodies in formalin-fixed and paraffin-embedded tissue. Lab Invest. 1999;79:619–620. [PubMed] [Google Scholar]
- 19.Sawada M, Hayes P, Matsuyama S. Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat Cell Biol. 2003;5:352–357. doi: 10.1038/ncb955. [DOI] [PubMed] [Google Scholar]
- 20.Di Giorgio FP, Carrasco M, Siao M, Maniatis T, Eggan K. Non–cell autonomous effect of glia on motor neurons in an embryonic stem cell–based ALS model. Nat Neurosci. doi: 10.1038/nn1885. advance online publication, 15 April 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, et al. Coincident thresholds of mutant protein for paralytic disease and protein aggregation caused by restrictively expressed superoxide dismutase cDNA. Neurobiol Dis. 2005;20:943–952. doi: 10.1016/j.nbd.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na+ current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol (Lond) 2005;563:843–854. doi: 10.1113/jphysiol.2004.074138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zona C, Pieri M, Carunchio I. Voltage-dependent sodium channels in spinal cord motor neurons display rapid recovery from fast inactivation in a mouse model of amyotrophic lateral sclerosis. J Neurophysiol. 2006;96:3314–3322. doi: 10.1152/jn.00566.2006. [DOI] [PubMed] [Google Scholar]
- 25.Ricart K, et al. Interactions between β-neuregulin and neurotrophins in motor neuron apoptosis. J Neurochem. 2006;97:222–233. doi: 10.1111/j.1471-4159.2006.03739.x. [DOI] [PubMed] [Google Scholar]
- 26.Gould TW, et al. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shin JY, et al. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howland DS, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci USA. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scheffler B, et al. Functional network integration of embryonic stem cell-derived astrocytes in hippocampal slice cultures. Development. 2003;130:5533–5541. doi: 10.1242/dev.00714. [DOI] [PubMed] [Google Scholar]
- 30.Lukas TJ, Luo WW, Mao H, Cole N, Siddique T. Informatics-assisted protein profiling in a transgenic mouse model of amyotrophic lateral sclerosis. Mol Cell Proteomics. 2006;5:1233–1244. doi: 10.1074/mcp.M500431-MCP200. [DOI] [PubMed] [Google Scholar]
- 31.Silva GA, Feeney C, Mills LR, Theriault E. A novel and rapid method for culturing pure rat spinal cord astrocytes on untreated glass. J Neurosci Methods. 1998;80:75–79. doi: 10.1016/s0165-0270(97)00198-2. [DOI] [PubMed] [Google Scholar]
- 32.Chalazonitis A, Kessler JA, Twardzik DR, Morrison RS. Transforming growth factor α, but not epidermal growth factor, promotes the survival of sensory neurons in vitro. J Neurosci. 1992;12:583–594. doi: 10.1523/JNEUROSCI.12-02-00583.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chalazonitis A, Crain SM, Kessler JA. Preferential cholinergic projections by embryonic spinal cord neurons within cocultured mouse superior cervical ganglia. Brain Res. 1988;458:231–248. doi: 10.1016/0006-8993(88)90466-0. [DOI] [PubMed] [Google Scholar]
- 34.Rideout HJ, Dietrich P, Wang Q, Dauer WT, Stefanis L. α-Synuclein is required for the fibrillar nature of ubiquitinated inclusions induced by proteasomal inhibition in primary neurons. J Biol Chem. 2004;279:46915–46920. doi: 10.1074/jbc.M405146200. [DOI] [PubMed] [Google Scholar]
- 35.Kaji K, Matsuo M. Aging of chick embryo fibroblasts in vitro. III Polyploid cell accumulation. Exp Cell Res. 1979;119:231–236. doi: 10.1016/0014-4827(79)90351-3. [DOI] [PubMed] [Google Scholar]
- 36.Przedborski S, et al. Increased superoxide dismutase activity improves survival of cultured postnatal midbrain neurons. J Neurochem. 1996;67:1383–1392. doi: 10.1046/j.1471-4159.1996.67041383.x. [DOI] [PubMed] [Google Scholar]
- 37.Pfaff SL, Mendelsohn M, Stewart CL, Edlund T, Jessell TM. Requirement for LIM homeobox gene Isl 1 in motor neuron generation reveals a motor neuron-dependent step in interneuron differentiation. Cell. 1996;84:309–320. doi: 10.1016/s0092-8674(00)80985-x. [DOI] [PubMed] [Google Scholar]
- 38.Ericson J, et al. Pax 6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997;90:169–180. doi: 10.1016/s0092-8674(00)80323-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.