

Abstract
Acid-sensing ion channel (ASIC)-1a, the major ASIC subunit with Ca2+ permeability, is highly expressed in the neurons of CNS. Activation of these channels with resultant intracellular Ca2+ accumulation plays a critical role in normal synaptic plasticity, learning/memory, and in acidosis-mediated glutamate receptor-independent neuronal injury. Here we demonstrate that the activities of ASICs in CNS neurons are tightly regulated by the redox state of the channels and that the modulation is ASIC1a subunit dependent. In cultured mouse cortical neurons, application of the reducing agents dramatically potentiated, whereas the oxidizing agents inhibited the ASIC currents. However, in neurons from the ASIC1 knock-out mice, neither oxidizing agents nor reducing reagents had any effect on the acid-activated current. In Chinese Hamster Ovary cells, redox-modifying agents only affected the current mediated by homomeric ASIC1a, but not homomeric ASIC1b, ASIC2a, or ASIC3. In current-clamp recordings and Ca2+-imaging experiments, the reducing agents increased but the oxidizing agents decreased acid-induced membrane depolarization and the intracellular Ca2+ accumulation. Site-directed mutagenesis studies identified involvement of cysteine 61 and lysine 133, located in the extracellular domain of the ASIC1a subunit, in the modulation of ASICs by oxidizing and reducing agents, respectively. Our results suggest that redox state of the ASIC1a subunit is an important factor in determining the overall physiological function and the pathological role of ASICs in the CNS.
Keywords: ASIC1a, oxidizing/reducing agent, patch-clamp, ischemia, neuronal injury, redox reagents
Introduction
Acid-sensing ion channels (ASICs), members of degenerin/epithelial sodium channel superfamily, are expressed highly in peripheral sensory and central neurons (Krishtal and Pidoplichko, 1981; Waldmann et al., 1997). They share a common structure feature with two transmembrane domains, short intracellular N and C termini, and a large cysteine-rich extracellular loop (Garcia-Anoveros et al., 1997; Waldmann and Lazdunski, 1998; Waldmann, 2001; Krishtal, 2003; Saugstad et al., 2004). Of six subunits cloned, ASIC1a, 1b, 2a, 2b, and 3 are expressed in sensory neurons, whereas only ASIC1a, 2a, and 2b are detected in central neurons (Waldmann et al., 1997; Baron et al., 2002; Duggan et al., 2002; Wemmie et al., 2002; Alvarez de la Rosa et al., 2003). Channels formed by different ASIC subunits have dramatic difference in pH sensitivity, kinetics, Ca2+ permeability, and physiological/pathological functions. In sensory neurons, for example, ASIC2a is likely involved in mechanosensation (Price et al., 2000) and ASIC3 in the sensation of cardiac pain during ischemia (Benson et al., 1999; Sutherland et al., 2001). In CNS neurons, ASIC1a has been shown to be involved in synaptic plasticity, learning and memory (Wemmie et al., 2002, 2003), and in acidosis-mediated, glutamate-independent neuronal injury (Xiong et al., 2004; Yermolaieva et al., 2004). Because current clinical trials using glutamate antagonists as the main neuroprotective agents have failed (Ikonomidou and Turski, 2002; Hoyte et al., 2004), these findings suggest that ASIC1a may represent a new therapeutic target for stroke patients (Xiong et al., 2004; Huang and McNamara, 2004; Benveniste and Dingledine, 2005). ASIC2a, in contrast, has been shown to be associated with neuronal survival after global ischemia (Johnson et al., 2001).
Redox status is known to affect the functions of various proteins (Slivka et al., 1987; Nanda et al., 1996). This oxidation/reduction-dependent modulation of protein function is important because the redox status changes dramatically in physiological/pathological conditions (Zangerle et al., 1992; Schulz et al., 2000). Particularly relevant, redox reagents have been shown to modulate the activities of both voltage-gated and ligand-gated ion channels (Aizenman et al., 1989; Kohr et al., 1994; DiChiara and Reinhart, 1997; Amato et al., 1999; Todorovic et al., 2001). Our preliminary experiments (Chu et al., 2004a) and a recent study by Andrey et al. (2005) have demonstrated that the activities of ASICs in peripheral sensory and central neurons are also modulated by the redox reagents. Because the overall ASIC currents in native neurons, particularly in sensory neurons, are mediated by various ASIC subunits, and the fact that activation of different ASIC subunits have dramatically different functional consequences, it is important to know which subunit of ASICs is involved in the modulation. Here we show that, in CNS neurons and in CHO cells expressing different ASICs, redox modulation is ASIC1a specific. Site-directed mutagenesis studies identified two amino acids (C61 and K133), located in the extracellular domain of the ASIC1a subunit, involved in the redox modulation. In addition to ASIC current, redox modulation has a dramatic effect on acid-induced increase of intracellular Ca2+ and membrane depolarization. Our studies suggest that targeting redox modulation site(s) on the ASIC1a subunit may be an alternative approach for developing an effective therapeutic strategy for stroke.
Materials and Methods
Primary cortical neuronal cultures.
The method for culturing mouse cortical neurons was essentially the same as the technique described previously (Chu et al., 2004b; Xiong et al., 2004). The use of mice for neuronal cell culture was approved by the Institutional Animal Care and Use Committee of Legacy Clinical Research and Technology Center. Briefly, time-pregnant (embryonic day 16) Swiss mice, and postnatal day 1 (P1) C57BL/6, ASIC1, or ASIC2 knock-out mice were anesthetized with halothane followed by decapitation. Brains were rapidly removed and placed in ice-cold Ca2+ and Mg2+-free Hank’s solution. Cerebral cortices were dissected and enzyme digested with 0.05% trypsin-EDTA for 10 min at 37°C, followed by trituration with fire-polished glass pipettes, and plated on poly-l-ornithine-coated culture dishes (35 mm in diameter), at a density of 1 × 106 cells per dish. Neurons were cultured with Neurobasal medium supplemented with B27 and maintained at 37°C in a humidified 5% CO2 atmosphere incubator. Cultures were fed twice per week and used for electrophysiological recording 12 ∼ 14 d after plating.
Acute isolation of mouse cortical neurons.
Cortical neurons were isolated according to the technique described previously (Xiong et al., 1999; Chu et al., 2004b). Briefly, 4-week-old Swiss mice were anesthetized with halothane and decapitated. The brain was removed and placed in ice-cold oxygenated extracellular fluid (ECF; see below), and subsequently sectioned at 400 μm with a microtome (VT 1000; Leica, Bannockburn, IL). The slices were then incubated in ECF containing 0.3 ∼ 0.5 mg/ml papain (from papaya latex; Sigma, St. Louis, MO) at room temperature for 30 min. All solutions were bubbled with 100% O2. Following the digestion, slices were washed three times in ECF and incubated in enzyme-free ECF for at least 30 min before dissociation. For dissociation of neurons, one slice was transferred into a 35 mm culture dish containing 2 ml of ECF, and the dish was placed on the stage of an inverted phase-contrast microscope. The cortical region of the slice was then cut out, and single cells were mechanically dissociated using two fire-polished glass pipettes or fine forceps. Electrophysiological recording of the isolated neurons began ∼15 min after the mechanical dissociation.
Electrophysiology.
Patch-clamp recordings were performed as described previously (Chu et al., 2004b; Xiong et al., 2004). Patch electrodes, with resistances of 2 ∼ 3 MΩ when filled with intracellular solution, were constructed from thin-walled borosilicated glass (1.5 mm diameter; World Precision Instruments, Sarasota, FL) on a two-stage puller (PP83; Narishige, Tokyo, Japan). A multibarrel perfusion system (SF-77; Warner Instruments, Hamden, CT) was used to achieve a rapid exchange of extracellular solutions (Chu et al., 2002, 2004b). Whole-cell currents or membrane potentials were recorded using Axopatch 1D amplifiers (Molecular Devices, Union City, CA). Data were filtered at 2 kHz and digitized at 5 Hz using a Digidata 1320 DAC unit (Molecular Devices). The on-line acquisition was done using pCLAMP software (version 8; Molecular Devices).
In general, ASICs were activated by acid perfusion every 2 min to allow for a complete recovery of the channels from desensitization. During each experiment, a voltage step of −10 mV from the holding potential was applied periodically to monitor cell capacitance and access resistance. Recordings in which access resistance or capacitance changed by >10% during the experiment were excluded for data analysis (Xiong et al., 1998; Chu et al., 2004b).
Solutions and chemicals.
Standard ECF contained the following (in mm): 140 NaCl, 5.4 KCl, 2.0 CaCl2, 1.0 MgCl2, 20 HEPES, and 10 glucose (pH 7.4; 320 ∼ 330 mOsm). For solutions with pH < 6.0, HEPES was replaced with MES for reliable pH buffering (Chu et al., 2002, 2004b). For voltage-clamp recordings, the pipette solution contained the following (mm): 140 CsF, 10 HEPES, 11 EGTA, 2 TEA, 1 CaCl2, 2 MgCl2, and 4 K2ATP (pH 7.3; 300 mOsm). For current-clamp recording, CsF in the pipette solution was replaced with KF. dl-Dithiothreitol (DTT), l-glutathione (reduced form, GSH), l-cysteine, 5,5′-dithio-bis(2-nitrobenzoic acid (DTNB), tri(2-carboxyethyl)phosphine hydrochloride (TCEP), N-ethylmaleimide (NEM), N-tris(hydroxymethyl)methylglycine (tricine), (5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (MK801), CNQX, nimodipine, ω-conotoxin MVIIC, and ZnCl2 were purchased from Sigma. [2-(Trimethylammonium)ethyl]methanethiosulfonate (MTSET) was purchased from Toronto Research Chemicals (Toronto, Ontario, Canada).
Transfection of Chinese hamster ovary cells.
Transfection of Chinese hamster ovary (CHO) cells with various ASIC subunits was described in detail previously (Chu et al., 2004b). Briefly, CHO cells were cultured in F12 medium (American Type Culture Collection, Manassas, VA) supplemented with 10% fetal bovine serum. At ∼50% confluence, cells were cotransfected with pcDNA3 constructors (Invitrogen, Carlsbad, CA) encoding individual ASICs and green fluorescent protein (GFP), using Fugene 6 transfection reagent (Roche Diagnostics, Indianapolis, IN). cDNA (0.75 μg) for individual ASIC and 0.25 μg cDNA for GFP were used for each culture dish. For coexpression of ASIC1a plus ASIC2a, equal amounts of cDNA (0.75 μg) for both subunits were used. All recordings were made 48–72 h after the transfection. GFP-positive cells were viewed under a fluorescent microscope for patch-clamp recording.
DNA preparation and site-directed mutagenesis.
Point mutations were made using the QuikChange Site-Directed Mutagenesis system (Stratagene, La Jolla, CA) (Chu et al., 2004b). All primers were obtained from Sigma-Genosys (The Woodlands, TX). All mutations were confirmed by restriction enzyme digest and sequence analysis. In all cases, the entire ASIC1a cDNA was sequenced to verify that nonspecific mutations were not introduced.
Ca2+ imaging.
Intracellular Ca2+ imaging was performed as described previously (Chu et al., 2004b; Xiong et al., 2004). Neurons grown on glass coverslips (25 mm in diameter) were washed three times with ECF and incubated with 5 μm fura-2 AM (Invitrogen, Eugene, OR) for ∼40 min at 22°C, followed by three washes and additional incubation in normal ECF for 30 min. Coverslips were then transferred to a perfusion chamber on the stage of an inverted microscope [Nikon (Tokyo, Japan) TE300]. Cells were illuminated using a xenon lamp (75 W; Nikon, Hawthorne, NY) and observed with a 40× UV fluor oil-immersion objective lens (Nikon). Video images were obtained using a cooled CCD camera (Sensys KAF 1401; Photometrics, Tucson, AZ). Digitized images were acquired, stored, and analyzed on a PC controlled by Axon Imaging Workbench (AIW) software (AIW2.1; Molecular Devices). The shutter and filter wheel (Lambda 10-2; Sutter Instrument, Novato, CA) were also controlled by AIW to allow for timed illumination of cells at either 340 nm or 380 nm excitation wavelength. Fluorescence was detected at an emission wavelength of 510 nm. Images (340/380 ratio) were analyzed by averaging pixel ratio values in circumscribed regions of cells in the field of view. The values were exported from AIW to SigmaPlot for additional analysis and plotting.
Data analysis.
Data are expressed as mean ± SEM. Student’s t test and ANOVA were used for statistical analysis where appropriate. The criteria of significance was set at p < 0.05.
Results
Reducing agents potentiate ASIC currents in cultured mouse cortical neurons
ASIC currents in cultured mouse cortical neurons were recorded as detailed in previous studies (Chu et al., 2004b; Xiong et al., 2004). To determine whether redox status modulates the activity of ASICs in these neurons, the effects of various redox-modifying reagents on ASIC currents were studied. We first tested the effect of DTT, a commonly used reducing agent (Aizenman et al., 1989; Kohr et al., 1994), on the ASIC current. After recordings of stable ASIC current activated by extracellular pH (pHo) reduction from 7.4 to 6.5, various concentrations of DTT were perfused onto the neurons. As shown in Figure 1A, the addition of DTT dose-dependently and dramatically potentiated the ASIC current in a reversible manner. A threshold concentration of 10 μm is required to produce detectable potentiation, and maximal potentiation was achieved by 3.0 mm DTT, with an approximately threefold increase in the peak amplitude of the current (control, −919.7 ± 121.3 pA; 3.0 mm DTT, −2851.1 ± 149.7 pA; n = 8; p < 0.001). Adding DTT (0.01–10.0 mm) alone to pH 7.4 solution did not activate any current on its own, and a coapplication of DTT (0.1 mm) with pH 6.5 for 10 s was not sufficient to induce notable potentiation of the ASIC currents (data not shown). In most neurons, the maximal potentiation by DTT can be reached within 2 ∼ 4 min after its perfusion. However, the recovery phase of the potentiation is much slower. In general, it took >30 min for the effect of DTT to be fully reversed after washout (n = 6) (Fig. 1B). The slow recovery of the DTT effect suggests that a chemical reaction may be involved in the potentiation of the ASIC current by DTT. This long-lasting potentiation, however, can be quickly reversed (within 2–4 min) by addition of the oxidizing agent DTNB (0.5 mm; n = 5) (Fig. 1C). Similarly, bath perfusion of NEM, an alkylating agent known to bind to thiol groups of redox modulatory sites (Creighton, 1984; Tang and Aizenman, 1993), also quickly reversed the persistent potentiation of the ASIC current by DTT, suggesting an involvement of critical sulfhydryl groups in the potentiation of the current by DTT (n = 6; p < 0.01) (Fig. 1D).
Figure 1.

Potentiation of ASIC currents in cultured mouse cortical neurons by reducing agents. A, Representative current traces and bar graphs showing the dose-dependent potentiation of ASIC current by DTT; n = 6–25. B, Time course of DTT potentiation of ASIC currents; n = 6. C, The persistent potentiation by DTT is quickly reversed by addition of the oxidizing agent DTNB (0.5 mm; n = 5). D, Representative traces and summary data showing a rapid reversal of DTT potentiation by the alkylating agent NEM (1.0 mm; n = 6). E, F, Representative traces and summary data showing the potentiation of ASIC current by the endogenous reducing agents GSH and cysteine. *p < 0.05; **p < 0.01.
We then determined whether the endogenous reducing agents such as glutathione (GSH, reduced form) and cysteine (Kohr et al., 1994; Todorovic et al., 2001) have the same effects as DTT. At 1.0 mm, GSH enhanced the peak amplitude of ASIC currents by 1.91 ± 0.14-fold (n = 15) (Fig. 1E), whereas cysteine increased the peak ASIC currents by 1.62 ± 0.13-fold (n = 7) (Fig. 1F). Similar to DTT, the persistent potentiation of the ASIC current by GSH and cysteine were quickly reversed by the addition of 0.5 mm DTNB (data not shown).
Oxidizing agent inhibits ASIC currents in cultured mouse cortical neurons
We then tested whether an oxidizing agent has an effect opposite to the reducing agents. Neurons were exposed to DTNB, a commonly used oxidizing reagent (Aronstam et al., 1978; Aizenman et al., 1989), at a concentration of 0.5 mm. Perfusion of DTNB did not affect the holding current but significantly inhibited the ASIC current in the majority of cortical neurons. In 25 of 36 neurons (69.4%), ∼4 min perfusion of 0.5 mm DTNB inhibited the ASIC current to 60% of the control value (0.60 ± 0.04; n = 25) (Fig. 2A,B). This inhibitory effect was fully reversible after the removal of DTNB. In these neurons, 100 or 300 μm Zn2+ did not potentiate the ASIC current (data not shown), as expected for homomeric ASIC1a channels (Chu et al., 2004b). In the remaining 11 neurons (30.5%), however, DTNB had no effect on the ASIC current (Fig. 2C,D), even with concentrations as high as 3.0 mm. In these neurons, addition of 100 or 300 μm Zn2+ significantly potentiated the ASIC currents (data not shown), as expected for ASIC2a containing channels (e.g., ASIC1a/2a) (Baron et al., 2001; Askwith et al., 2004; Chu et al., 2004b). Together, these data suggest that inhibition of the ASIC current by DTNB is likely ASIC1a subunit-dependent.
Figure 2.
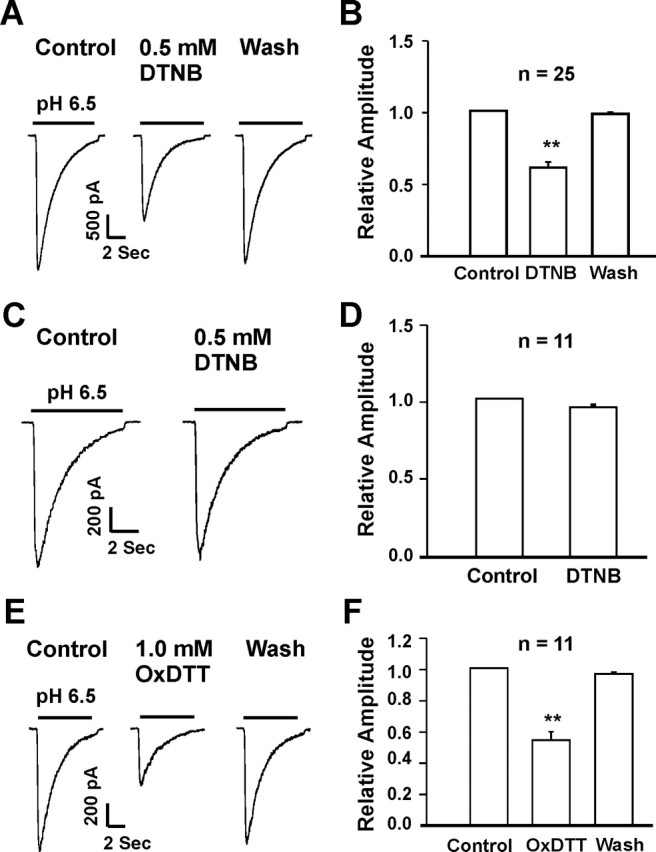
Inhibition of ASIC currents in cultured mouse cortical neurons by oxidizing agents. A, B, Representative current traces and summary data showing an inhibition of the ASIC current in ∼70% of the neurons by 0.5 mm DTNB; n = 25; **p < 0.01. C, D, Rpresentative traces and summary data showing the lack of DTNB inhibition in ∼30% of the neurons; n = 11; p > 0.05. E, F, Representative traces and summary data showing the inhibition of ASIC current by 1.0 mm OxDTT; n = 11; **p < 0.01.
Modulation of ASIC current by reducing agents requires chemical specificity
To further verify that potentiation of the ASIC current by DTT is attributable to redox modulation rather than a nonspecific binding or interaction, an oxidized form of DTT (OxDTT) was applied to neurons after recording of a stable ASIC current. Unlike the reduced form of DTT, OxDTT failed to enhance the ASIC current. Rather, 1.0 mm OxDTT induced an inhibition of the ASIC current to 58 ± 3% of the control (n = 11) (Fig. 2E,F). This data further suggests that redox-modulating property of DTT is required for its potentiation of the ASIC current.
Redox modulation of the ASIC current in acutely dissociated mouse cortical neurons
Prenatal cell cultures likely contain various immature neurons (Lendahl and McKay, 1990; Abdulla and Campbell, 1993). The properties of ion channels expressed in these neurons, and their modulation by redox reagents, may not be comparable with those expressed in mature and fully differentiated neurons. To address this concern, we also studied the effect of redox reagents on the ASIC current recorded in acutely isolated cortical neurons from adult mice. Neurons were isolated from 4-week-old mice, as described previously (Xiong et al., 1998). Similar to the cultured neurons, perfusion of 1.0 mm DTT increased the amplitude of the ASIC current (from −233.1 ± 52.9 to −347.9 ± 77.6 pA; n = 7; p < 0.01), whereas 0.5 mm DTNB inhibited the current (from −376.3 ± 51.8 to −265.9 ± 44.4 pA; n = 7; p < 0.05) (data not shown).
pH-dependent modulation of the ASIC current by redox reagents
To determine whether modulation of the ASIC current by redox reagents depends on the level of pHo, pH dose–response curves were constructed before and after the bath perfusion of DTT or DTNB. As shown in Figure 3, A and B, application of DTT induced a relatively larger potentiation of the ASIC current at higher pHo (e.g., 6.5) than at lower pHo (e.g., 5.0), resulting in a leftward shift of the pH dose–response curve (pH0.5 before DTT, 5.94 ± 0.08; after 1.0 mm DTT, 6.27 ± 0.03; n = 4; p < 0.05). The Hill coefficient was not significantly affected by DTT (before DTT, 0.94 ± 0.01; after DTT, 0.96 ± 0.01; n = 4; p > 0.05). This finding suggests that potentiation of the ASIC currents by DTT is likely mediated by an increase in the apparent affinity of ASICs to protons.
Figure 3.

Modulation of ASICs by redox reagents is pH dependent and voltage independent. A, Representative traces showing DTT potentiation of ASIC currents at different pHo levels. B, DTT potentiates the ASIC current by inducing a leftward shift of the pH dose–response curve. In the absence of DTT, pH0.5 is 5.94 ± 0.08; with 1.0 mm DTT, the pH0.5 is 6.27 ± 0.03; n = 4; *p < 0.05. C, Representative traces showing ASIC currents activated at different holding potentials ranging from −60 to +40 mV in the absence and presence of 1.0 mm DTT or 0.5 mm DTNB. Currents were activated by a pHo drop from 7.4 to 6.5. D, Current–voltage relationship (I–V curve) before and after DTT or DTNB; n = 5. Both curves were fit by a straight line with reversal potential at approximately +60 mV.
To determine whether the effect of redox reagents depends on the level of membrane potential, ASIC currents were activated at different holding potentials. As shown in Figure 3, C and D, addition of 1.0 mm DTT or 0.5 mm DTNB affected the ASIC current to a similar extent at membrane potentials ranging from −60 to +40 mV, indicating the lack of a voltage-dependent effect. In addition, the reversal potential of ASIC currents remained unchanged in the presence of DTT or DTNB, suggesting that redox modulation does not alter the ion selectivity of the channels.
Potentiation of the ASIC currents by reducing agents is not attributable to zinc chelation
It is known that most physiological solutions contain contaminating concentrations of zinc in the range of 20 ∼ 50 nm (Paoletti et al., 1997; Amar et al., 2001) or higher (Zheng et al., 1998; Wilkins and Smart, 2002). Our recent studies have demonstrated that zinc chelators such as TPEN and tricine dramatically potentiate the ASIC currents in mouse cortical neurons (Chu et al., 2004b). Because DTT and GSH may have some heavy metal chelating activity (Cornell and Crivaro, 1972; Paoletti et al., 1997; Wilkins and Smart, 2002), we have examined whether the effects of these agents on the ASIC currents were caused by zinc chelation. A 10 mm concentration of tricine, a heavy metal chelating agent, was added to the extracellular solution. With this concentration of tricine, free zinc in the extracellular solution is expected to be reduced to <1 nm (Amar et al., 2001; Wilkins and Smart, 2002), a concentration that has little inhibition of the ASIC current (Chu et al., 2004b). As expected, adding 10 mm tricine to the extracellular solution dramatically potentiated the ASIC current (data not shown). However, addition of 0.1 mm DTT or 1.0 mm GSH, in the presence of tricine, still potentiated the ASIC current, indicating Zn2+-independent effect (n = 8) (Fig. 4A,B).
Figure 4.

Potentiation of ASIC currents by reducing agents is not attributable to zinc chelation. A, B, Representative traces and summary data showing the potentiation of ASIC currents by 0.1 mm DTT and 1.0 mm GSH in the presence of zinc chelator tricine (10 mm); n = 8. C, D, Representative traces and summary data showing the potentiation of ASIC current by 0.1 and 0.5 mm TCEP, a reducing agent with little zinc chelating activity; n = 10; **p < 0.01.
To provide further evidence that potentiation of ASIC current by reducing agents was not caused by zinc chelation, we also studied the effect of tri (2-carboxyethyl) phosphine hydrochloride (TCEP), a reducing agent with little zinc chelating activity (Brohawn et al., 2003), on the ASIC currents. Similar to DDT and GSH, TCEP significantly potentiated the ASIC current at 0.1 and 0.5 mm (Fig. 4C,D).
Extracellular sites are involved in the modulation of the ASIC current by redox reagents
To address whether redox reagents modulate the ASIC currents via extracellular or intracellular redox site(s), we included 1.0 mm GSH in the intracellular solution and tested the effect of redox reagents applied to the extracellular solution. It is expected that, if the modulation of the ASIC current by reducing reagents is attributable to their effects on an intracellular site, inclusion of a high concentration of GSH in the pipette solution should preclude a further potentiation of the current by bath perfusion of the reducing agents. After formation of whole-cell configuration, up to 20 min was allowed for GSH to completely diffuse into neurons before bath application of DTT or GSH. As shown in Figure 5, A and B, with 1 mm GSH in the pipette solution, bath perfusion of GSH (1.0 mm) or DTT (0.1 mm) still potentiated the ASIC current, and the degree of potentiation is similar to that obtained without GSH in the pipette solution. This finding suggests that reducing agents potentiate ASIC currents through their interaction with an extracellular redox site. To provide additional evidence, we also included 0.5 mm DTNB in the pipette solution and tested the effect of 1.0 mm DTT or 0.5 mm DTNB on the ASIC current. Similar to the recordings without intracellular DTNB, bath perfusion of DTT potentiated, whereas perfusion of DTNB inhibited, the ASIC current (Fig. 5C,D). Together, these data strongly suggest that redox reagents modulate the ASIC activity through an extracellular redox site(s).
Figure 5.

Extracellular sites are involved in redox modulation of ASIC currents. A, B, Representative traces and summary data showing the potentiation of ASIC currents by extracellular GSH (1.0 mm; n = 6) and DTT (0.1 mm; n = 5) with the pipette solution containing 1.0 mm GSH. **p < 0.01. C, D, Representative traces and summary data showing the potentiation or inhibition of ASIC current by extracellular DTT (1.0 mm) or DTNB (0.5 mm) with the pipette solution containing 0.5 mm DTNB; n = 5; *p < 0.05. ICF, Intracellular fluid.
Redox modulation of ASIC currents in neurons from ASIC1 and ASIC2 knock-out mice
ASIC1a and ASIC2a are the major functional ASIC subunits expressed in mouse cortical neurons (Chu et al., 2004b; Xiong et al., 2004). However, activation of these two ASIC subunits seems to have opposite consequences for neuronal injury. Activation of ASIC1a is involved in acidosis-mediated ischemic neuronal death (Xiong et al., 2004), whereas increased expression of ASIC2a is known to be associated with neuronal survival (Johnson et al., 2001). For this reason, it is important to know which ASIC subunit is involved in the redox modulation of the ASIC current. To answer this question, we have studied the effect of redox reagents on ASIC currents in cortical neurons cultured from the ASIC1 or ASIC2 knock-out mice (Chu et al., 2004b).
ASIC currents in neurons from C57BL/6 mice, a wild-type control with the same genomic background as the ASIC1 knock-out mice, were potentiated by DTT (0.1 mm), GSH (1.0 mm), or cysteine (1.0 mm) and inhibited by DTNB (0.5 mm) or OxDTT (1.0 mm), similar to the current in neurons cultured from Swiss mice (Fig. 6A,B). This finding suggests that genomic background has no influence on the redox modulation of ASICs. In ASIC1−/− neurons, however, perfusion of 1.0 mm DTT, 1.0 mm GSH, 1.0 mm cysteine, 0.5 mm DTNB, or 1.0 mm OxDTT did not affect the ASIC current (Fig. 6C,D) (n = 8–9). In contrast to neurons from the ASIC1−/− mice, addition of 0.1 mm DTT, 1.0 mm GSH or 1.0 mm cysteine significantly potentiated (n = 7–12) (Fig. 6E,F), whereas 0.5 mm DTNB and 1.0 mm OxDTT inhibited the ASIC current in neurons from the ASIC2−/− mice (n = 7–12) (Fig. 6E,F). These data suggest that ASIC1a subunit is required for redox modulation of ASIC currents.
Figure 6.
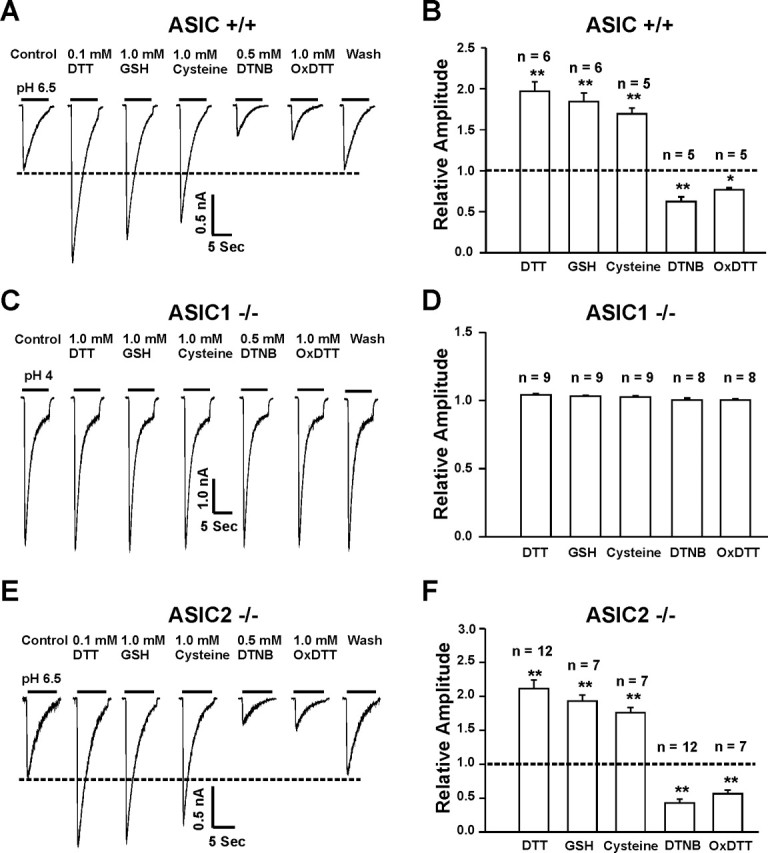
Redox modulation of ASIC currents in neurons from ASIC1−/− or ASIC2−/− mice. A, B, Representative traces and summary data showing the modulation of ASIC currents by the reducing or the oxidizing agents in cortical neurons from C57BL/6 mice, a wild-type control for the ASIC knock-out mice; n = 5–6. C, D, Representative traces and summary data showing a lack of modulation of ASIC currents in ASIC1−/− neurons by either reducing or oxidizing agents; n = 8–9. E, F, Representative traces and summary data showing the effect of redox reagents on ASIC currents in ASIC2−/− neurons; n = 7–12; *p < 0.05; **p < 0.01.
Subunit-specific modulation of ASIC currents by redox reagents in CHO cells
To further determine the role of each ASIC subunit in redox modulation of the ASIC current, we then studied the effect of redox reagents on ASIC currents mediated by various ASIC subunits expressed in CHO cells. Consistent with the findings in native neurons, bath perfusion of reducing agents DTT (1.0 mm), GSH (1.0 mm), or cysteine (1.0 mm) dramatically potentiated the current mediated by homomeric ASIC1a more than twofold (n = 8–15; p < 0.01), whereas DTNB (0.5 mm) or OxDTT (1.0 mm) inhibited the ASIC1a current by ∼50% (n = 8–15). In contrast, no effect was observed on currents mediated by homomeric ASIC1b (ASICβ), ASIC2a, or ASIC3 (n = 5–7) (Fig. 7C–H). To exclude the possibility that the lack of redox modulation of the ASIC2a current is attributable to the low pHo value used, as has been suggested for Zn2+ potentiation of the ASIC2a current (Baron et al., 2001), we also studied the effect of redox reagents on the ASIC2a current activated at a pHo of 5.0 (Fig. 7G). Again, no change of the ASIC2a current was induced by DTT, cysteine, or DTNB (n = 7). These data suggest that the lack of effect by redox reagents on the ASIC2a current is likely caused by a lack of redox modification rather than low pHo value used.
Figure 7.
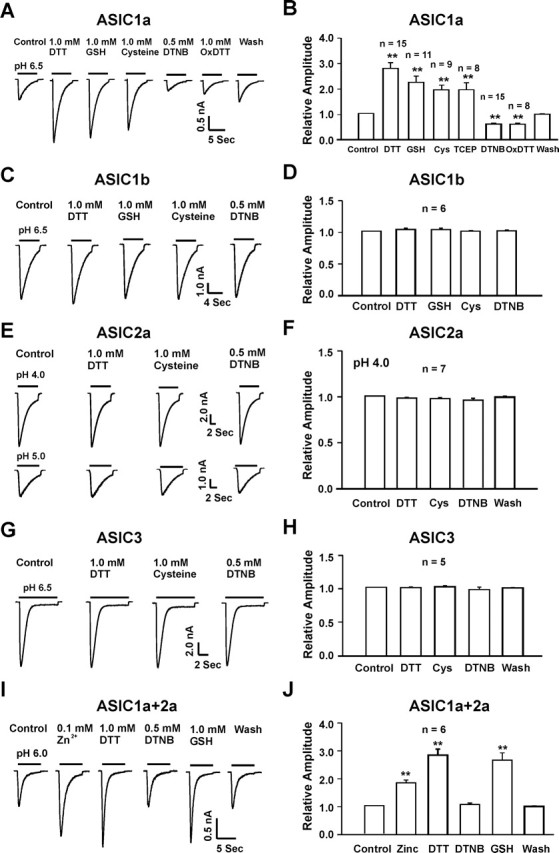
Redox reagents specifically modulate ASIC currents mediated by homomeric ASIC1a and heteromeric ASIC1a/2a without effect on currents mediated by homomeric ASIC1b, ASIC2a, or ASIC3. A, B, Representative traces and summary data showing the effect of reducing and oxidizing agents on homomeric ASIC1a currents in CHO cells; n = 8–15; **p < 0.01. C–H, Representative traces and summary data showing the lack of redox modulation of the ASIC current mediated by homomeric ASIC1b (C, D), ASIC2a (E, F), or ASIC3 (G, H); n = 5–7; p > 0.05. I, J, Representative traces and summary data showing a potentiation of ASIC1a/ASIC2a currents in CHO cells by Zn2+ and the reducing agents DTT or GSH, and a lack of modulation by the oxidizing agent DTNB; n = 6; **p < 0.01. Cys, Cysteine.
The effect of redox reagents on the ASIC1a/2a current was also studied in CHO cells. Two criteria, including pH0.5 value and Zn2+ potentiation, were used to ensure that the current studied was indeed mediated by the heteromeric ASIC1a/2a channels (Chu et al., 2004b). Similar to homomeric ASIC1a channels, current mediated by heteromeric ASIC1a/2a channels was potentiated by the reducing agents (Fig. 7I,J). However, unlike homomeric ASIC1a channels, current mediated by heteromeric ASIC1a/2a channels was not inhibited by the oxidizing agent DTNB (Fig. 7I,J). This finding is consistent with the lack of DTNB effect in some neurons, which may contain heteromeric ASIC1a/2a channels. It may also suggest that there could be more than one redox modulating site on the ASIC1a subunit: one involved in the potentiation by the reducing agents and the other involved in the inhibition by the oxidizing agents. Association of ASIC2a with the ASIC1a subunit does not seem to affect the sites for the reducing agents, but may occlude the sites for the oxidizing agents.
Involvement of extracellular cysteine and lysine in the redox modulation of the ASIC1a current
Site-directed mutagenesis was then used to identify the specific amino acid(s) involved in the redox modulation of the ASIC1a current. Previous studies have suggested that cysteine residues are commonly involved in the redox modulation of various ion channels (Creighton, 1984; Ruppersberg et al., 1991; Sullivan et al., 1994; Broillet, 2000; Choi et al., 2001; Lipton et al., 2002). In addition, residues such as methionine, tyrosine, phenylalanine, histidine, or lysine may also participate in the redox modulation of proteins (Thielges et al., 2005; Waring, 2005). To determine whether cysteine residues are involved in the redox modulation of ASIC1a channels, we first tested the effect of [2-(trimethylammonium)ethyl] methanethiosulfonate (MTSET), a methane-thiosulfonate agent known to modify the cysteine residues (Ruppersberg et al., 1991; Phillips et al., 2003), on the ASIC current. This agent can interact with free thiols rapidly and specifically to form mixed disulfides. Exposure of neurons to 1.0 mm MTSET for 3 min inhibited the ASIC1a current by ∼50% and abolished the subsequent inhibition by DTNB (n = 5; data not shown). Based on these findings and the fact that redox modulation is ASIC1a dependent, we focused on the cysteine residues specific to ASIC1a subunit and located in the extracellular domain between residues 61 and 175 (nonidentical extracellular region between ASIC1a and ASIC1b). Sequence analysis revealed two cysteine residues (C61 and C70) located close to the first transmembrane domain (Waldmann et al., 1997), as potential targets (Fig. 8A). To determine the effect of these residues in the redox modulation of the ASIC1a current, we mutated both cysteines to alanines. In addition, we also mutated C59, a cysteine residue located adjacent to the extracellular domain and conserved in ASIC1a and ASIC1b subunits, as a negative control. Other potential redox modulating residues that are specific to ASIC1a subunit, e.g., K133 and H173, were also mutated. Sequence alignment demonstrated that these residues are found only in ASIC1a but not ASIC1b, ASIC2a, or ASIC3 subunit in comparable positions (Fig. 8A). Noncharged residues were replaced with alanine (i.e., C59A, C61A, C70A, and H173A), whereas positive charged K133 was replaced with arginine (K133R). When expressed in CHO cells, all mutants had normal response to pHo drops with pH0.5 and amiloride blockade similar to the wild-type ASIC1a channels (data not shown).
Figure 8.
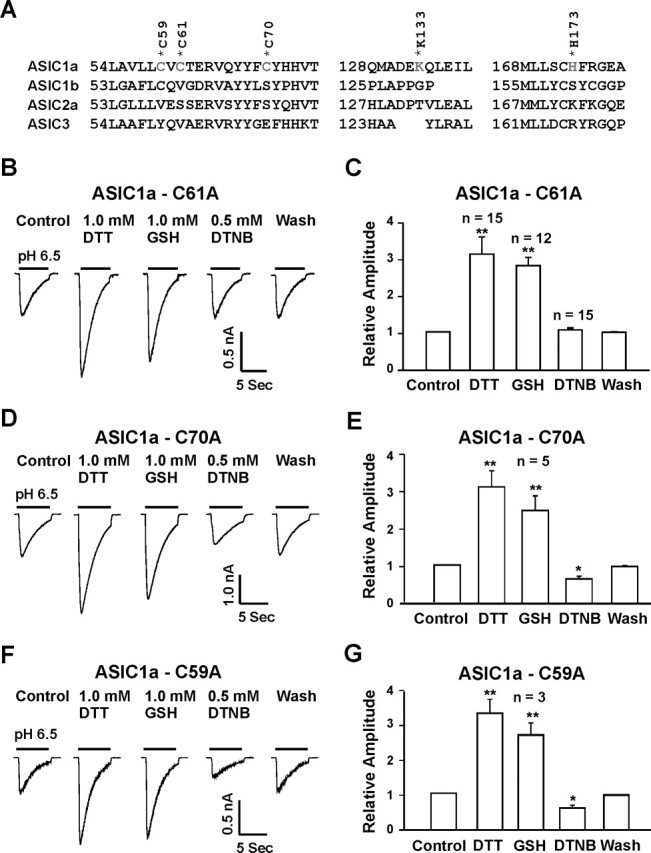
Identification of amino acid residues involved in redox modification. A, Alignment of ASIC1a, ASIC1b (ASICβ), ASIC2a, and ASIC3 identified potential redox modulatory sites present in ASIC1a but not conserved at homologous positions in ASIC1b, ASIC2a, and ASIC3 subunits. B, C, Representative traces and summary data showing the lack of inhibition of the ASIC current mediated by ASIC1a-C61A mutant by the oxidizing agent DTNB. D–G, Neither the C70A (n = 5) nor C59A (n = 3) mutation affected the potentiation or inhibition of the ASIC1a currents by reducing or oxidizing agents. *p < 0.05; **p < 0.01.
As shown in Figure 8, mutation of C61 to alanine did not affect the potentiation of ASIC currents by the reducing agents (Fig. 8B,C) (n = 12–15). However, this point mutation completely eliminated the inhibition of the currents by the oxidizing agent DTNB. In contrast, mutation of C70 (n = 5) or C59 (n = 3) affected neither the potentiation nor the inhibition of the ASIC currents by redox reagents (Fig. 8D–G). Similarly, mutation of H173 did not affect the modulation of the ASIC currents by redox reagents (Fig. 9A,B) (n = 5). Mutation of K133 to arginine, however, completely abolished the potentiation of ASIC currents by reducing agents, with no effect on the inhibition of the currents by DTNB (Fig. 9C,D) (n = 5). Double mutation (C61A and K133R) reduced the overall amplitude of the acid-activated current and completely eliminated the effects by both reducing and oxidizing agents (Fig. 9E,F).
Figure 9.
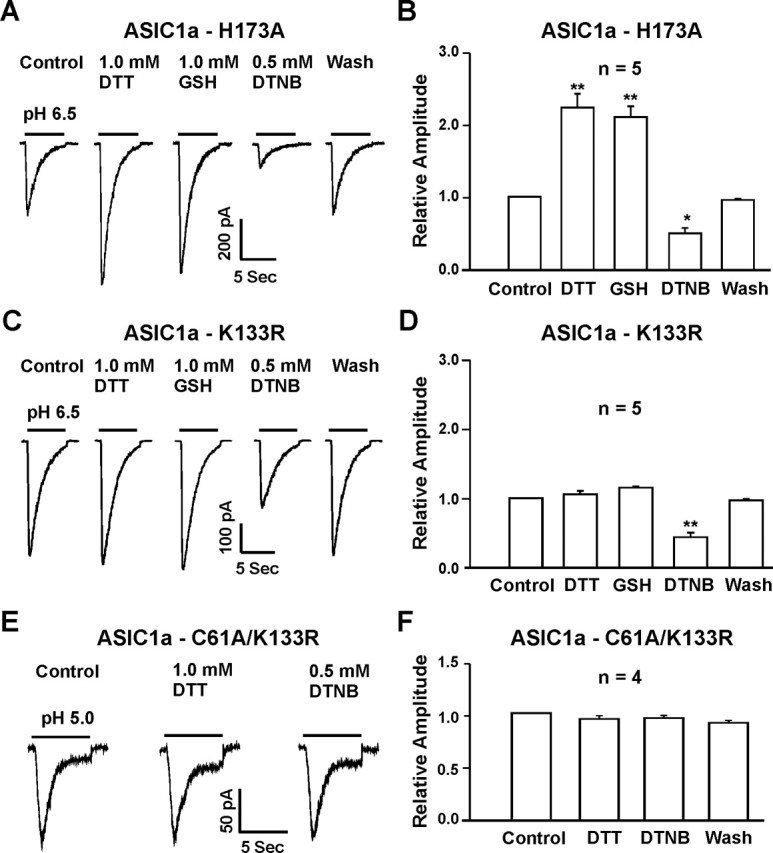
Lysine 133 mutation eliminated the potentiation of ASIC1a currents by reducing agents, whereas the C61A and K133R double mutation abolished the effect of both reducing and oxidizing agents on homomeric ASIC1a currents. A, B, Representative traces and summary data showing that the H173A mutation did not affect the potentiation or inhibition of ASIC1a currents by reducing or oxidizing agents; n = 5; *p < 0.05; **p < 0.01. C, D, Representative traces and summary data showing the lack of potentiation of ASIC1a–K133R currents by the reducing agents DTT and GSH, whereas DTNB inhibited the ASIC current; n = 5; **p < 0.01. E, F, Representative traces and summary data showing the lack of modulation by both reducing and oxidizing agents on currents mediated by the ASIC1a–C61A/K133R double mutation; n = 4; p > 0.05.
Effects of redox reagents on ASIC-mediated intracellular Ca2+ increase and membrane depolarization
Ca2+ entry through ASIC1a channels is largely responsible for acidosis-mediated glutamate-independent neuronal injury (Xiong et al., 2004). To know whether redox modulation of ASICs also affects the acid-induced Ca2+ entry into neurons, fluorescent Ca2+-imaging experiments were performed to study the effect of redox reagents on acid-induced increases in [Ca2+]i. For all experiments, the blockers of voltage-gated Ca2+ channels (e.g., 5 μm nimodipine and 3 μm ω-conotoxin MVIIC) and glutamate receptors (10 μm MK801 and 20 μm CNQX) were added to the solutions to exclude the possible secondary activation of these channels. As shown in Figure 10, A and B, bath perfusion of DTT (1.0 mm) did not affect the baseline [Ca2+]i; however, it dramatically potentiated the acid-induced increase of [Ca2+]i (340/380 nm ratio before DTT, 1.81 ± 0.15; after DTT, 3.30 ± 0.26; n = 10; p < 0.01) (Fig. 10A,B). In contrast, bath perfusion of the oxidizing agent DTNB inhibited the acid-induced increase of [Ca2+]i (340/380 nm ratio before DTNB: 2.02 ± 0.35; after DTNB 1.19 ± 0.16; n = 8; p < 0.05) (Fig. 10C,D).
Figure 10.

Effects of redox reagents on acid-induced increases in [Ca2+]i and membrane depolarization in cultured mouse cortical neurons. A, B, Representative 340/380 nm ratio and summary data demonstrating changes in [Ca2+]i induced by pHo decrease in the absence and presence of 1.0 mm DTT; n = 10; **p < 0.01. C, D, Representative 340/380 nm ratio and summary data demonstrating changes in [Ca2+]i induced by pHo decrease in the absence and presence of 0.5 mm DTNB; n = 8; *p < 0.05. E, F, Representative traces and summary data showing membrane depolarization induced by pHo reduction from 7.4 to 7.0 in the absence and presence of DTT (0.1 mm, n = 5); **p < 0.01. To block secondary activation of glutamate receptors and voltage-gated Ca2+ channels, 10 μm MK801, 20 μm CNQX, 5 μm nimodipine and 3 μm ω-conotoxin MVIIC were included in all solutions.
Our next experiment was to determine whether redox modulation of the ASICs has any impact on acid-induced membrane depolarization in mouse cortical neurons. Current-clamp experiments were performed to record the membrane potential in the presence of the blockers of voltage-gated Ca2+ channels and glutamate receptor-gated channels (Chu et al., 2004b). As shown in Figure 10, E and F, decreasing pHo from 7.4 to 7.0 induced a significant depolarization of membrane potential from −60 mV to −34.72 ± 1.46 mV (n = 5; p < 0.05). Bath perfusion of DTT (1 mm) dramatically potentiated acid-induced membrane depolarization to −5.07 ± 2.43 mV (n = 5; p < 0.01).
Discussion
Ca2+-permeable ASIC1a is one of the major ASIC subunits in CNS neurons. Activation of these channels is believed to be involved in normal brain function and in the pathology of ischemia. Here, we demonstrate that the redox status of the ASIC1a subunit is an important modulator for the function of ASICs. Because the redox status of proteins changes dramatically in both physiological and pathological processes, the regulation of ASICs by redox modification may have significant influence on the overall functions of these channels. Accordingly, targeting the redox modulation sites on these channels may offer a new approach for future neuroprotective interventions.
Similar to the study in sensory neurons (Andrey et al., 2005), reducing agents enhanced, whereas oxidizing agents inhibited the ASIC currents in CNS neurons. An additional finding that NEM, an alkylating agent known to irreversibly alkylate free thiol groups (Creighton, 1984; Tang and Aizenman, 1993), blocked the effects of redox reagents supports an involvement of critical sulfhydryl groups in the effects of redox reagents. The finding that both reducing and oxidizing agents have an effect on the currents suggests that the redox modulatory site(s) on ASICs (or closely associated proteins) exist in a dynamic equilibrium between fully reduced and oxidized states. The fact that the magnitude of potentiation by the reducing agents is severalfold greater than the magnitude of inhibition by the oxidizing agents may suggest that the redox site(s) of ASICs in mouse cortical neurons is closer to a fully oxidized state. Alternatively, there could be two or more separate sites responsible for the modulation of ASICs by oxidizing and reducing agents, respectively, and the impact of each site on the channel differs.
Different from our studies in mouse cortical neurons, the effect of redox reagents on the ASIC current in sensory neurons does not involve a change in the affinity of ASICs to protons (Andrey et al., 2005). This is likely attributable to different ASIC subunits expressed in sensory and CNS neurons. In CNS neurons, homomeric ASIC1a and heteromeric ASIC1a/ASIC2a are predominant (Askwith et al., 2004; Chu et al., 2004b), whereas in sensory neurons, ASIC currents are likely carried by a combination of ASIC1a, ASIC1b, ASIC2a, and ASIC3 (Waldmann and Lazdunski, 1998; Krishtal, 2003). Our studies in CHO cells clearly demonstrated that redox agents specifically modulate the currents mediated by homomeric ASIC1a or heteromeric ASIC1a/ASIC2a with little effect on other ASIC configurations. These data may suggest that, compared with CNS neurons, ASICs in sensory neurons are not the preferred target for the redox modulation. Indeed, the degree of potentiation of ASIC currents in rat trigeminal and DRG neurons (∼30%) by the reducing agents is far less than that in mouse cortical neurons (up to 200%).
Redox modulation of ASICs independent of zinc chelation
Homomeric ASIC1a and ASIC1a containing channels are tonically inhibited by nanomolar concentration of zinc, commonly present in physiological solutions (Chu et al., 2004b). Because reducing agents such as DTT may have some heavy metal chelating activity (Cornell and Crivaro, 1972; Paoletti et al., 1997; Wilkins and Smart, 2002), it was important to know whether the potentiation of ASIC currents was mediated by zinc chelation. Our finding that DTT and GSH continued to potentiate the currents in the presence of high concentration of zinc chelator strongly suggests that chelation of zinc is not responsible for the effect of these agents. In addition, TCEP, a reducing agent with little zinc binding activity (Brohawn et al., 2003), also potentiated the ASIC current, further supporting the claim that reducing agents potentiate the ASIC current through redox modification.
Redox modulation is ASIC1a dependent
ASIC1a and ASIC2a are the major functional ASICs in CNS neurons, and the acid-activated currents in these neurons are largely mediated by homomeric ASIC1a and heteromeric ASIC1a/ASIC2a channels. To determine the specific ASIC subunit involved in the redox modulation, we performed experiments in neurons cultured from the ASIC1−/− and ASIC2−/− mice. Our finding that redox reagents lost their effect in ASIC1−/− neurons strongly suggests that ASIC1a subunit is critical for the modulation. Consistent with these findings, redox reagents only affected the currents mediated by ASIC1a-containing channels expressed in CHO cells.
Cysteine 61 and lysine 133 are involved in the modulation of ASIC1a by redox reagents
The membrane-impermeable nature of GSH and DTNB suggests that the redox modulating site(s) are located on the extracellular domain of the channel. Furthermore, the fact that inclusion of a saturating concentration of GSH or DTNB in the intracellular solution did not preclude the ability of extracellular applied GSH and DTT to modulate the channels also supports the existence of an extracellular redox modulating site(s).
The sulfur moiety in cysteine residues is a primary target for redox modification (Ruppersberg et al., 1991; Sullivan et al., 1994; Broillet, 2000; Choi et al., 2001; Lipton et al., 2002). Other residues capable of redox alteration include methionine, tyrosine, phenylalanine, histidine, and lysine (Thielges et al., 2005; Waring, 2005). The multiple cysteine residues in the large extracellular domain of ASICs are potential targets for redox modulation. Because the redox reagents modulated the current mediated by ASIC1a without affecting the current mediated by ASIC1b, ASIC2a, or ASIC3, we have limited our search for redox modulation site(s) to the extracellular domain of ASIC1a not conserved in ASIC1b, ASIC2a, and ASIC3. Because ASIC1a and ASIC1b (ASICβ) only differ in the first 175 amino acids at the N terminal (Chen et al., 1998), the lack of redox modulation of the ASIC1b current further simplified our search. Taking all factors into consideration, only C61 and C70 were considered as the likely candidates involved in the redox modulation. Site-directed mutagenesis studies demonstrated that mutation of C61, but not C70, eliminated the inhibition of ASIC currents by the oxidizing agent DTNB. However, this mutation did not affect the potentiation of the current by the reducing agents. One possible explanation is that, under normal condition, C61 is in a fully reduced state, thus preventing additional modification by the reducing agents. In contrast, mutation of K133, a lysine residue also located in the extracellular domain specific to the ASIC1a subunit, eliminated the potentiation of ASIC currents by the reducing agents, without altering the effect by the oxidizing agent. It is likely that this site is in a fully oxidized state under the resting condition, thus occluding a further modification by the oxidizing agents. As expected, double mutation of C61 and K133 eliminated the effects by both oxidizing and reducing agents.
Interestingly, our previous studies have shown that K133 is also involved in high-affinity Zn2+ inhibition of the ASIC current (Chu et al., 2004b). The involvement of K133 in both Zn2+ inhibition and redox modulation of ASICs suggests that multiple modulators of ASICs may share a common molecular pathway. Additional investigation will determine how Zn2+ inhibition and redox modulation of ASICs share the same pathway.
Redox reagents are known to modulate the activities of other ligand-gated ion channels, including NMDA receptor-gated channels (Aizenman et al., 1989; Kohr et al., 1994; Sullivan et al., 1994). Similar to their effects on ASICs, reducing agents potentiate, whereas oxidizing agents inhibit, the NMDA currents. The redox modulation site or motif, in general, consists of several rather than a single amino acid and is defined by the tertiary/quaternary structure of the protein. For example, for NMDA channels, there are six cysteines comprising the redox modulation motif (Lipton et al., 2002). Although mutation of a single amino acid (e.g., C61 and K133) disrupts the redox modulation, these amino acids are likely part of a larger motif. Further studies are required to identify other potential residues, either conserved or nonconserved, that participate in the formation of redox modulation motif on ASICs.
Redox modulation of ASICs is physiologically and pathologically relevant
Endogenous redox compounds such as GSH and cysteine are present both in the periphery and in the CNS (Slivka et al., 1987; Olney et al., 1990; Nanda et al., 1996). For example, 1 ∼ 2 mm of GSH has been reported in the extracellular space of the brain (Slivka et al., 1987). It has also been demonstrated that GSH and cysteine can be released in the CNS during neurotransmission (Zangerle et al., 1992). In the present study, GSH dramatically enhanced ASIC currents at a concentration of 1.0 mm, suggesting that the physiological concentration of GSH is likely to alter the ASIC activity in vivo. Because activation of ASICs and subsequent membrane depolarization is known to facilitate excitatory neurotransmission (Wemmie et al., 2002), enhancement of the ASIC activities by endogenous reducing agents may participate in normal synaptic neurotransmission. Excessive activation of ASIC1a, resulting in high calcium influx, is associated with acidosis-mediated neuronal injury (Xiong et al., 2004). Modulation of the ASICs by redox modification is therefore expected to be involved in the pathology of brain ischemia. In support of this notion, the degree of acid-induced injury of cultured mouse cortical neurons was substantially potentiated by the presence of l-cysteine (Chu et al., unpublished observations). Together, our studies suggest that targeting the redox modulating sites of the ASIC1a subunit may provide an insight into development of novel therapeutic strategies for ischemic brain injury.
Footnotes
This work was supported by National Institutes of Health Grants R01NS42926 and R01NS47506 (Z.-G.X.), American Heart Association Grants 0230280N (Z.-G.X.) and 0465338Z (X.-P.C.), and grants from the Legacy Research Advisory Committee. We thank Roger P. Simon for critical comments on this manuscript, Joshua Seeds, Eric M. Kratzer, and Suzanne Zeitouni for technical support; J. A. Wemmie, M. P. Price, and M. J. Welsh (Howard Hughes Medical Institute, University of Iowa, Iowa City, IA) for ASIC knock-out mice; R. Waldmann and M. Lazdunski (Institut de Pharmacologie Moleculaire et Cellulaire, Centre National de la Recherche Scientifique, Valbonne, France) for ASIC clones, and E. W. McCleskey (Vollum Institute, Portland, OR) for CHO cells.
References
- Abdulla EM, Campbell IC (1993). In vitro tests of neurotoxicity. J Pharmacol Toxicol Methods 29:69–75. [DOI] [PubMed] [Google Scholar]
- Aizenman E, Lipton SA, Loring RH (1989). Selective modulation of NMDA responses by reduction and oxidation. Neuron 2:1257–1263. [DOI] [PubMed] [Google Scholar]
- Alvarez de la Rosa D, Krueger SR, Kolar A, Shao D, Fitzsimonds RM, Canessa CM (2003). Distribution, subcellular localization and ontogeny of ASIC1 in the mammalian central nervous system. J Physiol (Lond) 546:77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar M, Perin-Dureau F, Neyton J (2001). High-affinity Zn2+ block in recombinant N-methyl-d-aspartate receptors with cysteine substitutions at the Q/R/N site. Biophys J 81:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amato A, Connolly CN, Moss SJ, Smart TG (1999). Modulation of neuronal and recombinant GABAA receptors by redox reagents. J Physiol (Lond) 517:35–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrey F, Tsintsadze T, Volkova T, Lozovaya N, Krishtal O (2005). Acid sensing ionic channels: modulation by redox reagents. Biochim Biophys Acta 1745:1–6. [DOI] [PubMed] [Google Scholar]
- Aronstam RS, Abood LG, Hoss W (1978). Influence of sulfhydryl reagents and heavy metals on the functional state of the muscarinic acetylcholine receptor in rat brain. Mol Pharmacol 14:575–586. [PubMed] [Google Scholar]
- Askwith CC, Wemmie JA, Price MP, Rokhlina T, Welsh MJ (2004). ASIC2 modulates ASIC1 H+-activated currents in hippocampal neurons. J Biol Chem 279:18296–18305. [DOI] [PubMed] [Google Scholar]
- Baron A, Schaefer L, Lingueglia E, Champigny G, Lazdunski M (2001). Zn2+ and H+ are coactivators of acid-sensing ion channels. J Biol Chem 276:35361–35367. [DOI] [PubMed] [Google Scholar]
- Baron A, Waldmann R, Lazdunski M (2002). ASIC-like, proton-activated currents in rat hippocampal neurons. J Physiol (Lond) 539:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson CJ, Eckert SP, McCleskey EW (1999). Acid-evoked currents in cardiac sensory neurons: a possible mediator of myocardial ischemic sensation. Circ Res 84:921–928. [DOI] [PubMed] [Google Scholar]
- Benveniste M, Dingledine R (2005). Limiting stroke-induced damage by targeting an acid channel. N Engl J Med 352:85–86. [DOI] [PubMed] [Google Scholar]
- Brohawn SG, Miksa IR, Thorpe C (2003). Avian sulfhydryl oxidase is not a metalloenzyme: adventitious binding of divalent metal ions to the enzyme. Biochemistry 42:11074–11082. [DOI] [PubMed] [Google Scholar]
- Broillet MC (2000). A single intracellular cysteine residue is responsible for the activation of the olfactory cyclic nucleotide-gated channel by NO. J Biol Chem 275:15135–15141. [DOI] [PubMed] [Google Scholar]
- Chen CC, England S, Akopian AN, Wood JN (1998). A sensory neuron-specific, proton-gated ion channel. Proc Natl Acad Sci USA 95:10240–10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Chen HV, Lipton SA (2001). Three pairs of cysteine residues mediate both redox and Zn2+ modulation of the NMDA receptor. J Neurosci 21:392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XP, Miesch J, Johnson M, Root L, Zhu XM, Chen D, Simon RP, Xiong ZG (2002). Proton-Gated Channels in PC12 Cells. J Neurophysiol 87:2555–2561. [DOI] [PubMed] [Google Scholar]
- Chu XP, Close N, Saugstad JA, Simon RP, Xiong Z-G (2004a). Redox modulation of acid-sensing ion channels. Soc Neurosci Abstr 30:845–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu XP, Wemmie JA, Wang WZ, Zhu XM, Saugstad JA, Price MP, Simon RP, Xiong ZG (2004b). Subunit-dependent high-affinity zinc inhibition of acid-sensing ion channels. J Neurosci 24:8678–8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell NW, Crivaro KE (1972). Stability constant for the zinc-dithiothreitol complex. Anal Biochem 47:203–208. [DOI] [PubMed] [Google Scholar]
- Creighton TE (1984). Disulfide bond formation in proteins. Methods Enzymol 107:305–329. [DOI] [PubMed] [Google Scholar]
- DiChiara TJ, Reinhart PH (1997). Redox modulation of hslo Ca2+-activated K+ channels. J Neurosci 17:4942–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan A, Garcia-Anoveros J, Corey DP (2002). The PDZ domain protein PICK1 and the sodium channel BNaC1 interact and localize at mechanosensory terminals of dorsal root ganglion neurons and dendrites of central neurons. J Biol Chem 277:5203–5208. [DOI] [PubMed] [Google Scholar]
- Garcia-Anoveros J, Derfler B, Neville-Golden J, Hyman BT, Corey DP (1997). BNaC1 and BNaC2 constitute a new family of human neuronal sodium channels related to degenerins and epithelial sodium channels. Proc Natl Acad Sci USA 94:1459–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyte L, Barber PA, Buchan AM, Hill MD (2004). The rise and fall of NMDA antagonists for ischemic stroke. Curr Mol Med 4:131–136. [DOI] [PubMed] [Google Scholar]
- Huang Y, McNamara JO (2004). Ischemic stroke: “acidotoxicity” is a perpetrator. Cell 118:665–666. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Turski L (2002). Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol 1:383–386. [DOI] [PubMed] [Google Scholar]
- Johnson MB, Jin K, Minami M, Chen D, Simon RP (2001). Global ischemia induces expression of acid-sensing ion channel 2a in rat brain. J Cereb Blood Flow Metab 21:734–740. [DOI] [PubMed] [Google Scholar]
- Kohr G, Eckardt S, Luddens H, Monyer H, Seeburg PH (1994). NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron 12:1031–1040. [DOI] [PubMed] [Google Scholar]
- Krishtal O (2003). The ASICs: signaling molecules? Modulators? Trends Neurosci 26:477–483. [DOI] [PubMed] [Google Scholar]
- Krishtal OA, Pidoplichko VI (1981). A receptor for protons in the membrane of sensory neurons may participate in nociception. Neuroscience 6:2599–2601. [DOI] [PubMed] [Google Scholar]
- Lendahl U, McKay RD (1990). The use of cell lines in neurobiology. Trends Neurosci 13:132–137. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Takahashi H, Zhang D, Li W, Godzik A, Bankston LA (2002). Cysteine regulation of protein function–as exemplified by NMDA-receptor modulation. Trends Neurosci 25:474–480. [DOI] [PubMed] [Google Scholar]
- Nanda D, Tolputt J, Collard KJ (1996). Changes in brain glutathione levels during postnatal development in the rat. Brain Res Dev Brain Res 94:238–241. [DOI] [PubMed] [Google Scholar]
- Olney JW, Zorumski C, Price MT, Labruyere J (1990). L-cysteine, a bicarbonate-sensitive endogenous excitotoxin. Science 248:596–599. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Ascher P, Neyton J (1997). High-affinity zinc inhibition of NMDA NR1-NR2A receptors. J Neurosci 17:5711–5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips LR, Enkvetchakul D, Nichols CG (2003). Gating dependence of inner pore access in inward rectifier K(+) channels. Neuron 37:953–962. [DOI] [PubMed] [Google Scholar]
- Price MP, Lewin GR, McIlwrath SL, Cheng C, Xie J, Heppenstall PA, Stucky CL, Mannsfeldt AG, Brennan TJ, Drummond HA, Qiao J, Benson CJ, Tarr DE, Hrstka RF, Yang B, Williamson RA, Welsh MJ (2000). The mammalian sodium channel BNC1 is required for normal touch sensation. Nature 407:1007–1011. [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M (1991). Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature 352:711–714. [DOI] [PubMed] [Google Scholar]
- Saugstad JA, Roberts JA, Dong J, Zeitouni S, Evans RJ (2004). Analysis of the membrane topology of the acid-sensing ion channel 2a. J Biol Chem 279:55514–55519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JB, Lindenau J, Seyfried J, Dichgans J (2000). Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 267:4904–4911. [DOI] [PubMed] [Google Scholar]
- Slivka A, Spina MB, Cohen G (1987). Reduced and oxidized glutathione in human and monkey brain. Neurosci Lett 74:112–118. [DOI] [PubMed] [Google Scholar]
- Sullivan JM, Traynelis SF, Chen H-SV, Escobar W, Heinemann SF, Lipton SA (1994). Identification of two cysteine residues that are required for redox modulation of the NMDA subtype of glutamate receptor. Neuron 13:929–936. [DOI] [PubMed] [Google Scholar]
- Sutherland SP, Benson CJ, Adelman JP, McCleskey EW (2001). Acid-sensing ion channel 3 matches the acid-gated current in cardiac ischemia-sensing neurons. Proc Natl Acad Sci USA 98:711–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L-H, Aizenman E (1993). The modulation of N-methyl-d-aspartate receptors by redox and alkylating reagents in rat cortical neurones in vitro. J Physiol (Lond) 465:303–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thielges M, Uyeda G, Camara-Artigas A, Kalman L, Williams JC, Allen JP (2005). Design of a redox-linked active metal site: manganese bound to bacterial reaction centers at a site resembling that of photosystem II. Biochemistry 44:7389–7394. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic-Todorovic V, Meyenburg A, Mennerick S, Perez-Reyes E, Romano C, Olney JW, Zorumski CF (2001). Redox modulation of T-type calcium channels in rat peripheral nociceptors. Neuron 31:75–85. [DOI] [PubMed] [Google Scholar]
- Waldmann R (2001). Proton-gated cation channels–neuronal acid sensors in the central and peripheral nervous system. Adv Exp Med Biol 502:293–304. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Lazdunski M (1998). H(+)-gated cation channels: neuronal acid sensors in the ENaC/DEG family of ion channels. Curr Opin Neurobiol 8:418–424. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M (1997). A proton-gated cation channel involved in acid-sensing. Nature 386:173–177. [DOI] [PubMed] [Google Scholar]
- Waring P (2005). Redox active calcium ion channels and cell death. Arch Biochem Biophys 434:33–42. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH, Welsh MJ (2002). The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34:463–477. [DOI] [PubMed] [Google Scholar]
- Wemmie JA, Askwith CC, Lamani E, Cassell MD, Freeman JH Jr., Welsh MJ (2003). Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. J Neurosci 23:5496–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins ME, Smart TG (2002). Redox modulation of GABAA receptors obscured by Zn2+ complexation. Neuropharmacology 43:938–944. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Raouf R, Lu WY, Wang LY, Orser BA, Dudek EM, Browning MD, MacDonald JF (1998). Regulation of N-methyl-d-aspartate receptor function by constitutively active protein kinase C. Mol Pharmacol 54:1055–1063. [PubMed] [Google Scholar]
- Xiong ZG, Pelkey KA, Lu WY, Lu YM, Roder JC, MacDonald JF, Salter MW (1999). Src potentiation of NMDA receptors in hippocampal and spinal neurons is not mediated by reducing zinc inhibition. J Neurosci 19:RC37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, MacDonald JF, Wemmie JA, Price MP, Welsh MJ, Simon RP (2004). Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell 118:687–698. [DOI] [PubMed] [Google Scholar]
- Yermolaieva O, Leonard AS, Schnizler MK, Abboud FM, Welsh MJ (2004). Extracellular acidosis increases neuronal cell calcium by activating acid-sensing ion channel 1a. Proc Natl Acad Sci USA 101:6752–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangerle L, Cuenod M, Winterhalter KH, Do KQ (1992). Screening of thiol compounds: depolarization-induced release of glutathione and cysteine from rat brain slices. J Neurochem 59:181–189. [DOI] [PubMed] [Google Scholar]
- Zheng F, Gingrich MB, Traynelis SF, Conn PJ (1998). Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nat Neurosci 1:185–191. [DOI] [PubMed] [Google Scholar]