

Abstract
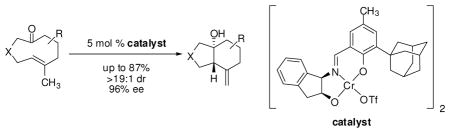
Highly enantio- and diastereoselective transannular ketone-ene reactions are catalyzed by a new chromium(III) triflate tridentate Schiff-base complex. Electronically unactivated keto-olefins undergo hetero-ene reactions at ambient temperature to afford enantioenriched bicyclic alcohols, common structural motifs in natural products. The kinetic resolution of a configurationally stable planar-chiral cyclodecenone is also described.
Transannular chemical reactions are noteworthy for generating structurally and stereochemically rich products from relatively simple precursors in a single transformation.1 Recently, we and others identified the first applications of chiral catalysts to promote transannular transformations, thereby achieving absolute stereocontrol in Diels–Alder, aldol, and Claisen-rearrangement reactions.2,3,4 Motivated by the power of ene-type reactions in organic synthesis, we became interested in extending the asymmetric catalytic transannular reaction concept to this important class of C–C bond-forming reactions (Figure 1).5,6
Figure 1.
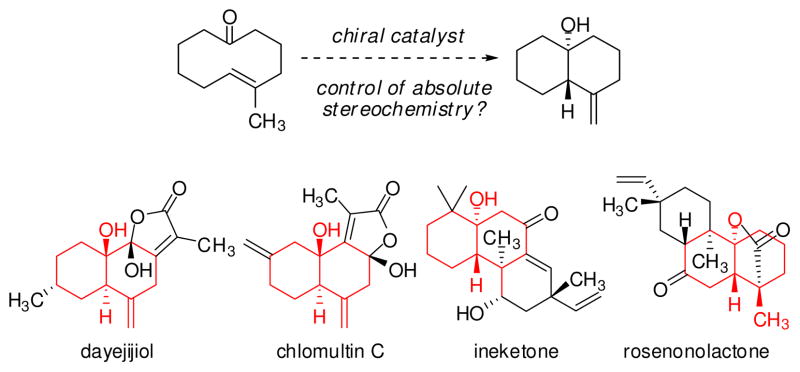
Proposed enantioselective catalytic transannular ketone-ene reaction and selected examples of natural products featuring trans-decalinol frameworks
Ketones are generally very poor reacting partners in Lewis acid-catalyzed processes, including hetero-ene reactions, and to date enantioselective catalytic ketone-ene reactions have only been achieved with highly electrophilic ketones bearing strongly electron-withdrawing substituents that often allow for two-point binding to the catalyst.7,8 Given that the transannular disposition of reacting partners can confer significant entropic advantage and corresponding reactivity enhancements, we considered the possibility of effecting enantioselective transannular ene reactions of electronically unactivated ketones. We were particularly interested in studying the intramolecular ketone-ene reaction of (E)-cyclodecenones, as the resulting products contain a decalinol framework that is prevalent in terpene natural products (Figure 1).9 This effort would necessarily take into consideration the known temperature-dependent planar chirality of medium-ring cyclic (E)-olefin substrates.10 In that context, Barriault has studied diastereoselective ketone-ene reactions of (E)-cyclodecenones and has shown that those cyclic structures were configurationally flexible under the elevated temperatures of the thermal reaction (140–220 °C).11 For efficient, enantioselective transannular ene reactions to be possible, the reaction must necessarily occur under conditions where interconversion of the enantiomeric conformers of the substrate takes place. Herein, we report highly diastereo- and enantioselective transannular ketone-ene reactions catalyzed by a new chromium(III) tridentate Schiff-base complex.
5-methyl-(E)-cyclodecenone 1a was chosen as a model substrate and was readily synthesized from cyclohexene oxide in four steps (Scheme 1).12 A copper-catalyzed epoxide-opening with isopropenyl magnesium bromide and a subsequent Swern oxidation afforded unconjugated enone 2. Vinylmagnesium bromide addition provided the desired trans-substituted cyclohexanol 3 in 20:1 dr and 55% isolated yield over the three steps. Exposure of the divinyl alcohol to potassium hydride and 18-crown-6 promoted an anionic oxy-Cope rearrangement and yielded cyclodecenone 1a as the exclusive olefin isomer.13
Scheme 1.

Synthesis of Cyclodecenone 1a
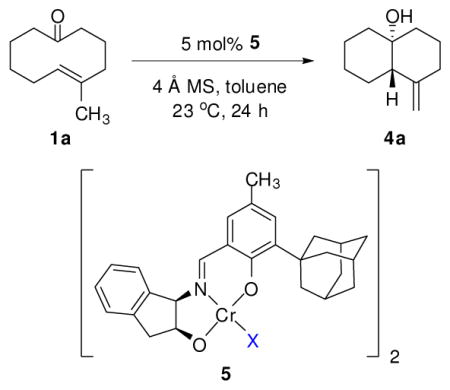
Chiral chromium(III) tridentate Schiff-base complexes,14 which have been shown to activate aldehydes and quinones through single-point binding, were uniquely effective in catalyzing the ketone-ene reaction of 1a (Table 1).15 For example, in the presence of dimeric chromium chloride complex 5a,16 trans-decalinol 5a was obtained in modest enantioenrichment, excellent diastereoselectivity, and as a single olefin regioisomer (entry 2). Pronounced effects of the catalyst counterion on the reaction outcome, were observed, with reactivity increasing steadily with decreasing coordinating ability of the counterion. Catalysts 5e and 5f, bearing PF6− and SbF6− counterions, respectively, promoted complete conversion within 24 h, albeit with diminished enantioselectivities (entries 7 and 8).17,18 Complexes bearing sulfonate counterions were somewhat less reactive, but induced significantly improved enantioselectivities (entries 3 and 4), with triflate complex 5c identified as the optimal catalyst. The high substrate conversion along with high product enantioenrichment confirmed that substrate 1a is configurationally dynamic under the reaction conditions.19 The use of activated desiccant was found to be essential for catalytic activity, as the corresponding reaction conducted in the absence of activaed 4 Å molecular sieves afforded significantly lower conversion (entry 5).20
Table 1.
Catalyst Evaluation for the Enantioselective Transannular Ketone-Ene Reaction of Cyclodecenone 1a.a
| ||||
|---|---|---|---|---|
| entry | catalyst | conv (%)b | drc | 4a ee (%)d |
| 1 | none | <1 | – | – |
| 2 | 5a (X = Cl) | 50 | >19:1 | 60 |
| 3 | ent-5b (X = OTs) | 67 | >19:1 | −84 |
| 4 | 5c (X = OTf) | 76 | >19:1 | 93 |
| 5e | 5c (X = OTf) | 6 | >19:1 | n.d. |
| 6 | 5d (X = NTf2) | 98 | >19:1 | 46 |
| 7 | 5e (X = PF6) | 100 | >19:1 | 50 |
| 8 | 5f (X = SbF6) | 100 | >19:1 | 56 |
Reactions were performed on a 0.2 mmol scale with 5 mol % catalyst 5 (10 mol % based on Cr) and in the presence of powdered 4 Å molecular sieves at 23 °C in anhydrous toluene ([1a] = 4 M).
Determined by GC analysis of the crude reaction mixtures using dodecane as an internal standard.
Determined by 1H NMR analysis of the crude reaction mixtures.
Determined by GC analysis using commercial chiral columns.
Reaction performed in the absence of powdered 4 Å molecular sieves. n.d = not determined.
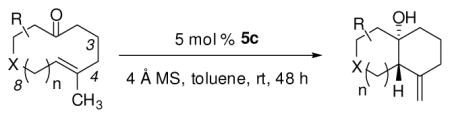
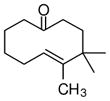
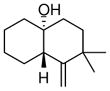
The substrate scope of the enantioselective transannular ketone-ene reaction with catalyst 5c was evaluated (Table 2).21 Full conversion of cyclodecenone 1a was achieved by extending the reaction time to 48 h, and product 4a was obtained in 81% yield and 93% ee. While gem-dimethyl-substituted trans-decalinols 4b and 4c, were accessed in high yield and enantioselectivity (entries 2 and 3), the closely analogous product 4d was obtained in low yield and as a racemate (entry 4). Analysis of the chair-chair conformations of these substrates provides a plausible exaplanation (Figure 2).22 Only cyclodecenone 1d possesses a syn-pentane relationship between its methyl substituents, and the pseudo-axial methyl substituent at C3 is also likely to interfere with complexation of the Lewis acidic catalyst
Table 2.
Substrate Scope of the Enantioselective Transannular Ketone-Ene Reactiona
| |||||
|---|---|---|---|---|---|
| entry | substrate | productb | yield (%)c | drd | ee(%)e |
| 1 |
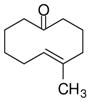 1a |
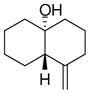 4a |
81 | >19:1 | 93 |
| 2 |
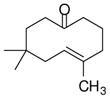 1b |
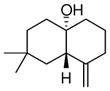 4b |
96 | >19:1 | 94 |
| 3 |
 1c |
 4c |
84 | >19:1 | 94 |
| 4 |
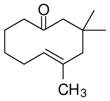 1d |
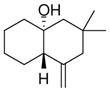 4d |
32f | >19:1 | 0 |
| 5 |
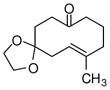 1e |
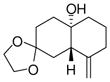 4e |
87 | >19:1 | 96 |
| 6 |
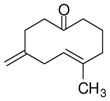 1f |
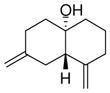 4f |
62 | >19:1 | 94 |
| 7g |
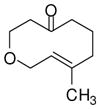 6 |
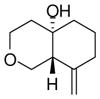 7 |
13 | >19:1 | 49 |
| 8g |
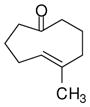 8 |
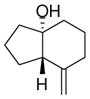 9 |
18 | >19:1 | 68 |
Reactions were performed on a 0.2 mmol scale with 5 mol % catalyst 5c (10 mol % based on Cr) and in the presence of powdered 4 Å molecular sieves at 23 °C in anhydrous toluene ([substrate] = 4 M). Unless otherwise noted, reactions showed complete conversion after 48 h and the bicyclic alcohol product was obtained as a single regioisomer.
The absolute configuration of 4a was determined by X-ray crystallographic analysis of the corresponding p-bromobenzoate. The stereochemistry of all other products is assigned by analogy.
Isolated yield of the ketone-ene products following purification by flash chromatography
Determined by 1H NMR analysis of the crude reaction mixtures.
Determined by GC analysis using commercial chiral columns.
Combined yield of 4d and an inseparable regioisomeric product. This reaction went to 70% conversion after 48 h.
Reaction time was 24 h.
Figure 2.
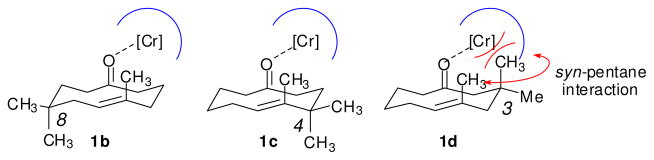
Conformational analysis to account for the low reactivity observed for cyclodecenone 1d relative to 1b and 1c.
With these substitution effects in mind, we probed more highly functionalized substrates (entries 5 and 6). The acid-sensitive acetal 1e and unconjugated diene 1f both proved to be effective substrates, affording the corresponding ene products in high enantioselectivities and good yields. Additionally, ether 6 and cyclononenone 8 underwent enantioselective ketone-ene reactions to afford the corresponding bicyclic alcohol products, although in diminished yields and enantioselectivities.
Tetrasubstituted alkene 10 proved much less reactive than trisubstituted olefins 1a–f under the catalytic conditions, undergoing only 19% conversion after 24 h at 50°C. The transannular ketone-ene reaction afforded trans-decalinol 11, bearing a quaternary stereocenter, in 12% yield and 73% ee (Scheme 2). Cyclodecenone (+)-10 was recovered in 69% yield and in 10% ee, confirming that this substrate undergoes racemization slowly under the catalytic conditions, and that complex 5c had induced a measurable kinetic resolution.23
Scheme 2.
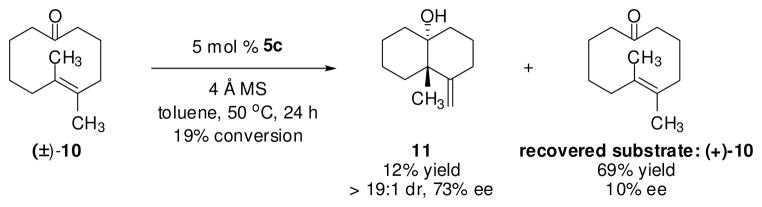
Kinetic Resolution of Planar Chiral Cyclodecenone (±)-10 by a Cr(III)-Catalyzed Transannular Ketone-Ene Reaction
In conclusion, we have demonstrated that chiral chromium(III) tridentate Schiff-base complex 5c catalyzes transannular ketone-ene reactions of medium-sized cyclic keto-(E)-olefins in high diastereo- and enantioselectivity to access fused bicyclic alcohols. Significantly, the transannular strategy allows electronically unactivated ketones to be engaged as substrates in a chiral Lewis acid-catalyzed process. This approach is most effective in the case of (E)-cyclodecenones that lack β-substitution, as the reactive components are held in close proximity and in proper alignment for the productive ketone-ene reaction.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-43214), and by predoctoral fellowship support to N.S.R from Boehringer Ingelheim. We thank Dr. Shao-Liang Zheng at the Center for Crystallographic Studies at Harvard University for his help with X-ray data collection and structure determination.
Footnotes
Supporting Information Available Complete experimental procedures, characterization data, 1H and 13C NMR spectra of all ketone-ene substrates and products, and crystallographic data for the p-bromo-benzoate of 4a. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews on the application of transannular reactions to the synthesis of natural products, see: Marsault E, Toró A, Nowak P, Deslongchamps P. Tetrahedron. 2001;57:4243.Clarke PA, Reeder AT, Winn J. Synthesis. 2009;5:691.
- 2.a) Balskus EP, Jacobsen EN. Science. 2007;317:1736. doi: 10.1126/science.1146939. [DOI] [PubMed] [Google Scholar]; b) Chandler CL, List B. J Am Chem Soc. 2008;130:6737. doi: 10.1021/ja8024164. [DOI] [PubMed] [Google Scholar]; c) Jaschinski T, Hiersemann M. Org Lett. 2012;14:4114. doi: 10.1021/ol3017676. [DOI] [PubMed] [Google Scholar]
- 3.a) Hodgson DM, Lee GP, Marriott RE, Thompson AJ, Wisedale R, Witheringon J Chem Soc Perkin Trans 1. 1998:2151. [Google Scholar]; b) Hodgson DM, Robinson LA. Chem Commun. 1999:309. [Google Scholar]
- 4.For examples of enantioselective transannular transformations that utilize stoichiometric or super-stoichiometric chiral reagents, see: Inoue M, Sato T, Hirama M. Angew Chem, Int Ed. 2006;45:4848. doi: 10.1002/anie.200601358.Inoue M, Lee N, Kasuya S, Sato T, Hirama M. J Org Chem. 2007;72:3065. doi: 10.1021/jo0700474.Knopff O, Kuhne J, Fehr C. Angew Chem, Int Ed. 2007;46:1307. doi: 10.1002/anie.200604518.
- 5.For a general review of asymmetic ene reactions, see: Mikami K, Shimizu M. Chem Rev. 1992;92:1021.
- 6.For reviews on enantioselective, catalytic carbonyl-ene reactions, see: Mikami K, Terada M. In: Comprehensive Asymmetric Catalysis. 3. Jacobsen EN, Pfaltz A, Yamamoto H, editors. Springer; Berlin: 1999. p. 1143.Clarke ML, France MB. Tetrahedron. 2008;64:9003.
- 7.Examples of Brønsted acid catalyzed enantioselective ketone-ene reaction of α,α,α-trifluoropyruvates: Clarke ML, Jones CES, France MB. Beilstein J Org Chem. 2007;3:24. doi: 10.1186/1860-5397-3-24.Rueping M, Thiessmann T, Kuenkel A, Koenigs RM. Angew Chem, Int Ed. 2008;47:6798. doi: 10.1002/anie.200802139.
- 8.For selected examples of metal-catalyzed enantioselective carbonyl-ene reactions of α-ketoester and diketones, see: Evans DA, Tregay SW, Burgey CS, Paras NA, Vojkovsky T. J Am Chem Soc. 2000;122:7936.Yang D, Yang M, Zhu N-Y. Org Lett. 2003;5:3749. doi: 10.1021/ol035486d.Mikami K, Aikawa K, Kainuma S, Kawakami Y, Saito T, Sayo N, Kumobayashi H. Tetrahedron: Asymmetr. 2004;15:3885.Doherty S, Knight JG, Smyth CH, Harrington RW, Clegg W. J Org Chem. 2006;71:9751. doi: 10.1021/jo062023n.Mikami K, Kawakami Y, Akiyama K, Aikawa K. J Am Chem Soc. 2007;129:12950. doi: 10.1021/ja076539f.Zhao J-F, Tsui H-Y, Wu P-J, Lu J, Loh T-P. J Am Chem Soc. 2008;130:16492. doi: 10.1021/ja807501a.Luo H-K, Woo Y-L, Schumann H, Jacob C, van Meurs M, Yang H-Y, Tan Y-T. Adv Synth Catal. 2010;352:1356.Zheng K, Yang Y, Zhao J, Yin C, Lin L, Liu X, Feng X. Chem Eur J. 2010;16:9969. doi: 10.1002/chem.201001412.Zhao Y-J, Li B, Tan L-JS, Shen Z-L, Loh T-P. J Am Chem Soc. 2010;132:10242. doi: 10.1021/ja104119j.
- 9.For a recent review of natural sesquiterpenoids, see: Fraga BM. Nat Prod Rep. 2011;28:1580. doi: 10.1039/c1np00046b.
- 10.Nakazaki M, Yamamoto K, Naemura K. In: Stereochemistry, Topics in Current Chemistry. Vogtle F, Weber E, editors. Vol. 125. Springer; Berlin: 1984. p. 1. [Google Scholar]
- 11.a) Sauer ELO, Hooper J, Woo T, Barriault L. J Am Chem Soc. 2007;129:2112. doi: 10.1021/ja066830f. [DOI] [PubMed] [Google Scholar]; b) Sauer ELO, Barriault L. J Am Chem Soc. 2004;126:8670. doi: 10.1021/ja048301m. [DOI] [PubMed] [Google Scholar]
- 12.Divinyl alcohol3has been prepared by Barriault and coworkers. The route shown in Scheme 1 is modified version of the one reported: Warrington JM, Yap GPA, Barriault L. Org Lett. 2000;2:663. doi: 10.1021/ol005502w.
- 13.Evans DA, Golob AM. J Am Chem Soc. 1975;97:4765. [Google Scholar]
- 14.a) Dossetter AG, Jamison TF, Jacobsen EN. Angew Chem, Int Ed. 1999;38:2398. doi: 10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; b) Gademann K, Chavez DE, Jacobsen EN. Angew Chem, Int Ed. 2002;41:3059. doi: 10.1002/1521-3773(20020816)41:16<3059::AID-ANIE3059>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]; c) Ruck RT, Jacobsen EN. J Am Chem Soc. 2002;124:2882. doi: 10.1021/ja025588j. [DOI] [PubMed] [Google Scholar]; d) Joly GD, Jacobsen EN. Org Lett. 2002;4:1795. doi: 10.1021/ol0258785. [DOI] [PubMed] [Google Scholar]; e) Ruck RT, Jacobsen EN. Angew Chem, Int Ed. 2003;42:4771. doi: 10.1002/anie.200351591. [DOI] [PubMed] [Google Scholar]; f) Chavez DE, Jacobsen EN. Org Lett. 2003;5:2563. doi: 10.1021/ol034883l. [DOI] [PubMed] [Google Scholar]; g) Jarvo ER, Lawrence BM, Jacobsen EN. Angew Chem, Int Ed. 2005;44:6043. doi: 10.1002/anie.200502176. [DOI] [PubMed] [Google Scholar]; h) Grachan ML, Tudge MT, Jacobsen EN. Angew Chem, Int Ed. 2008;47:1469. doi: 10.1002/anie.200704439. [DOI] [PubMed] [Google Scholar]
- 15.See Supporting Information for the results of a screen of chiral Lewis acids.
- 16.Chavez DE, Jacobsen EN. Org Synth. 2005;82:34. See also refs. 14a,b,f. [Google Scholar]
- 17.Catalyst 5f has been shown to induce superior reactivity and enantioselectivity relative to catalyst 5a in hetero-Diels–Alder reactions. See refs. 14a and 16.
- 18.The transannular ketone-ene reaction conducted with complex 5f afforded minor olefin byproducts (<10%). This outcome is an indication of diminished regioselectivity with this more Lewis acidic catalyst and suggests that this catalyst may promote a stepwise ketone-ene reaction.
- 19.Studies on related substrates suggest that 1a does not exhibit planar chirality at 23 °C: Cope AC, Banholzer K, Keller H, Pawson BA, Whang JJ, Winkler HJS. J Am Chem Soc. 1965;87:3644.Westen HH. Helv, Chim Acta. 1964;47:575.Binsch G, Roberts JD. J Am Chem Soc. 1965;87:5158.Tomooka K, Ezawa T, Inoue H, Uehara K, Igawa K. J Am Chem Soc. 2011;133:1754. doi: 10.1021/ja1092375.
- 20.The requirement for desiccant is a common feature of all reactions catalyzed by chromium(III) tridentate Schiff-base complexes (see ref. 14). The crystal structures obtained of this class of catalysts indicate that metal centers have octahedral geometry and contain water molecules to complete the coordination sphere. It has been proposed that the desiccant removes a water molecule from the chromium center allowing for coordination of the substrate carbonyl group.
- 21.Substrates were synthesized via anionic or palladium-catalyzed oxy-Cope rearrangements of divinyl alcohols: a) Ref. 13 Bluthe N, Malacria M, Gore J. Tetrahedron Lett. 1983;24:1157.
- 22.Chair-like transition structures that are consistent with reaction outcomes have been determined computationally for diastereoselective thermal ketone-ene reactions of (E)-cyclodecenones: Terada Y, Yamamura S. Tetrahedron Lett. 1979;20:1623.
- 23.A kinetic resolution of planar chiral cyclic ethers has been achieved through an enantioselective transannular [2,3]-Wittig rearrangement: Tomooka K, Komine N, Fujiki D, Nakai T, Yanagitsuru S. J Am Chem Soc. 2005;127:12182. doi: 10.1021/ja053347g.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.