

Abstract
Gli-similar (Glis) 1–3 proteins constitute a sub-family of Krüppel-like zinc finger proteins that are closely related to members of the Gli family. Glis proteins have been implicated in several pathologies, including cystic kidney disease, diabetes, hypothyroidism, fibrosis, osteoporosis, psoriasis, and cancer. In humans, a mutation in the Glis2 gene has been linked to the development of nephronophthisis (NPHP), a recessive cystic kidney disease, while mutations in Glis3 lead to an extended multi-system phenotype that includes the development of neonatal diabetes, polycystic kidneys, congenital hypothyroidism, and facial dysmorphism. Glis3 has also been identified as a risk locus for type-1 and type-2 diabetes and additional studies have revealed a role for Glis3 in pancreatic endocrine development, β-cell maintenance, and insulin regulation. Similar to Gli1-3, Glis2 and 3 have been reported to localize to the primary cilium. These studies appear to suggest that Glis proteins are part of a primary cilium-associated signaling pathway(s). It has been hypothesized that Glis proteins are activated through post-translational modifications and subsequently translocate to the nucleus where they regulate transcription by interacting with Glis binding sites in the promoter regions of target genes. This chapter will summarize the current state of knowledge regarding mechanisms of action of the Glis family of proteins, their physiological functions, as well as their roles in disease.
Keywords: Gli-similar proteins, diabetes, cystic kidney disease, primary cilium, pancreas, insulin, β cells, epithelial-mesenchymal transition, iPS cells
Introduction
Krüppel-like zinc finger proteins, so named for their similarity to the Drosophila segmentation gene product Krüppel (Preiss et al., 1985), belong to a large and evolutionarily conserved family of transcriptional regulators implicated in a broad range of cellular processes including proliferation, apoptosis, differentiation, and development (Pearson et al., 2008). The characteristic feature of the Krüppel-like family is the presence of two or more classical Cys2/His2 zinc fingers that are separated by the conserved consensus sequence TGEKP(Y/F)X (Dang et al., 2000). The zinc fingers constitute a DNA binding domain that recognizes specific DNA elements in the promoter/regulatory regions of target genes (Kaczynski et al., 2003). Outside of the zinc finger region there is relatively little homology amongst family members. Krüppel-like zinc finger proteins exhibit diverse roles during embryonic development and in the adult and are implicated in a variety of pathologies.
Gli and Zic constitute two closely-related subfamilies of Krüppel-like zinc finger protein that contain five Cys2/His2 zinc finger motifs and function as activators and/or repressors of gene transcription. Gli and Zic proteins are essential for normal embryonic development and have been implicated in a number of human diseases and cancers (Kasper et al., 2006; Merzdorf, 2007; Mo et al., 1997; Ruiz i Altaba, 1999; Sasaki et al., 1999). Gli-similar (Glis) proteins, the focus of this review, are closely related to the Gli and Zic sub-families (Kang et al., 2010).
Glis1 was independently identified by two laboratories employing a yeast two-hybrid screen against a mouse kidney cDNA library using the ligand-binding domain of the retinoid-related orphan receptor, RORγ, as bait (Kim et al., 2002) or by sequence homology to the Gli zinc finger domain (Nakashima et al., 2002). Subsequently, two additional family members exhibiting a high degree of homology to the Glis1 zinc finger domain were identified and referred to as Glis2 and Glis3 (Kim et al., 2003; Lamar et al., 2001; Zhang and Jetten, 2001; Zhang et al., 2002). Like the members of the Gli and Zic sub-families, Glis1–3 share a highly homologous zinc-finger domain consisting of five Cys2/His2 motifs. The zinc finger domain of Glis1 is 58% and 93% identical to that of Glis2 and Glis3 respectively (Kim et al., 2002; Kim et al., 2003); however, Glis family members exhibit little sequence homology outside of their zinc finger domains. The Glis proteins are conserved across species with homologues for Glis3 identified in Drosophila and Oryzias latipes and a high degree of homology existing between the human and mouse proteins (Furlong et al., 2001; Hashimoto et al., 2009).
The human GLIS1-3 genes are located on chromosomes 1p32.3, 16p13.3, and 9p24.2, and encode for proteins that are approximately 65.9, 55.7, and 90 kD in size, respectively. Glis1-3 are expressed in a temporal and spatial manner during embryonic development and in the adult are expressed in a tissue- and cell type-specific manner. Under normal circumstances, all three Glis family members are most abundantly expressed in the kidney. Glis1 is further expressed in the placenta, brown adipose tissue, brain, thymus, colon, and testis (Kim et al., 2002), while Glis2 expression was also detected in the lung, prostate, colon, brain, liver, heart and intestine (Zhang and Jetten, 2001; Zhang et al., 2002). In addition to the kidney, Glis3 expression was detected in the pancreas, thyroid, thymus, uterus, ovary, brain and lung (Kim et al., 2003; Senee et al., 2006).
Characterization of Glis-null mouse models has identified several biological functions associated with the Glis proteins. For example, mice deficient in Glis2 develop nephronophthisis (NPHP), characterized by renal atrophy and fibrosis involving defects in epithelial mesenchymal transition (EMT) within renal tubules (Attanasio et al., 2007; Kim et al., 2008; Kang et al., 2010). Glis3-null mice develop neonatal diabetes, hypothyroidism, and polycystic kidney disease (Kang et al., 2009b; Watanabe et al., 2009). This chapter will review what is currently known about the mechanisms of action of Glis proteins, their physiological function, and their roles in disease.
Mechanism of Action of Glis Proteins
Sub-cellular localization
Examination of fluorescent-tagged Glis proteins by confocal microscopy revealed that these proteins were predominantly localized to the nuclear compartment (Kim et al., 2002; Kim et al., 2003; Zhang et al., 2002). However, the mechanisms by which Glis proteins are directed to the nucleus are not yet fully understood. Analyses of several putative bipartite nuclear localization sequences within the Glis proteins, including a motif within Glis3 overlapping ZF5 that shares homology with the bona fide nuclear localization signal (NLS) of the Drosophila homologue of Gli, Cubitus interruptus (Ci) (Wang and Holmgren, 1999), demonstrated that these sequences are not required for nuclear localization (Beak et al., 2008; Kim et al., 2002; Zhang et al., 2002). Deletion analyses suggested that the region containing ZF4 is essential for the nuclear localization of Glis3, while ZF3 is essential for Glis2 nuclear localization (Beak et al., 2008; Vasanth et al., 2011). Furthermore, while disruption of the tetrahedral configuration of ZF4 abated Glis3 nuclear localization, Glis2 localization was unaffected by corresponding mutations. These observations indicated that the nuclear localization of Glis2 and Glis3 seems to be dependent upon their zinc finger domains. Future studies are required to understand the mechanisms by which these regions mediate nuclear localization.
DNA binding
In addition to their role in nuclear localization, the zinc finger domains of the Glis proteins are essential for the interaction of Glis proteins with DNA. The Glis zinc finger domains are comprised of five tandem Cys2His2 zinc fingers. Cys2His2 zinc fingers, the most common DNA binding domain found in eukaryotes, each form a ββα structure and maintain a tetrahedral configuration stabilized by a zinc ion that interacts with the four cysteine and histidine residues (Brayer and Segal, 2008; Frankel et al., 1987; Lee et al., 1989). The specificity of the zinc fingers for defined DNA sequences is mediated by the alpha helices of each finger, which fits within the major groove of the DNA and recognizes overlapping short stretches of nucleotides (Elrod-Erickson et al., 1996). Thus, modularly composed zinc fingers can be capable of recognizing a relatively diverse array of DNA elements. The sequence 5′-(G/C)TGGGGGG(A/C) was identified as the optimal Glis binding site (GlisBS) for Glis3 in vitro (Beak et al., 2008). The tetrahedral configuration of each ZF was required for DNA binding. Given the great degree of homology between the zinc finger domains of Gli and Glis proteins, it came as little surprise that the Glis proteins were also capable of interacting with the consensus Gli binding site (GBS), 5′-GACCACCCA in vitro (Kim et al., 2002; Kim et al., 2003; Lamar et al., 2001; Nakashima et al., 2002; Ruppert et al., 1988; Vasanth et al., 2011). However, both Glis3 and GLI1 bound the GlisBS with a higher affinity than the GBS (Beak et al., 2008). The fact that Gli and Glis proteins can both bind the same elements allows for the possibility of cross-talk between the Gli and Glis signaling pathways.
Binding of Glis proteins to specific DNA elements is likely not only determined by the specificity of individual residues within the alpha-helices of the zinc fingers, but also affected by the co-factors that are part of the Glis transcription regulatory complex and the promoter context of the GlisBS. In addition, posttranslational modifications of Glis proteins might influence their binding specificity for GlisBS. The latter is supported by a recent study showing that the binding of Glis2 to GlisBS was abrogated by a phosphomimetic mutation of Ser245 within the loop of ZF3 both in vitro and in cultured cells (Vasanth et al., 2011). The specific kinase(s) involved in the potential phosphorylation of Glis2 and its relevance to the physiological function of Glis2 in vivo have yet to be determined.
Transcriptional regulation by Glis proteins
The ability of Glis proteins to function as transcription factors requires not only the DNA-binding domain, but also a transcriptional activation or repressor domain (Kang et al., 2010). In that regard, several distinct regions outside the zinc finger domain that mediate transcriptional regulation by Glis1–3 have been characterized and are depicted in figure 1 (Beak et al., 2007; Beak et al., 2008; Kang et al., 2009a; Kang et al., 2010; Kim et al., 2002; Kim et al., 2003; Nakashima et al., 2002). Glis3 was able to induce transactivation of a reporter gene under the control of the consensus GlisBS or GBS, whereas Glis1 and Glis2 were unable to activate the reporter gene very effectively. A potent transactivation domain (TAD) was identified within the C-terminus of Glis1 (Kim et al., 2002). However, the transactivation by Glis1 required the removal of the N-terminus, suggesting the presumed presence of an N-terminal repressor domain. In contrast, full-length Glis1 was found to potently activate a reporter under the control of the mouse insulin 2 (mIns2) promoter in both HEK293 cells and INS1 (832/13) cells (Zeruth et al., 2011) suggesting that Glis1 activity is dependent on the cell type and the promoter context. Furthermore, Ca2+-dependent calmodulin kinase IV (CaMKIV), which has been reported to phosphorylate transcription factors such as Serum Response Factor (SRF) and CAATT enhancer binding protein (CEBPα) enhancing their transcriptional activity (Marshall et al., 2003; Miranti et al., 1995), increased the activation capabilities of Glis1 approximately 4-fold (Kim et al., 2002). The significance of CaMKIV on the transcriptional activity of Glis1 in vivo is unknown and elicits further study; however, given the noted ability of CaMKIV to phosphorylate and activate the ubiquitous co-activator, CREB binding protein (CBP), it is possible that the enhancement of Glis1 transactivation function by CaMKIV involves the activation of CBP or another co-factor (Soderling, 1999).
Figure 1. Schematic representation of Glis1-3 protein structure.

The primary structure of Glis proteins and their identified domains are shown (ZFD = zinc finger domain; TAD = transactivation domain; NCR = N-terminal conserved region that is shared with GLI proteins). Bold lines represent regions within Glis proteins required for their interaction with C-terminal binding protein 1 (CtBP1); p120 catenin (p120ctn), Suppressor of Fused (SuFu), Cullin 3 (Cul3), and transcriptional coactivator with PDZ-binding motif (Taz) as indicated. The potential phosphorylation site, Serine245, is specified in Glis2. Numbers indicate the amino acid position.
A putative TAD and a potential repressor domain were also identified by monohybrid analyses within the N-terminus of Glis2 between amino acids 71 and 137 and extending into ZF1, respectively (Zhang et al., 2002). While Glis2 was incapable of activating a (GlisBS)6 controlled reporter, exogenous full-length Glis2 was capable of activating a reporter controlled by the mIns2 promoter in HEK293 cells (Vasanth et al., 2011; Zeruth et al., 2011). Maximal induction of the mIns2 promoter required the N- and C-terminus of Glis2 suggesting that both regions may be involved in the binding and recruitment of transcriptional mediators. In contrast, Glis2 was able to repress GLI1-mediated activation of a (GliBS)6 driven reporter. This was likely due to competition between Glis2 and GLI1 for GliBS binding rather than Glis2 repressor activity given that the DNA binding domain of Glis2 was sufficient to repress GLI1-mediated activation, while a DNA binding mutant of Glis2 had no effect.
Glis3 is able to induce transactivation of a (GlisBS)6-driven reporter as well as reporters under control of the mIns2 and FGF18 promoters in various cellular contexts (Beak et al., 2007; Kang et al., 2009b; Yang et al., 2009; Zeruth et al., 2011). A potent TAD was identified within the C-terminus of Glis3, which is indispensable for Glis3-mediated transactivation (Kang et al., 2010; Kim et al., 2003). N-terminal deletions of Glis3 increase its activation of the mIns2 promoter that attained a maximum with the ΔN302 mutant and decreased with subsequent deletions suggesting the presence of a regulatory motif(s) within the N-terminus (Kang et al., 2009b; Kim et al., 2003; Zeruth et al., 2011). The molecular basis by which the N-terminus of Glis3 regulates Glis3 transactivation function is currently unknown. It is possible that deletions within the N-terminus eliminate a repressor or protein interaction domain. Alternatively, truncation of the N-terminus may lead to a conformational change that enhances interactions with cofactors and/or increases DNA binding affinity.
Interacting partners
Transcriptional regulation by transcription factors is mediated through their interaction with co-activator and co-repressor complexes. A number of proteins that interact with Glis proteins and mediate or modulate their transcriptional activity have been identified (figure 1). Amongst these, C-terminal binding protein 1 (CtBP1) has been reported to interact with both the N-and C-terminus of Glis2 (Kim et al., 2005). CtBP1 interacts with and functions as a co-repressor for a number of transcription factors. It mediates transcriptional repression by recruiting proteins with various histone modifying enzymatic activities, including histone deacetylases (HDACs) and histone lysine methyl transferases (Chinnadurai, 2007). In fact, HDAC3 has been identified as part of a Glis2-CtBP1 complex and may contribute to the ability of Glis2 to act as a transcriptional repressor (Kim et al., 2005).
Glis2 has also been reported to interact with p120 catenin (p120ctn) and to promote its nuclear localization (Hosking et al., 2007). Interaction with p120ctn was found to induce a Src kinase-dependent cleavage of Glis2 between zinc fingers 4 and 5. P120ctn has been reported to be bound to E-cadherin at cell-cell contacts and can also be associated with microtubules. Over-expression of E-cadherin resulted in a reduction of Glis2 cleavage, while the induction of microtubule depolymerization enhanced Glis3 cleavage. Taken together, this suggests that p120ctn must be free in the cytosol to interact with Glis2 (Hosking et al., 2007). While the physiological function of its interaction with Glis2 is currently unknown, p120ctn, a member of the Armadillo family of proteins, has emerged as a regulator of RhoGTPases and E-cadherin stability as well as a regulator of the transcriptional activity of the transcription factor Kaiso (Daniel, 2007; Reynolds, 2007; Xiao et al., 2007). Because of its association with Kaiso, p120ctn has also been implicated as a modulator of the Wnt signaling pathway (Kim et al., 2004; Na et al., 2007; Park et al., 2005) and therefore raises the possibility that Glis2 may be linked to Wnt signaling. The connection between Glis2 and p120ctn is interesting in the light that both proteins have been implicated in the control of epithelial-mesenchymal transition (EMT).
Further reinforcing the possibility of Glis2 cross-talk with the Wnt pathway, Glis2 interacts with the armadillo repeats of β-catenin via its first zinc finger (Kim et al., 2007). β-catenin is an integral component of the canonical Wnt signaling pathway and in combination with T-cell factor/Lymphoid enhancer factor (TCF/LEF), positively regulates Wnt target genes (Akiyama, 2000). β-catenin also regulates cell adhesion and migration through interactions with the cytoplasmic domains of cadherins and is therefore implicated in a number of cancers (Gavert and Ben-Ze’ev, 2007). Glis2 acts as a negative regulator of β-catenin and subsequently inhibits TCF/LEF signaling and the β-catenin-TCF/LEF mediated activation of cyclin D1. Because of the essential role β-catenin plays in Wnt signaling, its interaction with Glis2 suggests cross-talk between the Wnt and Glis signaling pathways.
Recently, Glis3 was reported to interact with the tumor suppressor and negative regulator of Hedgehog (Hh) signaling, Suppressor of Fused (SUFU), via a YGH motif (Zeruth et al., 2011). This motif is part of a 58 amino acid region within the Glis3 N-terminus that exhibits high levels of homology to a corresponding region in the Ci/Gli proteins. A putative degron was identified within this conserved region of the Glis3 N-terminus upstream of the SUFU interaction motif, a loss of which resulted in a significant increase in Glis3 protein levels. Glis3 protein levels are stabilized by the proteasome inhibitor, MG132, suggesting that Glis3 is targeted for proteolytic degradation by the 26S proteasome. In fact, the E3-ubiquitin ligase scaffolding protein, Cullin 3 (Cul3) was shown to associate with the Glis3 N-terminus and its over-expression enhanced Glis3 polyubiquitination in cultured cells. SUFU, conversely, inhibited Glis3-Cul3 interaction and decreased the level of Glis3 polyubiquitination, thereby stabilizing Glis3 protein levels (Zeruth et al., 2011). Cul3 is typically associated with ubiquitin ligases by means of BTB-domain containing proteins that target specific proteins for degradation. To date, a BTB-domain containing protein that mediates interaction of Glis3 and Cul3 has not yet been identified.
In addition to stabilizing Glis3, SUFU has been shown to modulate Glis3-mediated transactivation of the Ins2 promoter. SUFU also interacts with the C-terminus of Glis2 through a disparate mechanism, although the effects of the interaction are unknown and do not appear to influence Glis2 protein stability or transactivation function. SUFU has been reported to interact with members of the GLI family and shown to regulate the stability and processing of GLI2 and GLI3 into repressor or activator forms (Barnfield et al., 2005; Cheng and Bishop, 2002; Humke et al., 2010; Svard et al., 2006; Wang et al., 2010). In the absence of signaling, SUFU restrains Gli3 in the cytoplasm, promoting its processing into a repressor, while initiation of hedgehog signaling triggers the dissociation of SUFU and promotes the translocation of the activated form into the nucleus. SUFU has also been reported to interact with β-catenin to negatively regulate TCF/LEF signaling (Meng et al., 2001). The interaction of Glis2–3 with SUFU yields the possibility of cross-talk between the Glis, hedgehog, and Wnt signaling pathways.
Glis3 has further been reported to interact with the key insulin transcriptional regulatory factors, pancreatic duodenal homeobox 1 (Pdx1), v-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA), and NeuroD1 resulting in synergistic activation of Ins2 (Yang et al., 2009). Transcriptional regulation and β-cell specific expression of the insulin 2 gene occurs via a 600 bp promoter region upstream of the transcriptional start site (Edlund et al., 1985; German et al., 1992; Hanahan, 1985; Melloul et al., 2002; Ohneda et al., 2000a; Walker et al., 1983). The most notable elements within this regulatory region are the A, E, and C-boxes, which bind Pdx1, β2/NeuroD1, and MafA, respectively (Melloul et al., 2002; Ohneda et al., 2000a). These transcription factors interact to form a stable DNA-binding complex capable of recruiting additional co-factors and transcriptional machinery (Ohneda et al., 2000a; Ohneda et al., 2000b; Peshavaria et al., 1997; Petersen et al., 1994). The precise dynamics of Glis3 interaction with insulin transcriptional regulators and the role Glis3 plays in the maintenance of adult pancreatic endocrine cells is not fully understood and requires further investigation.
Recent studies have demonstrated that Glis3 is able to interact with the transcriptional co-regulator, transcriptional coactivator with PDZ-binding motif (TAZ, also known as WWTR1) through a PPXY site within the Glis3 C-terminus (Kang et al., 2009a). TAZ interaction with Glis3 modestly enhances Glis3-mediated activation of a (GlisBS)6 controlled reporter. The interaction between Glis3 and TAZ and the observation that loss of either TAZ or Glis3 expression lead to polycystic kidney disease suggest a possible common link in the development of this phenotype (Chan et al., 2008; Hossain et al., 2007; Kang et al., 2009a; Makita et al., 2008; Senee et al., 2006). TAZ is part of the Hippo signaling pathway (Lei et al., 2008) that has been implicated in the regulation of cell proliferation, EMT, and planar cell polarity (PCP), cellular processes that have been implicated in the formation of polycystic kidneys. Thus, disruption in Glis3-TAZ interaction might result in defects in these functions and be causally involved in the development of polycystic kidneys.
Glis signaling and the primary cilium
In addition to the nucleus, both Glis2 and Glis3 have been reported to localize to the primary cilium (Attanasio et al., 2007; Hashimoto et al., 2009; Kang et al., 2009a; Kang et al., 2010). Thus far, no evidence has emerged linking Glis1 to the primary cilium. The primary cilium, a microtubule-based, hair-like projection present on nearly all mammalian cells, extends from the apical surface and functions as a sensory organelle (Goetz and Anderson, 2010). It serves as a signaling hub for an increasing number of distinct signaling pathways such as phototransduction, mechano-, osmo-, and chemosensation. Key components of the canonical hedgehog, Wnt, platelet-derived growth factor, and PCP pathways have also been shown to localize within the primary cilium, which has been demonstrated as indispensable for their proper signaling (Berbari et al., 2009). Moreover, certain G protein-coupled receptors (GPCRs), including somatostatin receptor 3 (Sstr3), melanin-concentrating hormone receptor 1 (Mchr1), and serotonin receptor 6 (Htr6), have been reported to localize to the ciliary membrane in various cell types (Berbari et al., 2008a; Iwanaga et al., 2011; Stanic et al., 2009). The trafficking of many signaling receptors to the ciliary membrane appears to be mediated by the BBsome, a large protein complex containing specific Bardet-Biedl syndrome (BBS) proteins (Berbari et al., 2008b; Domire et al., 2010; Goetz and Anderson, 2010; Nachury et al., 2010). Once inside the ciliary compartment, many essential ciliary proteins utilize a modified system of microtubule associated transport termed intraflagellar transport (IFT). Anterograde transport of proteins toward the cilium tip requires the kinesin II motor proteins, while retrograde transport back toward the cilium base requires dynein motor proteins (Blacque et al., 2008; Goetz and Anderson, 2010; Rosenbaum and Witman, 2002). Since many of the proteins necessary for cilia construction utilize IFT, disruption of IFT components inhibits ciliogenesis or results in the deformation of cilia (Pedersen and Rosenbaum, 2008). Furthermore, disruption of IFT proteins affects cilia related signaling pathways such as several aspects of sonic hedgehog (Shh)-Gli signaling, including the movement of the downstream effectors, Gli2–3, suggesting that they depend on a functional IFT (Haycraft et al., 2005; Liu et al., 2005; May et al., 2005; Qin et al., 2011). Whether or not Glis proteins associate with IFT motors or whether IFT is required for proper Glis signaling is currently unknown and requires further investigation.
It appears likely that Glis proteins are part of a primary cilium-associated signaling pathway and although the functional significance of Glis protein accumulation in the primary cilium has not yet been established, the role that the primary cilium plays in Shh/Gli signaling has been fairly well characterized (Goetz and Anderson, 2010). In the absence of Shh, its receptor Patched (Ptch1) is localized to the primary cilium and prevents ciliary localization of Smoothened (Smo). Shh binding to Ptch1 results in its exit from the ciliary compartment and relieves repression of Smo, allowing it to enter the cilium (Corbit et al., 2005; Rohatgi et al., 2007). The mutually exclusive presence of Ptch1 or Smo in the cilium regulates the processing of Gli3 into its activator or repressor forms (Haycraft et al., 2005; Huangfu et al., 2003; Liu et al., 2005). Modified Gli proteins then exit the cilium and translocate to the nucleus where they regulate the transcription of target genes. We proposed previously that the primary cilium might play a similar role in the regulation of Glis. The activity of Glis proteins might be regulated by external signals received through receptors (e.g. G protein-coupled receptors) localized to the primary ciliary membrane that control Glis activity by promoting their proteolytic cleavage or regulating other posttranslational modifications. After activation, Glis proteins might then translocate to the nucleus where they regulate the transcription of target genes and subsequently influence various physiological processes (figure 2). Defects in the structure or synthesis of the primary cilium and abnormalities in primary cilium-associated signaling pathways have been implicated in several pathologies collectively referred to as ciliopathies, including disorders with various sensory defects and cystic renal diseases (Fliegauf et al., 2007). Because of their connection with the primary cilium, it is not surprising that defective Glis signaling is associated with the development of several ciliopathies.
Figure 2. Schematic representation of Glis signaling and its role in disease.

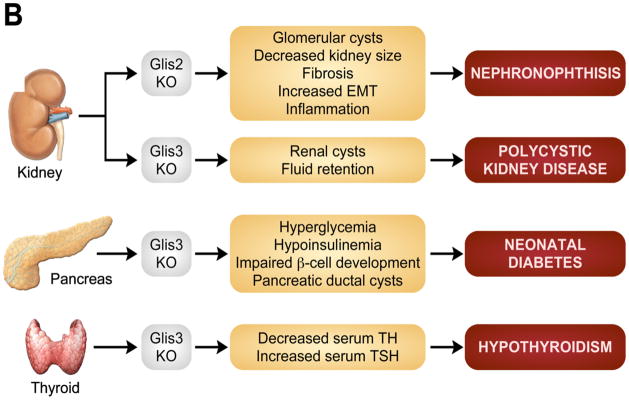
Glis2 and 3 localize to the primary cilium and defective Glis signaling is associated with the development of several ciliopathies. (A) An as of yet unidentified upstream signal may regulate the sub-cellular localization and activation of Glis proteins. During activation in the primary cilium, Glis proteins may undergo post-translational modifications or proteolytic processing and upon translocation to the nucleus, regulate the transcription of target genes through interactions with Glis binding sites (GlisBS) and transcriptional coregulators (X and Y). (B) Disruption of Glis signaling in the kidney, pancreas, and thyroid leads to the development of nephronophthisis and polycystic kidney disease, neonatal diabetes, and hypothyroidism, respectively.
Role of Glis Proteins in Renal Physiology and Pathology
Glis2, nephronophthisis, and mesenchymal-epithelial transition
All three Glis proteins are highly expressed in adult kidney and exhibit a temporal and spatial pattern of expression during embryonic kidney development. At E9.0 of murine development, the ureteric bud develops from budding of the caudal portion of the mesonephric Wolffian duct and invades the metanephric mesenchyme (Michos, 2009; Wilson, 2008). This is followed by extensive branching of the ureteric bud, giving rise to the renal collecting tubules. At E11.5, the mesenchymal cells condense and undergo mesenchymal-epithelial transition (MET) to form comma and S-shaped bodies that by E13.5 begin to develop into the glomeruli and renal tubules. Reciprocal crosstalk between the metanephric mesenchyme and the ureteric bud plays an important role in the control of MET and ureteric bud branching. During murine metanephric development, Glis2 is most highly expressed in the ureteric bud, whereas mesenchymal tissue, comma- and S-shaped bodies expressed at low levels or were devoid of Glis2 (Attanasio et al., 2007; Zhang et al., 2002). In adult kidneys, Glis2 was detected in the epithelial cells of all segments of the renal tubule and in the epithelial cells of Bowman’s capsule, but not in glomeruli, endothelial, or mesenchymal cells (Attanasio et al., 2007). Recent studies reported that loss of Glis2 function in humans and mice leads to the development of NPHP, a recessive cystic kidney disease that is the most common genetic cause of end-stage renal disease in children and young adults (Hildebrandt and Zhou, 2007). NPHP is a characterized by tubular atrophy, fibrosis and glomerular cyst formation which progressively leads to dramatic changes in renal architecture and ultimately end-stage kidney disease. Through positional cloning, mutations in nine distinct genes (NPHP1–9) have been linked to NPHP (Hildebrandt and Zhou, 2007; Simms et al., 2011). Mutations in the NPHP1 gene account for approximately 21% of all NPHP cases, whereas mutations in NPHP2–9 each account for less than 9% of cases. Mutations that abolish the splice site within exon 5 of Glis2, which is also referred to as NPHP7, were linked to NPHP in three patients from a single family (Attanasio et al., 2007). Similarly, Glis2-null mice develop renal atrophy and tubulo-interstitial fibrosis characteristic of NPHP at an early age that is accompanied by increased inflammation. The severity of the symptoms progress with age as evidenced by increased blood urea nitrogen (BUN) and creatinine levels and the development of proteinuria, which ultimately lead to renal failure and premature death (Attanasio et al., 2007; Kim et al., 2008).
The progressive tubulointerstitial fibrosis associated with NPHP7 is accompanied by increased accumulation of extra-cellular matrix (ECM) components produced by myofibroblasts. The increase in myofibroblasts during fibrosis may occur through several mechanisms, including differentiation of fibroblasts into myofibroblast, EMT, infiltration of fibrocytes, or a combination of the three processes. In EMT, epithelial cells transdifferentiate into myofibroblasts that migrate into the interstitium (Liu, 2009; Wada et al., 2007; Wynn, 2007; Zeisberg and Kalluri, 2004). Gene expression profile analysis of kidneys from Glis2-null and wild-type mice revealed increased expression of a number of genes that play a critical role in EMT, including transforming growth factor β (Tgfβ), vimentin, matrix metallopeptidase 14 (Mmp14), connective tissue growth factor (Ctgf), Snail, and Slug (Attanasio et al., 2007; Kim et al., 2008). These data suggested that the induction of renal fibrosis in Glis2-null mice is mediated through EMT in renal tubule epithelial cells. These observations further implied that Glis2 may act as a repressor of EMT and EMT-related gene expression. Additional research is required to determine the precise molecular mechanism by which Glis2 regulates the maintenance of normal renal structure and function and to determine its role in controlling EMT.
Glis3 and polycystic kidney disease
Glis3 deficiency in humans and mice is associated with an extended multi-system phenotype that includes the development of polycystic kidney disease (PKD) (Dimitri et al., 2011; Kang et al., 2009b; Senee et al., 2006; Taha et al., 2003; Watanabe et al., 2009). Furthermore, a mutation in the pc gene, an ortholog of Glis3, has been associated with the development of polycystic kidneys in medaka (Hashimoto et al., 2009; Kang et al., 2010).
PKD is a common heritable kidney disease characterized by the formation of large fluid-filled cysts in the glomerulus, renal tubules, and collecting ducts. Autosomal dominant PKD, the most common PKD in humans, results from mutations in PKD1 and PKD2, which encode polycystin-1 and -2, respectively (Gallagher et al., 2010; Harris and Torres, 2009). Autosomal recessive PKD, which is mostly associated with young children, is caused by mutations in PKHD1, which encodes polyductin/fibrocystin (Al-Bhalal and Akhtar, 2008; Bergmann et al., 2004). At E14.5 of kidney development, Glis3 mRNA is highly expressed in the branches of the ureteric bud of the metanephros (Kim et al., 2003), while in the adult mouse kidney it is expressed in the epithelia of the collecting ducts, renal tubules, and Bowman’s capsule (Hashimoto et al., 2009; Kang et al., 2009a). Histological analyses of Glis3-null mouse kidneys showed dilation of Bowman’s spaces as early as E14.5. Loss of Glis3 does not appear to affect the extent of ureteric bud branching and the renal phenotype became more pronounced at later stages of mouse development. By postnatal day 3 (PND3), major glomerular cysts were observed along with the formation of cystic renal tubules and dilation of collecting ducts (Kang et al., 2009a). These observations indicate that Glis3 plays a critical role in the maintenance of normal renal architecture and function (Kang et al., 2010).
Links between Glis signaling, the primary cilium, and renal disease
The molecular mechanisms that lead to cyst formation are still not fully understood; however, it has become evident that there is a strong causal relationship between the primary cilium and cystic renal diseases as defects in the formation of the primary cilium or in primary cilium-associated signaling pathways result in the development of cystic kidney diseases, including, PKD and NPHP (Hildebrandt et al., 2011; Hildebrandt and Zhou, 2007; Yoder, 2007). Moreover, many proteins involved in NPHP and PKD, including NPHP1–5 and polycystins 1 and 2, have been localized to the primary cilium. Thus, the development of PKD and NPHP due to Glis3 and Glis2 deficiency, respectively, is consistent with an association between Glis signaling pathways and the primary cilium (figure 2). Although a reduction in the percentage of primary cilium-containing cells was observed in renal cysts, Glis3-deficiency does not result in the loss of the primary cilium in renal tubule epithelial cells, implying that Glis3 is not required for ciliogenesis (Hashimoto et al., 2009; Kang et al., 2009a; Kim et al., 2008). The reduction in primary cilium-containing cells is likely a consequence rather than a cause of renal cyst formation.
Although the primary cilium plays a critical role in the regulation of many developmental processes (Bisgrove and Yost, 2006; Eggenschwiler and Anderson, 2007), the function of the primary cilium and its relationship to the development of cystic kidney disease are not fully understood. A role for the primary cilia in sensing fluid flow has been suggested by a number of laboratories (Nauli and Zhou, 2004; Praetorius and Spring, 2001; Schwartz et al., 1997) and mechano-sensation by the primary cilium has been shown to result in increased levels of intracellular Ca2+ concentration both in cultured cells and in collecting ducts of kidney (Nauli et al., 2003). Alternatively, the primary cilium contributes to the control of proper kidney development and maintenance by regulating cell proliferation, PCP, and oriented cell division (OCD). Several protein kinases, including the mTOR, MAPK, and Akt pathways, and Wnt signaling have been implicated in these processes (McNeill, 2009; Menezes and Germino, 2009).
PCP, which is defined as the polarization of a field of cells within the plane of a sheet, has been implicated in postnatal development of nephrons as well as in cystic kidney diseases (Fischer and Pontoglio, 2009). Disruption of PCP genes, including Vang-like 2 (Vangl2) and Four-jointed box 1 (Fjx1), has been shown to result in the development of tubular cysts. Kidney tubules undergo extensive proliferation in the developing kidney. The elongation of the tubule in the longitudinal direction is driven by cell division along the axis of the tubule, a process referred to as OCD. Non-directional (mis-oriented) cell division has been proposed as a mechanism of cyst formation; however, cyst formation has been observed at stages of renal development before cells undergo OCD suggesting that additional mechanisms may be involved (Karner et al., 2009; Nishio et al., 2010). Defects in non-canonical Wnt signaling have been reported to result in cystic kidney disease (Lancaster and Gleeson, 2010). Whether defects in PCP, OCD, and Wnt signaling play a role in the cystogenic phenotype associated with deficiencies Glis2 and Glis3, requires further study.
Role of Glis3 in Pancreas Physiology and Pathology
Development of diabetes
In humans, mutations in the gene encoding GLIS3 are associated with a rare syndrome characterized by neonatal diabetes and congenital hypothyroidism with 8 affected cases from 5 families reported to date (Dimitri et al., 2011; Senee et al., 2006; Kang et al., 2010). Depending on the nature of the mutation, additional features include hepatic fibrosis, congenital glaucoma, polycystic kidney disease, facial dysmorphism, bilateral sensorineural deafness, and osteopenia (Dimitri et al., 2011; Senee et al., 2006; Taha et al., 2003). According to several human genome-wide association studies (GWAS), GLIS3 has also been linked to aberrant glucose regulation and reduced β-cell function, and was identified as a risk locus for both type-1 and type-2 diabetes (Barker et al., 2011; Barrett et al., 2009; Boesgaard et al., 2010; Dupuis et al., 2010; Hu et al., 2010). In accordance with the human studies, two independent laboratories have reported that Glis3-null mice developed neonatal diabetes characterized by hyperglycemia and hypoinsulinemia (Kang et al., 2009b; Watanabe et al., 2009). These findings suggest that GLIS3 contributes to the maintenance of endocrine functions in the pancreas, while aberrant GLIS3 function is associated with the development of diabetes (including neonatal, type-1, and type-2). Thus, Glis3-null mice provide an excellent functional model system in which to study the biological role of Glis3 in diabetes.
Pancreatic β-cell development and maintenance
The pancreas exhibits important exocrine and endocrine functions. It contains 3 major cell compartments: the acini, which produce digestive enzymes, the ducts through which the secretory enzymes are transported into the gastrointestinal tract, and the islets of Langerhans (Leung, 2010). The islets, produce and secrete various hormones, including glucagon, insulin, pancreatic polypeptide, somatostatin, and ghrelin from α, β, γ, δ, and ε cells, respectively. Although islets compose a small portion (roughly 5–10%) of the pancreas, they are crucial for the maintenance of pancreas function and endocrine hormone signaling, including the control of glucose homeostasis. Glis2 and Glis3 are highly expressed in pancreatic islets, specifically in the insulin-producing β-cells. Glis3 is also highly expressed in the pancreatic ducts (Senee et al., 2006; Kang et al., 2009b; Watanabe et al., 2009).
The mouse pancreas develops from two buds of the foregut into the dorsal and ventral pancreas at approximately embryonic day 9 (E9), and then forms a single organ after rotation and fusion between E13 and E14 (Guney and Gannon, 2009; Pan and Wright, 2011). The major components of the mature pancreas are not discernible until E16.5. For example, islet-like structures are formed at E16.5 and continue to develop and mature until 2–3 weeks after birth. Several genetically modified mouse models have been used to identify the hierarchy of the signaling pathways and transcription factors critical for pancreatic lineage determination (Jorgensen et al., 2007; Murtaugh and Melton, 2003). At E9, epithelial cells of the dorsal and ventral pancreas, regarded as pancreatic progenitors, express Pdx1, Nkx2.2, pancreatic transcription factor 1a (Ptfla), and carboxypeptidase A1 (Cpa1) (Burlison et al., 2008; Zhou et al., 2007). In fact, Pdx1-null mice fail to develop a pancreas and die shortly after birth (Jonsson et al., 1994). Notch signaling is also crucial for pancreas development at multiple stages. Under normal circumstances, reduced Notch signaling induces the expression of neurogenin 3 (Ngn3)-positive endocrine progenitor cells from pancreatic progenitors, which subsequently become committed to the various endocrine lineages (Apelqvist et al., 1999; Guney and Gannon, 2009; Pan and Wright, 2011). Ngn3-deficient embryos expressed Pdx1+ pancreatic progenitor cells at E11.5 but lack endocrine cells at birth (Gradwohl et al., 2000). Finally, transcription factors including NeuroD1, Pax6, Pax4, Nkx2.2, Nxk6.1 play a critical role in the further development of endocrine progenitors into pancreatic β-cells (Bernardo et al., 2008; Guney and Gannon, 2009; Pan and Wright, 2011).
Glis3-null mice die within one week after birth likely due to neonatal diabetes. Islet size and the expression of several β-cell markers, including insulin, glucose transporter type 2 (Glut2), MafA, and Nkx6.1, were dramatically decreased in Glis3-null mice. Other hormones including somatostatin and pancreatic polypeptide were also significantly reduced in pancreas of Glis3-null mice, while the expression of acini-specific genes was not changed (Kang et al., 2009b). The depletion of β-cells was not due to increased apoptosis, as the number of apoptotic cells was not changed in the pancreas of Glis3-null compared to wild-type mice (Kang et al., 2009b; Watanabe et al., 2009). Given that the expression of Pdx1 and Cpa1 was not changed in the pancreas of Glis3-null compared to wild-type embryos at E13.5 and E14.5, Glis3 does not appear to play a critical role in early pancreatic progenitor cell maintenance or development. However, the expression of Ngn3 was significantly decreased in Glis3-null compared to wild-type pancreas at E15.5, PND0 and PND3 (Kang et al., 2009b). As Ngn3 is known to be a key transcription factor for the development of endocrine progenitor cells, these data suggest that Glis3 plays a critical role in the maintenance or proliferation of endocrine progenitor cells and/or in the specification or development of endocrine cells, particularly β-cells (figure 3) (Kang et al., 2010).
Figure 3. Schematic representation of the role of Glis3 in pancreatic cell lineage determination.

Pancreatic progenitor cells, expressing Pdx1, Ptf1a, and Cpa1, can differentiate into exocrine, ductal, and proendocrine cell lineages. The expression of Glis3 is induced during the second transition of pancreas development and remains highly expressed in endocrine progenitor cells, ductal cells, and in mature β-cells where Glis3 can regulate insulin gene expression. In mice, loss of Glis3 inhibits the generation of β-cells and leads to the development of neonatal diabetes.
Hh signaling molecules including Indian hedgehog (Ihh), Desert hedgehog (Dhh), and Ptch1 are expressed in developing pancreas of embryos. A recent study showed that Hh signaling molecules including Smo, Ptch1, and the downstream effector, Gli, localized to the primary cilia in pancreatic epithelium and that activated Hh signaling induced the expansion of ductal and endocrine progenitor cells, suggesting that a certain level of Hh signaling is required for the development of endocrine cells (Cervantes et al., 2010; Chen et al., 2009; Tukachinsky et al., 2010). In fact, activation of Hh signaling induced Pdx1-dependent insulin expression (Thomas et al., 2001; Thomas et al., 2000) and the expression of insulin was decreased in pancreas-specific Smo-null mice (Lau and Hebrok, 2010). Given that genes encoding Hh signaling proteins including Ptch1, Smo, Dhh and Ihh, showed similar expression pattern with Glis3 in islets and ducts of adult pancreas, there could be a possibility for cross-talk between Hh and Glis3.
Glis-mediated Regulation of Insulin
Many transcription factors involved in pancreatic development also play a critical role in the regulation of insulin gene expression. For example, Pdx1, NeuroD, MafA, and Pax4/6 regulate the expression of the insulin gene through their binding the A-box, E-box, C-box, and C2 element of the insulin promoter, respectively (Cerf, 2006; Melloul et al., 2002). Given the role of Glis3 in β-cell development and that the expression of Ins2 was dramatically decreased in pancreas of Glis3-null mice at PND3 and in Glis3-null embryos as early as E15.5, Glis3 may also directly regulate insulin gene expression in mature β-cells (figure 3).
Ins2 expression in rat insulinoma 832/13 cells is markedly increased by Glis3 over-expression and decreased when Glis3 is knocked down by siRNA. Furthermore, Glis3 binds to the mIns2 promoter in these cells as indicated by chromatin immunoprecipitation (Yang et al., 2009). Indeed, promoter analysis showed that the human INS and mouse Ins2 genes contain 2 well-conserved GlisBS within 600 base pairs upstream of the transcription start site. In vitro DNA binding assays and cell-based reporter gene assays provided further support that Glis3 directly regulates the expression of human and mouse insulin genes through these 2 GlisBS sequences (Kang et al., 2009b; Yang et al., 2009). Glis2 can bind a similar GlisBS as Glis3. Glis2 activated the mouse mIns2 promoter in vitro in non-beta cells through the same 2 GlisBS as Glis3 (Vasanth et al., 2011). Thus, Glis2, which is expressed in mouse pancreatic islets, may also play a role in insulin regulation in the mouse pancreas. Glis2-null mice did not show any diabetic or abnormal pancreatic phenotype; however, it is possible that Glis3 may compensate for the loss of Glis2 function in pancreas of Glis2-null mice.
Glis3 is also reported to physically and functionally interact with other known regulators of insulin gene expression, including MafA, NeuroD, and Pdx1. Glis3 co-immunoprecipitated with MafA, NeuroD, and Pdx1 in insulinoma 832/13 cells and cell-based reporter assays indicated that Pdx1 and, to a lesser degree, MafA had synergistic effects with Glis3 on insulin promoter activity (Yang et al., 2009). Taken together, these results reveal potential for cross-talk between Glis2, Glis3, and other known regulators of insulin gene expression. Further examination is required to determine the mechanism by which Glis2 and Glis3 may directly regulate insulin gene expression.
Even though insulin has been identified as a direct Glis3 target gene, Glis3 may also regulate insulin expression by indirect mechanisms. The expression of MafA was significantly decreased in the pancreas of Glis3-null mice at PND3 and in insulinoma 832/13 cells when Glis3 is knocked down by siRNA (Kang et al., 2009b; Yang et al., 2009). Although MafA is known to regulate the expression of Ins2, MafA-null mice do not exhibit neonatal diabetes. At an early age, no difference was observed in insulin content in the pancreas of MafA-null compared to wild-type mice, but MafA-null mice were intolerant to glucose challenge and developed diabetes with age. In addition, glucose-stimulated insulin secretion (GSIS) was impaired in the islets of MafA-null compared to wild-type mice (Zhang et al., 2005). These results suggest that MafA may be more critical for insulin secretion than insulin production. Abcc8, another gene decreased in PND3 pancreas of Glis3-null mice, encodes the sulfonylurea receptor 1 (Sur1), which plays a role in the regulation of insulin secretion (Aguilar-Bryan et al., 1995; Kang et al., 2009b; Koehn et al., 2008). These results indicate that in addition to its function in the control of insulin gene expression, Glis3 may also play a role, either directly or indirectly, in regulating insulin secretion. In addition to the continual investigation into the role that Glis3 plays in insulin regulation, further research is needed to identify additional Glis3 target genes in order to elucidate the full scope of Glis3 biological function in the pancreas.
Glis Functions in Other Tissues
Glis1 function in the epidermis
Although Glis1 is expressed in several tissues, the physiological functions of this Glis family member are still poorly understood (Kang et al., 2010). The characterization of Glis1-null mice did not reveal any obvious phenotype. In the skin, Glis1 mRNA expression was detected in the dermal papilla, but not in normal human epidermis (Nakanishi et al., 2006; Nakashima et al., 2002). However, Glis1 expression was significantly induced in psoriatic epidermis where its expression was associated with the suprabasal, differentiated layers. Glis1 is also expressed in mouse skin treated with the tumor promoter phorbol-12-myristate-13-acetate (PMA) and induced in normal human epidermal keratinocytes (NHEK) cells in culture by the addition of PMA and IFNγ, both inflammatory mediators and inducers of epidermal differentiation. The overexpression of Glis1 in NHEK cells resulted in the increased expression of several markers of epidermal differentiation, including S100 calcium binding protein A9 (S100A9), kallikrein 7 (KLK7), small proline-rich protein (SPRR), involucrin, and transglutaminase 1 (Nakanishi et al., 2006). The induction of differentiation markers by Glis1 is consistent with its expression in the suprabasal, differentiated layers of hyperplastic epidermis. Taken together, these observations suggest that Glis1 promotes differentiation of epidermal keratinocytes and may play a role in aberrant epidermal differentiation associated with psoriatic skin.
Promotion of iPS cell generation by Glis1
A recent study showed that Glis1 is highly expressed in the unfertilized egg and in one-cell embryos, but is rapidly down-regulated in two-cell embryos and blastocysts (Maekawa et al., 2011). Moreover, expression of Glis1 was very low in embryonic stem cells (ES) cells. Maekawa et al. further demonstrated that expression of Glis1 significantly promoted the generation of induced pluripotent stem (iPS) cells. The generation of iPS cells has been achieved by the transgenic expression of the transcription factors Oct3/4, Sox2, and Klf4; however, conversion has been inefficient. Co-expression of Glis1 greatly enhanced the generation of iPS cells from both mouse and human fibroblasts. Glis1 was shown to associate with Klf4, Oct3/4 and Sox2 protein complexes. Gene expression profiling showed that Glis1 induced the expression of several genes that have been reported to enhance iPS generation, including estrogen-related receptor β (Esrrb), several Wnt ligands, lin-28 homologue A (Lin28a), the homeobox transcription factor Nanog, Mycn, and Mycl1, while the expression of Myc was suppressed. Interestingly, Glis1 also enhanced expression of forkhead box A2 (Foxa2), which has been reported to antagonize EMT. Mesenchymal-epithelial transition (MET) is required for the reprogramming of fibroblasts into IPS cells (Li et al., 2010). Thus, Glis1 might stimulate somatic cell reprogramming by promoting MET. These observations indicate that Glis1 enhance iPS generation by inducing several pro-reprogramming pathways.
Role for Glis2 in neurogenesis
At embryonic day E9.5, Glis2 is expressed in the neural tube, cranial ganglia and dorsal root ganglia (Lamar et al., 2001). By E10.5, Glis2 is expressed in the intermediate zone of the hindbrain and spinal cord and in the dorsal root ganglia. A similar pattern of Glis2 expression was observed in the chick embryo in the intermediate zone where newly post-mitotic neurons are located. In Xenopus, Glis2 expression corresponds to two midline regions that contain precursors of primary neurons. Overexpression of Glis2 in neuronal progenitors in the spinal cord of chick embryos was shown to promote the neuronal differentiation. Likewise, ectopic expression of Glis2 in Xenopus induced the expression of several neuron-specific markers. Expression of the neurogenin 1 (Ngn1) was shown to induce Glis2 suggesting that Glis2 functions downstream of Ngn1 (Lamar et al., 2001). Together, these observations suggest that Glis2 plays a role in the regulation of neurogenesis.
Glis3 and osteogenesis
A reduction in bone formation leads to reduced bone density and an increased risk of fractures. Several studies have indicated a role for Glis3 in osteogenesis. Human patients with aberrant Glis3 expression develop osteopenia with thoracolumbar lordosis and multiple rib fractures (Dimitri et al., 2011). Moreover, Glis3 was highly expressed in human osteoblasts and induced during osteoblast differentiation of mesenchymal stem cells (Beak et al., 2007). Ectopic expression of Glis3 in the multipotent cell line C3H10T1/2 acts synergistically with bone morphogenic protein 2 (BMP2) to promote osteoblast differentiation as measured by the increased levels of alkaline phosphatase activity and osteopontin and osteocalcin expression. In contrast, Glis3 expression inhibited adipocyte differentiation in C3H10T1/2 cells. Gene expression profiling identified a number of additional genes that were induced or down-regulated by Glis3 in this cell system. Interestingly, the expression of fibroblast growth factor 18 (FGF18), which has a positive role in osteogenesis, was significantly increased. EMSA and reporter gene analysis revealed that Glis3 regulates FGF18 by binding a GlisBS in proximal promoter region (Beak et al., 2007). These observations suggest that FGF18 is direct target gene of Glis3 and indicates that Glis3 is an important modulator of osteogenesis. Interestingly, the primary cilium also mediates important functions in osteoblast differentiation and normal bone development as well as in the generation of osteopenia (Malone et al., 2007; Xiao et al., 2008; Xiao and Quarles, 2010) Further insights into the role of the Glis3 signaling pathway in osteoblast differentiation might lead to novel approaches for the treatment of osteoporosis.
Glis3 and hypothyroidism
In addition to diabetes and polycystic kidney disease, patients with aberrant Glis3 expression also develop congenital hypothyroidism that is accompanied by reduced levels of T3 and T4 and elevated blood levels of thyroid stimulating hormone (TSH) and thyroglobulin (Dimitri et al., 2011; Senee et al., 2006; Taha et al., 2003). Similarly, hypothyroidism was observed in Glis3-null mice (Watanabe et al., 2009). About 85% of this disorder is caused by abnormal thyroid gland development; however, histological examination of thyroid gland of Glis3-null mice suggested that Glis3 does not affect thyroid gland development. Some patients responded well to T4 treatment, while others did not. While all patients with Glis3 mutations profiled thus far display thyroid dysfunction, the inconsistent clinical features make it difficult to establish a causative mechanism. Further analysis is required to determine whether the development of hypothyroidism is related to mis-regulation of the hypothalamus-pituitary-thyroid axis and/or involves changes in iodine or thyroid hormone metabolism.
Glis3 and cancer
Increased Glis3 expression has been detected in several different cancer cell types, while no links have yet been made between tumorigenesis and Glis1 or Glis2 (Kang et al., 2010). Increased expression of Glis3 has been reported in ependymomas with high proliferation indices and poor patient prognosis (Lukashova-v Zangen et al., 2007). Amplification of Glis3 was observed in proneural glioblastomas (Cooper et al., 2010). Glis3 was also highly expressed in chromophobe renal cell carcinomas (Yusenko and Kovacs, 2009). Analysis of the role of Glis3 in cell fate and tumor metastasis could provide additional information regarding a link between Glis3 expression and cancer progression.
Conclusion
It is clear that Glis transcription factors play a critical role in the regulation of several physiological processes and are implicated in various pathologies. Mutations in Glis2 have been linked to NPHP, an end-stage renal disease characterized by renal atrophy and fibrosis. This appears to involve induction of EMT in tubule epithelial cells. Mutations in Glis3 lead to the development of diabetes, hypothyroidism, polycystic kidney disease and several other abnormalities. Glis3 was shown to play a key role in endocrine lineage determination in the pancreas and is required for β-cell development. In addition, Glis3 has a role in the regulation of insulin gene expression. Although the consensus GlisBS and several target genes have been identified, little is known about the proteins that mediate transcriptional regulation by Glis proteins. Moreover, the mechanisms by which Glis activity is controlled are not well understood. Several reports have indicated that Glis2 and Glis3 localize to the primary cilium suggesting that Glis proteins are part of a primary cilium-mediated signaling pathway. This is consistent with studies showing that loss of Glis2 or Glis3 is linked to several ciliopathies, such as NPHP and polycystic kidney disease. The activities of Glis proteins may be regulated by external signals that interact with membrane-bound receptors in the primary cilium. Reception of such signals may result in a post-translational modification of Glis proteins and their subsequent translocation into the nucleus where they regulate the transcription of target genes. Elucidation of the different stages of the Glis signaling pathway will be crucial to understanding the biological functions of Glis proteins and to the discovery of therapeutic opportunities for the treatment of diabetes and cystic kidney disease.
Acknowledgments
The authors would like to thank Drs. Zhengyu Yin and Christina Teng (NIEHS) for their comments on the manuscript. This research was supported by the Intramural Research Program of the NIEHS, NIH (Z01-ES-100485).
References
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, Boyd AE, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Akiyama T. Wnt/beta-catenin signaling. Cytokine Growth Factor Rev. 2000;11:273–282. doi: 10.1016/s1359-6101(00)00011-3. [DOI] [PubMed] [Google Scholar]
- Al-Bhalal L, Akhtar M. Molecular basis of autosomal recessive polycystic kidney disease (ARPKD) Adv Anat Pathol. 2008;15:54–58. doi: 10.1097/PAP.0b013e31815e5295. [DOI] [PubMed] [Google Scholar]
- Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, Hrabe de Angelis M, Lendahl U, Edlund H. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400:877–881. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- Attanasio M, Uhlenhaut NH, Sousa VH, O’Toole JF, Otto E, Anlag K, Klugmann C, Treier AC, Helou J, Sayer JA, Seelow D, Nurnberg G, Becker C, Chudley AE, Nurnberg P, Hildebrandt F, Treier M. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet. 2007;39:1018–24. doi: 10.1038/ng2072. [DOI] [PubMed] [Google Scholar]
- Barker A, Sharp SJ, Timpson NJ, Bouatia-Naji N, Warrington NM, Kanoni S, Beilin LJ, Brage S, Deloukas P, Evans DM, Grontved A, Hassanali N, Lawlor DA, Lecoeur C, Loos RJ, Lye SJ, McCarthy MI, Mori TA, Ndiaye NC, Newnham JP, Ntalla I, Pennell CE, St Pourcain B, Prokopenko I, Ring SM, Sattar N, Visvikis-Siest S, Dedoussis GV, Palmer LJ, Froguel P, Smith GD, Ekelund U, Wareham NJ, Langenberg C. Association of genetic Loci with glucose levels in childhood and adolescence: a meta-analysis of over 6,000 children. Diabetes. 2011;60:1805–1812. doi: 10.2337/db10-1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnfield PC, Zhang X, Thanabalasingham V, Yoshida M, Hui CC. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation. 2005;73:397–405. doi: 10.1111/j.1432-0436.2005.00042.x. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, Plagnol V, Pociot F, Schuilenburg H, Smyth DJ, Stevens H, Todd JA, Walker NM, Rich SS Type 1 Diabetes Genetics Consortium. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41:703–707. doi: 10.1038/ng.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beak JY, Kang HS, Kim YS, Jetten AM. Kruppel-like zinc finger protein Glis3 promotes osteoblast differentiation by regulating FGF18 expression. J Bone Miner Res. 2007;22:1234–1244. doi: 10.1359/jbmr.070503. [DOI] [PubMed] [Google Scholar]
- Beak JY, Kang HS, Kim YS, Jetten AM. Functional analysis of the zinc finger and activation domains of Glis3 and mutant Glis3(NDH1) Nucleic Acids Res. 2008;36:1690–1702. doi: 10.1093/nar/gkn009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, Johnson AD, Lewis JS, Askwith CC, Mykytyn K. Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol Biol Cell. 2008a;19:1540–1547. doi: 10.1091/mbc.E07-09-0942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K. Bardet-Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc Natl Acad Sci U S A. 2008b;105:4242–4246. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, O’Connor AK, Haycraft CJ, Yoder BK. The primary cilium as a complex signaling center. Curr Biol. 2009;19:R526–R535. doi: 10.1016/j.cub.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann C, Senderek J, Kupper F, Schneider F, Dornia C, Windelen E, Eggermann T, Rudnik-Schoneborn S, Kirfel J, Furu L, Onuchic LF, Rossetti S, Harris PC, Somlo S, Guay-Woodford L, Germino GG, Moser M, Buttner R, Zerres K. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD) Hum Mutat. 2004;23:453–463. doi: 10.1002/humu.20029. [DOI] [PubMed] [Google Scholar]
- Bernardo AS, Hay CW, Docherty K. Pancreatic transcription factors and their role in the birth, life and survival of the pancreatic beta cell. Mol Cell Endocrinol. 2008;294:1–9. doi: 10.1016/j.mce.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Bisgrove BW, Yost HJ. The roles of cilia in developmental disorders and disease. Development. 2006;133:4131–4143. doi: 10.1242/dev.02595. [DOI] [PubMed] [Google Scholar]
- Blacque OE, Cevik S, Kaplan OI. Intraflagellar transport: from molecular characterisation to mechanism. Front Biosci. 2008;13:2633–2652. doi: 10.2741/2871. [DOI] [PubMed] [Google Scholar]
- Boesgaard TW, Grarup N, Jorgensen T, Borch-Johnsen K, Hansen T, Pedersen O. Variants at DGKB/TMEM195, ADRA2A, GLIS3 and C2CD4B loci are associated with reduced glucose-stimulated beta cell function in middle-aged Danish people. Diabetologia. 2010;53:1647–1655. doi: 10.1007/s00125-010-1753-5. [DOI] [PubMed] [Google Scholar]
- Brayer KJ, Segal DJ. Keep your fingers off my DNA: protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem Biophys. 2008;50:111–131. doi: 10.1007/s12013-008-9008-5. [DOI] [PubMed] [Google Scholar]
- Burlison JS, Long Q, Fujitani Y, Wright CV, Magnuson MA. Pdx-1 and Ptf1a concurrently determine fate specification of pancreatic multipotent progenitor cells. Dev Biol. 2008;316:74–86. doi: 10.1016/j.ydbio.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerf ME. Transcription factors regulating beta-cell function. Eur J Endocrinol. 2006;155:671–679. doi: 10.1530/eje.1.02277. [DOI] [PubMed] [Google Scholar]
- Cervantes S, Lau J, Cano DA, Borromeo-Austin C, Hebrok M. Primary cilia regulate Gli/Hedgehog activation in pancreas. Proc Natl Acad Sci U S A. 2010;107:10109–10114. doi: 10.1073/pnas.0909900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W, Zeng Q, Hong W. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008;68:2592–2598. doi: 10.1158/0008-5472.CAN-07-2696. [DOI] [PubMed] [Google Scholar]
- Chen MH, Wilson CW, Li YJ, Law KK, Lu CS, Gacayan R, Zhang X, Hui CC, Chuang PT. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009;23:1910–1928. doi: 10.1101/gad.1794109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SY, Bishop JM. Suppressor of Fused represses Gli-mediated transcription by recruiting the SAP18-mSin3 corepressor complex. Proc Natl Acad Sci U S A. 2002;99:5442–5447. doi: 10.1073/pnas.082096999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnadurai G. Transcriptional regulation by C-terminal binding proteins. Int J Biochem Cell Biol. 2007;39:1593–607. doi: 10.1016/j.biocel.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Cooper LA, Gutman DA, Long Q, Johnson BA, Cholleti SR, Kurc T, Saltz JH, Brat DJ, Moreno CS. The proneural molecular signature is enriched in oligodendrogliomas and predicts improved survival among diffuse gliomas. PLoS One. 2010;5:e12548. doi: 10.1371/journal.pone.0012548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Dang DT, Pevsner J, Yang VW. The biology of the mammalian Kruppel-like family of transcription factors. Int J Biochem Cell Biol. 2000;32:1103–1121. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JM. Dancing in and out of the nucleus: p120(ctn) and the transcription factor Kaiso. Biochim Biophys Acta. 2007;1773:59–68. doi: 10.1016/j.bbamcr.2006.08.052. [DOI] [PubMed] [Google Scholar]
- Dimitri P, Warner JT, Minton JA, Patch AM, Ellard S, Hattersley AT, Barr S, Hawkes D, Wales JK, Gregory JW. Novel GLIS3 mutations demonstrate an extended multisystem phenotype. Eur J Endocrinol. 2011;164:437–443. doi: 10.1530/EJE-10-0893. [DOI] [PubMed] [Google Scholar]
- Domire JS, Green JA, Lee KG, Johnson AD, Askwith CC, Mykytyn K. Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell Mol Life Sci. 2010 doi: 10.1007/s00018-010-0603-4. (In press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, Lindgren CM, Magi R, Morris AP, Randall J, Johnson T, Elliott P, Rybin D, Thorleifsson G, Steinthorsdottir V, Henneman P, Grallert H, Dehghan A, Hottenga JJ, Franklin CS, Navarro P, Song K, Goel A, Perry JR, Egan JM, Lajunen T, Grarup N, Sparso T, Doney A, Voight BF, Stringham HM, Li M, Kanoni S, Shrader P, Cavalcanti-Proenca C, Kumari M, Qi L, Timpson NJ, Gieger C, Zabena C, Rocheleau G, Ingelsson E, An P, O’Connell J, Luan J, Elliott A, McCarroll SA, Payne F, Roccasecca RM, Pattou F, Sethupathy P, Ardlie K, Ariyurek Y, Balkau B, Barter P, Beilby JP, Ben-Shlomo Y, Benediktsson R, Bennett AJ, Bergmann S, Bochud M, Boerwinkle E, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Bottcher Y, Brunner E, Bumpstead SJ, Charpentier G, Chen YD, Chines P, Clarke R, Coin LJ, Cooper MN, Cornelis M, Crawford G, Crisponi L, Day IN, de Geus EJ, Delplanque J, Dina C, Erdos MR, Fedson AC, Fischer-Rosinsky A, Forouhi NG, Fox CS, Frants R, Franzosi MG, Galan P, Goodarzi MO, Graessler J, Groves CJ, Grundy S, Gwilliam R, Gyllensten U, Hadjadj S, Hallmans G, Hammond N, Han X, Hartikainen AL, Hassanali N, Hayward C, Heath SC, Hercberg S, Herder C, Hicks AA, Hillman DR, Hingorani AD, Hofman A, Hui J, Hung J, Isomaa B, Johnson PR, Jorgensen T, Jula A, Kaakinen M, Kaprio J, Kesaniemi YA, Kivimaki M, Knight B, Koskinen S, Kovacs P, Kyvik KO, Lathrop GM, Lawlor DA, Le Bacquer O, Lecoeur C, Li Y, Lyssenko V, Mahley R, Mangino M, Manning AK, Martinez-Larrad MT, McAteer JB, McCulloch LJ, McPherson R, Meisinger C, Melzer D, Meyre D, Mitchell BD, Morken MA, Mukherjee S, Naitza S, Narisu N, Neville MJ, Oostra BA, Orru M, Pakyz R, Palmer CN, Paolisso G, Pattaro C, Pearson D, Peden JF, Pedersen NL, Perola M, Pfeiffer AF, Pichler I, Polasek O, Posthuma D, Potter SC, Pouta A, Province MA, Psaty BM, Rathmann W, Rayner NW, Rice K, Ripatti S, Rivadeneira F, Roden M, Rolandsson O, Sandbaek A, Sandhu M, Sanna S, Sayer AA, Scheet P, Scott LJ, Seedorf U, Sharp SJ, Shields B, Sigurethsson G, Sijbrands EJ, Silveira A, Simpson L, Singleton A, Smith NL, Sovio U, Swift A, Syddall H, Syvanen AC, Tanaka T, Thorand B, Tichet J, Tonjes A, Tuomi T, Uitterlinden AG, van Dijk KW, van Hoek M, Varma D, Visvikis-Siest S, Vitart V, Vogelzangs N, Waeber G, Wagner PJ, Walley A, Walters GB, Ward KL, Watkins H, Weedon MN, Wild SH, Willemsen G, Witteman JC, Yarnell JW, Zeggini E, Zelenika D, Zethelius B, Zhai G, Zhao JH, Zillikens MC, Borecki IB, Loos RJ, Meneton P, Magnusson PK, Nathan DM, Williams GH, Hattersley AT, Silander K, Salomaa V, Smith GD, Bornstein SR, Schwarz P, Spranger J, Karpe F, Shuldiner AR, Cooper C, Dedoussis GV, Serrano-Rios M, Morris AD, Lind L, Palmer LJ, Hu FB, Franks PW, Ebrahim S, Marmot M, Kao WH, Pankow JS, Sampson MJ, Kuusisto J, Laakso M, Hansen T, Pedersen O, Pramstaller PP, Wichmann HE, Illig T, Rudan I, Wright AF, Stumvoll M, Campbell H, Wilson JF, Bergman RN, Buchanan TA, Collins FS, Mohlke KL, Tuomilehto J, Valle TT, Altshuler D, Rotter JI, Siscovick DS, Penninx BW, Boomsma DI, Deloukas P, Spector TD, Frayling TM, Ferrucci L, Kong A, Thorsteinsdottir U, Stefansson K, van Duijn CM, Aulchenko YS, Cao A, Scuteri A, Schlessinger D, Uda M, Ruokonen A, Jarvelin MR, Waterworth DM, Vollenweider P, Peltonen L, Mooser V, Abecasis GR, Wareham NJ, Sladek R, Froguel P, Watanabe RM, Meigs JB, Groop L, Boehnke M, McCarthy MI, Florez JC, Barroso I. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund T, Walker MD, Barr PJ, Rutter WJ. Cell-specific expression of the rat insulin gene: evidence for role of two distinct 5′ flanking elements. Science. 1985;230:912–916. doi: 10.1126/science.3904002. [DOI] [PubMed] [Google Scholar]
- Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elrod-Erickson M, Rould MA, Nekludova L, Pabo CO. Zif268 protein-DNA complex refined at 1.6 A: a model system for understanding zinc finger-DNA interactions. Structure. 1996;4:1171–80. doi: 10.1016/s0969-2126(96)00125-6. [DOI] [PubMed] [Google Scholar]
- Fischer E, Pontoglio M. Planar cell polarity and cilia. Semin Cell Dev Biol. 2009;20:998–1005. doi: 10.1016/j.semcdb.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- Frankel AD, Berg JM, Pabo CO. Metal-dependent folding of a single zinc finger from transcription factor IIIA. Proc Natl Acad Sci U S A. 1987;84:4841–4845. doi: 10.1073/pnas.84.14.4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong EE, Andersen EC, Null B, White KP, Scott MP. Patterns of gene expression during Drosophila mesoderm development. Science. 2001;293:1629–1633. doi: 10.1126/science.1062660. [DOI] [PubMed] [Google Scholar]
- Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:118–130. doi: 10.1053/j.ackd.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavert N, Ben-Ze’ev A. beta-Catenin signaling in biological control and cancer. J Cell Biochem. 2007;102:820–828. doi: 10.1002/jcb.21505. [DOI] [PubMed] [Google Scholar]
- German MS, Moss LG, Wang J, Rutter WJ. The insulin and islet amyloid polypeptide genes contain similar cell-specific promoter elements that bind identical beta-cell nuclear complexes. Mol Cell Biol. 1992;12:1777–1788. doi: 10.1128/mcb.12.4.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradwohl G, Dierich A, LeMeur M, Guillemot F. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U S A. 2000;97:1607–1611. doi: 10.1073/pnas.97.4.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guney MA, Gannon M. Pancreas cell fate. Birth Defects Res C: Embryo Today. 2009;87:232–248. doi: 10.1002/bdrc.20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D. Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature. 1985;315:115–122. doi: 10.1038/315115a0. [DOI] [PubMed] [Google Scholar]
- Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321–337. doi: 10.1146/annurev.med.60.101707.125712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Miyamoto R, Watanabe N, Shiba D, Ozato K, Inoue C, Kubo Y, Koga A, Jindo T, Narita T, Naruse K, Ohishi K, Nogata K, Shin IT, Asakawa S, Shimizu N, Miyamoto T, Mochizuki T, Yokoyama T, Hori H, Takeda H, Kohara Y, Wakamatsu Y. Polycystic kidney disease in the medaka (Oryzias latipes) pc mutant caused by a mutation in the Gli-Similar3 (glis3) gene. PLoS One. 2009;4:e6299. doi: 10.1371/journal.pone.0006299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18:1855–1871. doi: 10.1681/ASN.2006121344. [DOI] [PubMed] [Google Scholar]
- Hosking CR, Ulloa F, Hogan C, Ferber E, Figueroa A, Gevaert K, Birchmeier W, Briscoe J, Fujita Y. The Transcriptional Repressor Glis2 Is a Novel Binding Partner for p120 Catenin. Mol Biol Cell. 2007;18:1918–1927. doi: 10.1091/mbc.E06-10-0941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossain Z, Ali SM, Ko HL, Xu J, Ng CP, Guo K, Qi Z, Ponniah S, Hong W, Hunziker W. Glomerulocystic kidney disease in mice with a targeted inactivation of Wwtr1. Proc Natl Acad Sci U S A. 2007;104:1631–1636. doi: 10.1073/pnas.0605266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C, Zhang R, Wang C, Wang J, Ma X, Hou X, Lu J, Yu W, Jiang F, Bao Y, Xiang K, Jia W. Variants from GIPR, TCF7L2, DGKB, MADD, CRY2, GLIS3, PROX1, SLC30A8 and IGF1 are associated with glucose metabolism in the Chinese. PLoS One. 2010;5:e15542. doi: 10.1371/journal.pone.0015542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010;24:670–682. doi: 10.1101/gad.1902910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanaga T, Miki T, Takahashi-Iwanaga H. Restricted expression of somatostatin receptor 3 to primary cilia in the pancreatic islets and adenohypophysis of mice. Biomed Res. 2011;32:73–81. doi: 10.2220/biomedres.32.73. [DOI] [PubMed] [Google Scholar]
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- Jorgensen MC, Ahnfelt-Ronne J, Hald J, Madsen OD, Serup P, Hecksher-Sorensen J. An illustrated review of early pancreas development in the mouse. Endocr Rev. 2007;28:685–705. doi: 10.1210/er.2007-0016. [DOI] [PubMed] [Google Scholar]
- Kaczynski J, Cook T, Urrutia R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003;4:206. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HS, Beak JY, Kim YS, Herbert R, Jetten AM. Glis3 is associated with primary cilia and Wwtr1/TAZ and implicated in polycystic kidney disease. Mol Cell Biol. 2009a;29:2556–2569. doi: 10.1128/MCB.01620-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HS, Kim YS, ZeRuth G, Beak JY, Gerrish K, Kilic G, Sosa-Pineda B, Jensen J, Foley J, Jetten AM. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol Cell Biol. 2009b;29:6366–6379. doi: 10.1128/MCB.01259-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HS, ZeRuth G, Lichti-Kaiser K, Vasanth S, Yin Z, Kim YS, Jetten AM. Gli-similar (Glis) Krüppel-like zinc finger proteins: insights into their physiological functions and critical roles in neonatal diabetes and cystic renal disease. Histol Histopath. 2010;25:1481–1496. doi: 10.14670/hh-25.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karner CM, Chirumamilla R, Aoki S, Igarashi P, Wallingford JB, Carroll TJ. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat Genet. 2009;41:793–799. doi: 10.1038/ng.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper M, Regl G, Frischauf AM, Aberger F. GLI transcription factors: mediators of oncogenic Hedgehog signalling. Eur J Cancer. 2006;42:437–445. doi: 10.1016/j.ejca.2005.08.039. [DOI] [PubMed] [Google Scholar]
- Kim SC, Kim YS, Jetten AM. Kruppel-like zinc finger protein Gli-similar 2 (Glis2) represses transcription through interaction with C-terminal binding protein 1 (CtBP1) Nucleic Acids Res. 2005;33:6805–6815. doi: 10.1093/nar/gki985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Park JI, Spring CM, Sater AK, Ji H, Otchere AA, Daniel JM, McCrea PD. Non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor and p120-catenin. Nat Cell Biol. 2004;6:1212–1220. doi: 10.1038/ncb1191. [DOI] [PubMed] [Google Scholar]
- Kim YS, Kang HS, Herbert R, Beak JY, Collins JB, Grissom SF, Jetten AM. Kruppel-like zinc finger protein Glis2 is essential for the maintenance of normal renal functions. Mol Cell Biol. 2008;28:2358–2367. doi: 10.1128/MCB.01722-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Kang HS, Jetten AM. The Kruppel-like zinc finger protein Glis2 functions as a negative modulator of the Wnt/beta-catenin signaling pathway. FEBS Lett. 2007;581:858–864. doi: 10.1016/j.febslet.2007.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, Lewandoski M, Perantoni AO, Kurebayashi S, Nakanishi G, Jetten AM. Identification of Glis1, a novel Gli-related, Kruppel-like zinc finger protein containing transactivation and repressor functions. J Biol Chem. 2002;277:30901–30913. doi: 10.1074/jbc.M203563200. [DOI] [PubMed] [Google Scholar]
- Kim YS, Nakanishi G, Lewandoski M, Jetten AM. GLIS3, a novel member of the GLIS subfamily of Kruppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res. 2003;31:5513–5525. doi: 10.1093/nar/gkg776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehn J, Fountoulakis M, Krapfenbauer K. Multiple drug resistance associated with function of ABC-transporters in diabetes mellitus: molecular mechanism and clinical relevance. Infect Disord Drug Targets. 2008;8:109–118. doi: 10.2174/187152608784746510. [DOI] [PubMed] [Google Scholar]
- Lamar E, Kintner C, Goulding M. Identification of NKL, a novel Gli-Kruppel zinc-finger protein that promotes neuronal differentiation. Development. 2001;128:1335–1346. doi: 10.1242/dev.128.8.1335. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Gleeson JG. Cystic kidney disease: the role of Wnt signaling. Trends Mol Med. 2010;16:349–360. doi: 10.1016/j.molmed.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau J, Hebrok M. Hedgehog signaling in pancreas epithelium regulates embryonic organ formation and adult beta-cell function. Diabetes. 2010;59:1211–1221. doi: 10.2337/db09-0914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Gippert GP, Soman KV, Case DA, Wright PE. Three-dimensional solution structure of a single zinc finger DNA-binding domain. Science. 1989;245:635–637. doi: 10.1126/science.2503871. [DOI] [PubMed] [Google Scholar]
- Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, Guan KL. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426–2436. doi: 10.1128/MCB.01874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung PS. Physiology of the pancreas. Adv Exp Med Biol. 2010;690:13–27. doi: 10.1007/978-90-481-9060-7_2. [DOI] [PubMed] [Google Scholar]
- Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, Qin B, Xu J, Li W, Yang J, Gan Y, Qin D, Feng S, Song H, Yang D, Zhang B, Zeng L, Lai L, Esteban MA, Pei D. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Liu Y. New Insights into Epithelial-Mesenchymal Transition in Kidney Fibrosis. J Am Soc Nephrol. 2009;21:212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashova-v Zangen I, Kneitz S, Monoranu CM, Rutkowski S, Hinkes B, Vince GH, Huang B, Roggendorf W. Ependymoma gene expression profiles associated with histological subtype, proliferation, and patient survival. Acta Neuropathol. 2007;113:325–337. doi: 10.1007/s00401-006-0190-5. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Yamaguchi K, Nakamura T, Shibukawa R, Kodanaka I, Ichisaka T, Kawamura Y, Mochizuki H, Goshima N, Yamanaka S. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature. 2011;474:225–229. doi: 10.1038/nature10106. [DOI] [PubMed] [Google Scholar]
- Makita R, Uchijima Y, Nishiyama K, Amano T, Chen Q, Takeuchi T, Mitani A, Nagase T, Yatomi Y, Aburatani H, Nakagawa O, Small EV, Cobo-Stark P, Igarashi P, Murakami M, Tominaga J, Sato T, Asano T, Kurihara Y, Kurihara H. Multiple renal cysts, urinary concentration defects, and pulmonary emphysematous changes in mice lacking TAZ. Am J Physiol Renal Physiol. 2008;294:F542–553. doi: 10.1152/ajprenal.00201.2007. [DOI] [PubMed] [Google Scholar]
- Malone AM, Anderson CT, Stearns T, Jacobs CR. Primary cilia in bone. J Musculoskelet Neuronal Interact. 2007;7:301. [PubMed] [Google Scholar]
- Marshall J, Dolan BM, Garcia EP, Sathe S, Tang X, Mao Z, Blair LA. Calcium channel and NMDA receptor activities differentially regulate nuclear C/EBPbeta levels to control neuronal survival. Neuron. 2003;39:625–639. doi: 10.1016/s0896-6273(03)00496-3. [DOI] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- McNeill H. Planar cell polarity and the kidney. J Am Soc Nephrol. 2009;20:2104–2111. doi: 10.1681/ASN.2008111173. [DOI] [PubMed] [Google Scholar]
- Melloul D, Marshak S, Cerasi E. Regulation of insulin gene transcription. Diabetologia. 2002;45:309–326. doi: 10.1007/s00125-001-0728-y. [DOI] [PubMed] [Google Scholar]
- Menezes LF, Germino GG. Polycystic kidney disease, cilia, and planar polarity. Methods Cell Biol. 2009;94:273–297. doi: 10.1016/S0091-679X(08)94014-0. [DOI] [PubMed] [Google Scholar]
- Meng X, Poon R, Zhang X, Cheah A, Ding Q, Hui CC, Alman B. Suppressor of fused negatively regulates beta-catenin signaling. J Biol Chem. 2001;276:40113–40119. doi: 10.1074/jbc.M105317200. [DOI] [PubMed] [Google Scholar]
- Merzdorf CS. Emerging roles for zic genes in early development. Dev Dyn. 2007;236:922–940. doi: 10.1002/dvdy.21098. [DOI] [PubMed] [Google Scholar]
- Michos O. Kidney development: from ureteric bud formation to branching morphogenesis. Curr Opin Genet Dev. 2009;19:484–490. doi: 10.1016/j.gde.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranti CK, Ginty DD, Huang G, Chatila T, Greenberg ME. Calcium activates serum response factor-dependent transcription by a Ras- and Elk-1-independent mechanism that involves a Ca2+/calmodulin-dependent kinase. Mol Cell Biol. 1995;15:3672–3684. doi: 10.1128/mcb.15.7.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo R, Freer AM, Zinyk DL, Crackower MA, Michaud J, Heng HH, Chik KW, Shi XM, Tsui LC, Cheng SH, Joyner AL, Hui C. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development. 1997;124:113–123. doi: 10.1242/dev.124.1.113. [DOI] [PubMed] [Google Scholar]
- Murtaugh LC, Melton DA. Genes, signals, and lineages in pancreas development. Annu Rev Cell Dev Biol. 2003;19:71–89. doi: 10.1146/annurev.cellbio.19.111301.144752. [DOI] [PubMed] [Google Scholar]
- Na J, Lykke-Andersen K, Torres Padilla ME, Zernicka-Goetz M. Dishevelled proteins regulate cell adhesion in mouse blastocyst and serve to monitor changes in Wnt signaling. Dev Biol. 2007;302:40–49. doi: 10.1016/j.ydbio.2006.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachury MV, Seeley ES, Jin H. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol. 2010;26:59–87. doi: 10.1146/annurev.cellbio.042308.113337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi G, Kim YS, Nakajima T, Jetten AM. Regulatory role for Kruppel-like zinc-finger protein Gli-similar 1 (Glis1) in PMA-treated and psoriatic epidermis. J Invest Dermatol. 2006;126:49–60. doi: 10.1038/sj.jid.5700018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M, Tanese N, Ito M, Auerbach W, Bai C, Furukawa T, Toyono T, Akamine A, Joyner AL. A novel gene, GliH1, with homology to the Gli zinc finger domain not required for mouse development. Mech Dev. 2002;119:21–34. doi: 10.1016/s0925-4773(02)00291-5. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, Elia AE, Lu W, Brown EM, Quinn SJ, Ingber DE, Zhou J. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- Nauli SM, Zhou J. Polycystins and mechanosensation in renal and nodal cilia. Bioessays. 2004;26:844–856. doi: 10.1002/bies.20069. [DOI] [PubMed] [Google Scholar]
- Nishio S, Tian X, Gallagher AR, Yu Z, Patel V, Igarashi P, Somlo S. Loss of oriented cell division does not initiate cyst formation. J Am Soc Nephrol. 2010;21:295–302. doi: 10.1681/ASN.2009060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohneda K, Ee H, German M. Regulation of insulin gene transcription. Semin Cell Dev Biol. 2000a;11:227–233. doi: 10.1006/scdb.2000.0171. [DOI] [PubMed] [Google Scholar]
- Ohneda K, Mirmira RG, Wang J, Johnson JD, German MS. The homeodomain of PDX-1 mediates multiple protein-protein interactions in the formation of a transcriptional activation complex on the insulin promoter. Mol Cell Biol. 2000b;20:900–911. doi: 10.1128/mcb.20.3.900-911.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, Wright C. Pancreas organogenesis: from bud to plexus to gland. Dev Dyn. 2011;240:530–565. doi: 10.1002/dvdy.22584. [DOI] [PubMed] [Google Scholar]
- Park JI, Kim SW, Lyons JP, Ji H, Nguyen TT, Cho K, Barton MC, Deroo T, Vleminckx K, Moon RT, McCrea PD. Kaiso/p120-catenin and TCF/beta-catenin complexes coordinately regulate canonical Wnt gene targets. Dev Cell. 2005;8:843–854. doi: 10.1016/j.devcel.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Pearson R, Fleetwood J, Eaton S, Crossley M, Bao S. Kruppel-like transcription factors: a functional family. Int J Biochem Cell Biol. 2008;40:1996–2001. doi: 10.1016/j.biocel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- Peshavaria M, Henderson E, Sharma A, Wright CV, Stein R. Functional characterization of the transactivation properties of the PDX-1 homeodomain protein. Mol Cell Biol. 1997;17:3987–3996. doi: 10.1128/mcb.17.7.3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen HV, Serup P, Leonard J, Michelsen BK, Madsen OD. Transcriptional regulation of the human insulin gene is dependent on the homeodomain protein STF1/IPF1 acting through the CT boxes. Proc Natl Acad Sci U S A. 1994;91:10465–10469. doi: 10.1073/pnas.91.22.10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- Preiss A, Rosenberg UB, Kienlin A, Seifert E, Jackle H. Molecular genetics of Kruppel, a gene required for segmentation of the Drosophila embryo. Nature. 1985;313:27–32. doi: 10.1038/313027a0. [DOI] [PubMed] [Google Scholar]
- Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc Natl Acad Sci U S A. 2011;108:1456–1461. doi: 10.1073/pnas.1011410108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds AB. p120-catenin: Past and present. Biochim Biophys Acta. 2007;1773:2–7. doi: 10.1016/j.bbamcr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–316. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–25. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development. 1999;126:3205–3216. doi: 10.1242/dev.126.14.3205. [DOI] [PubMed] [Google Scholar]
- Ruppert JM, Kinzler KW, Wong AJ, Bigner SH, Kao FT, Law ML, Seuanez HN, O’Brien SJ, Vogelstein B. The GLI-Kruppel family of human genes. Mol Cell Biol. 1988;8:3104–3113. doi: 10.1128/mcb.8.8.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–3924. doi: 10.1242/dev.126.17.3915. [DOI] [PubMed] [Google Scholar]
- Schwartz EA, Leonard ML, Bizios R, Bowser SS. Analysis and modeling of the primary cilium bending response to fluid shear. Am J Physiol. 1997;272:F132–138. doi: 10.1152/ajprenal.1997.272.1.F132. [DOI] [PubMed] [Google Scholar]
- Senee V, Chelala C, Duchatelet S, Feng D, Blanc H, Cossec JC, Charon C, Nicolino M, Boileau P, Cavener DR, Bougneres P, Taha D, Julier C. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet. 2006;38:682–687. doi: 10.1038/ng1802. [DOI] [PubMed] [Google Scholar]
- Simms RJ, Hynes AM, Eley L, Sayer JA. Nephronophthisis: a genetically diverse ciliopathy. Int J Nephrol. 2011;2011:527137. doi: 10.4061/2011/527137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- Stanic D, Malmgren H, He H, Scott L, Aperia A, Hokfelt T. Developmental changes in frequency of the ciliary somatostatin receptor 3 protein. Brain Res. 2009;1249:101–112. doi: 10.1016/j.brainres.2008.10.024. [DOI] [PubMed] [Google Scholar]
- Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, Ericson J, Toftgard R, Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Taha D, Barbar M, Kanaan H, Williamson Balfe J. Neonatal diabetes mellitus, congenital hypothyroidism, hepatic fibrosis, polycystic kidneys, and congenital glaucoma: a new autosomal recessive syndrome? Am J Med Genet A. 2003;122A:269–273. doi: 10.1002/ajmg.a.20267. [DOI] [PubMed] [Google Scholar]
- Thomas MK, Lee JH, Rastalsky N, Habener JF. Hedgehog signaling regulation of homeodomain protein islet duodenum homeobox-1 expression in pancreatic beta-cells. Endocrinology. 2001;142:1033–1040. doi: 10.1210/endo.142.3.8007. [DOI] [PubMed] [Google Scholar]
- Thomas MK, Rastalsky N, Lee JH, Habener JF. Hedgehog signaling regulation of insulin production by pancreatic beta-cells. Diabetes. 2000;49:2039–2047. doi: 10.2337/diabetes.49.12.2039. [DOI] [PubMed] [Google Scholar]
- Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191:415–428. doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasanth S, Zeruth G, Kang HS, Jetten AM. Identification of Nuclear Localization, DNA Binding, and Transactivating Mechanisms of Kruppel-like Zinc Finger Protein Gli-Similar 2 (Glis2) J Biol Chem. 2011;286:4749–4759. doi: 10.1074/jbc.M110.165951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Sakai N, Matsushima K, Kaneko S. Fibrocytes: a new insight into kidney fibrosis. Kidney Int. 2007;72:269–273. doi: 10.1038/sj.ki.5002325. [DOI] [PubMed] [Google Scholar]
- Walker MD, Edlund T, Boulet AM, Rutter WJ. Cell-specific expression controlled by the 5′-flanking region of insulin and chymotrypsin genes. Nature. 1983;306:557–561. doi: 10.1038/306557a0. [DOI] [PubMed] [Google Scholar]
- Wang C, Pan Y, Wang B. Suppressor of fused and Spop regulate the stability, processing and function of Gli2 and Gli3 full-length activators but not their repressors. Development. 2010;137:2001–2009. doi: 10.1242/dev.052126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QT, Holmgren RA. The subcellular localization and activity of Drosophila cubitus interruptus are regulated at multiple levels. Development. 1999;126:5097–5106. doi: 10.1242/dev.126.22.5097. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Hiramatsu K, Miyamoto R, Yasuda K, Suzuki N, Oshima N, Kiyonari H, Shiba D, Nishio S, Mochizuki T, Yokoyama T, Maruyama S, Matsuo S, Wakamatsu Y, Hashimoto H. A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett. 2009;583:2108–2113. doi: 10.1016/j.febslet.2009.05.039. [DOI] [PubMed] [Google Scholar]
- Wilson PD. Mouse models of polycystic kidney disease. Curr Top Dev Biol. 2008;84:311–350. doi: 10.1016/S0070-2153(08)00606-6. [DOI] [PubMed] [Google Scholar]
- Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest. 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao K, Oas RG, Chiasson CM, Kowalczyk AP. Role of p120-catenin in cadherin trafficking. Biochim Biophys Acta. 2007;1773:8–16. doi: 10.1016/j.bbamcr.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Zhang S, Magenheimer BS, Luo J, Quarles LD. Polycystin-1 regulates skeletogenesis through stimulation of the osteoblast-specific transcription factor RUNX2-II. J Biol Chem. 2008;283:12624–12634. doi: 10.1074/jbc.M710407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao ZS, Quarles LD. Role of the polycytin-primary cilia complex in bone development and mechanosensing. Ann N Y Acad Sci. 2010;1192:410–421. doi: 10.1111/j.1749-6632.2009.05239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Chang BH, Samson SL, Li MV, Chan L. The Kruppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res. 2009;37:2529–2538. doi: 10.1093/nar/gkp122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18:1381–1388. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- Yusenko MV, Kovacs G. Identifying CD82 (KAI1) as a marker for human chromophobe renal cell carcinoma. Histopathology. 2009;55:687–695. doi: 10.1111/j.1365-2559.2009.03449.x. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Kalluri R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med. 2004;82:175–181. doi: 10.1007/s00109-003-0517-9. [DOI] [PubMed] [Google Scholar]
- Zeruth GT, Yang XP, Jetten AM. Modulation of the transactivation function and stability of Kruppel-like Zinc finger protein Gli-similar 3 (Glis3) by suppressor of fused. J Biol Chem. 2011;24:22077–22089. doi: 10.1074/jbc.M111.224964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, Kudo T, Engel JD, Yamamoto M, Takahashi S. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. 2005;25:4969–4976. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Jetten AM. Genomic structure of the gene encoding the human GLI-related, Kruppel-like zinc finger protein GLIS2. Gene. 2001;280:49–57. doi: 10.1016/s0378-1119(01)00764-8. [DOI] [PubMed] [Google Scholar]
- Zhang F, Nakanishi G, Kurebayashi S, Yoshino K, Perantoni A, Kim YS, Jetten AM. Characterization of Glis2, a novel gene encoding a Gli-related, Kruppel-like transcription factor with transactivation and repressor functions. Roles in kidney development and neurogenesis. J Biol Chem. 2002;277:10139–10149. doi: 10.1074/jbc.M108062200. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Law AC, Rajagopal J, Anderson WJ, Gray PA, Melton DA. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell. 2007;13:103–114. doi: 10.1016/j.devcel.2007.06.001. [DOI] [PubMed] [Google Scholar]