

Abstract
Multiple receptors within the innate immune system have evolved to recognize nucleic acids as signatures of viral infection. It is believed that this specificity is essential for viral detection, as viruses often lack other invariant features that can serve as suitable targets for innate receptors. One such innate receptor, TLR9, has been implicated in the detection of many dsDNA viruses. In this study, we investigate the detection of murine gammaherpesvirus 68 (MHV68) by TLR9. We find that the genomic DNA of the murine CMV, a very potent inducer of innate responses. Genome-wide analysis of the number of stimulatory versus nonstimulatory CpG motifs present in the genome of each virus reveals that the MHV68 genome contains only a fraction of the number of immunostimulatory motifs present in murine CMV. Notably, MHV68 appears to have selectively suppressed the number of stimulatory motifs through cytosine to thymine conversion. These data suggest that certain viruses may have evolved and modified their genomic content to avoid recognition by nucleic acid-sensing receptors of the innate immune system.
The innate immune response represents the first line of defense against pathogens. The activation of this response relies on the efficient recognition of conserved features of pathogens by a variety of innate immune receptors referred to as pattern recognition receptors (PRRs). Four families of PRRs are currently known, including TLRs, retinoic acid inducible gene I (RIG-I)-like receptors, nucleotide-binding oligomerization domain-like receptors, and HIN-200 family members (1, 2). PRRs link the recognition of viral and microbial ligands to activation of both the innate and adaptive immune system, thereby playing a vital role in the host response to infection (3, 4). Microbial ligands recognized by TLRs 1/2, 2/6, 4, and 5 represent biochemical products that are present in pathogens but absent from the host (e.g., LPS, peptidoglycan, β-glucans) (5). Viruses, however, because they replicate within our own cells, often lack the unique biochemical products that are typically targeted by these TLRs. Instead, the innate immune system uses the recognition of nucleic acids as one indicator of viral infection (6, 7). Distinct pathways recognizing viral nucleic acid have evolved in mammals. TLR3, TLR7, TLR8, and TLR9 detect various forms of viral nucleic acid in endosomal compartments (8). The DExD/H box helicases RIG-I, MDA-5, and LGP2 recognize viral RNA in the cytoplasm (8), and cytosolic DNA is sensed by receptors like DAI/ZBP1 (9, 10), AIM2 (11), RNA polymerase III (which generates an RNA intermediate that is sensed through RIG-I) (12, 13), as well as other as-yet unidentified receptors. These pathways all lead to induction of type I IFN (IFN-I) and/or inflammatory cytokines, yet their signal transduction pathways and their roles in antiviral immunity appear distinct (14, 15). The features of nucleic acid that are recognized vary for each receptor, and in some cases the precise sequence motifs or structural elements remain poorly defined. In recent years, various strategies by which viruses modulate innate immune responses have been identified (16).
TLR9 has been implicated in the recognition of different families of DNA viruses (17), including herpesviruses (18–25), poxviruses (26), adenoviruses (27), and most recently Torquetenoviruses (28). However, how this receptor discriminates between self and viral DNA is not yet fully understood. The localization of TLR9 in endolysosomes, vesicles normally devoid of self-nucleic acid but where viruses are degraded upon phagocytosis, is believed to account in part for this discrimination (29). TLR9 present in endolysosomes has undergone proteolytic cleavage of its ectodomain, and only the cleaved receptor is able to recruit its signaling adapter, MyD88 (30, 31). Additionally, TLR9 is thought to recognize unmethylated CpG dinucleotide motifs in DNA, a feature that is enriched in viruses but suppressed in host genomes (7, 17). The nature of the residues flanking the CpG motifs, as well as the methylation status of the cytosine, can also influence recognition by TLR9 (32).
An essential role for TLR9 has been demonstrated in the response to α-herpesviruses (HSV-1 and HSV-2) (18, 20, 23, 33) and betaherpesviruses (murine CMV; MCMV) (19, 21, 22, 24). Cells from TLR9-deficient mice are impaired in their ability to recognize these viruses, and TLR9-deficient mice are highly susceptible to MCMV infection (19, 21, 22). The importance of TLR9 in controlling gammaherpesvirus infection is less evident. In two recent studies with murine gammaherpesvirus 68 (MHV68), a mouse model for gammaherpesvirus infection, no acute replication defect was observed in MyD88−/− or TLR9−/− mice after intranasal infection (34, 35). However, viral titers in TLR9−/− mice were significantly increased after i.p. infection with MHV68. In TLR9−/− but not MyD88−/− mice, latent viral load was also only increased upon i.p. but unaffected after intranasal infection (34, 35). Surprisingly, latency establishment was reduced in MyD88−/− mice after intranasal infection, suggesting that TLR signaling is important rather than detrimental for these events (35). An understanding of innate responses to MHV68 is further complicated by conflicting data from in vitro dendritic cell (DC) infection studies. In general, MHV68 only infects immature DCs, the majority of which become latently infected (36, 37). Furthermore, GM-CSF–derived DCs, a homogeneous population with mostly CD11c+ CD11b+ conventional DCs (cDCs) (38), respond poorly when exposed to MHV68 as evident by the lack of cytokine secretion or upregulation of cell surface activation markers such as CD86 or major histocompatibility molecules (36, 37, 39, 40). Whether this is a result of an active immune evasion mechanism is still unclear. Previous studies have not distinguished between uninfected, lytically infected, or latently infected DCs when assessing whether MHV68 can prevent activation of infected cDCs after stimulation with TLR ligands (36, 39, 40). Notably, FLT3-ligand–derived DCs, which are a heterogeneous population composed of 70–80% CD11c+ CD11b+ cDCs and 20–30% CD11c+ B220+ plasmacytoid DCs (pDCs) (38), become activated when infected with MHV68, though IFN-I production is un-detectable (40). The authors conclude that MHV68-induced activation of cDCs requires the presence of pDCs and that the virus does not actively prevent activation of either pDCs or cDCs when stimulated with exogenous TLR ligands. The inability to detect IFN-I upon MHV68 infection in vitro or in vivo is particularly surprising given the fact that IFNR−/− mice are highly susceptible to infection with MHV68 (41, 42).
In this study, we show that viral immune evasion mechanisms, as well as inefficient recognition by TLR9, contribute to the poor innate immune response to MHV68.
Materials and Methods
Mice and viruses
C57BL/6 mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Experiments were conducted with 6- to 12-wk-old mice in accordance with institutional guidelines for animal care and use. MHV68 was obtained from the American Type Culture Collection (VR-1465) and propagated on BHK-21 cells. Viral titers were determined in a standard plaque assay on NIH 3T3 cells. BAC-derived Smith strain MCMV (MW97.01) was obtained from Dr. David Raulet (University of California, Berkeley) and was first described by Wagner and colleagues (43). MCMV was propagated and titered on NIH 3T3 cells. Viral titers reflect the titer of the stock at the time of the infection (i.e., after freeze-thawing). Virus containing supernatants were cleared of cellular debris by low-speed centrifugation for 20 min. Virus was subsequently concentrated by centrifugation at 24,000 × g for 2 h and purified over a 30% sucrose cushion by centrifugation at 85,000 × g. Pelleted virus was resuspended in PBS and stored at −80°C. BHK-21 and NIH 3T3 cells were grown in DMEM supplemented with 10% FBS and 100 units/ml penicillin and 100 μg/ml streptomycin. Mice were injected i. p. with MHV68 or MCMV (2 × 106 PFU per mouse) in 200-μl volumes and sera collected at the indicated time points. Intraperitoneal lavage was performed with 2 ml PBS per mouse.
Isolation and culture of DC
Bone marrow cells were isolated from the femurs and tibiae of C57BL/6 or MyD88−/− mice, and RBCs were removed by ammonium chloride lysis. To derive cDC, bone marrow cells were plated into 24-well plates at 7 × 105 cells per well in RPMI 1640 medium supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 100 μM MEM non-essential amino acids, 1 mM sodium pyruvate, 2 mM l-glutamine, 50 μM 2-mercaptoethanol, and GM-CSF for 7 d. Cells were fed every other day by adding 50% fresh medium containing GM-CSF. By day 7, cultures contained 70–80% CD11c+ CD11b+ cDCs (data not shown). To derive pDCs, bone marrow cells were plated into 24-well plates at 1 × 106 cells per well in RPMI 1640 medium supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 100 μM MEM nonessential amino acids, 1 mM sodium pyruvate, 2 mM l-glutamine, 50 μM 2-mercaptoethanol, and 25 ng/ml recombinant mouse FLT3-ligand (R&D) for 8 d. Cells were fed on day 4 by adding 50% fresh medium containing FLT3-ligand. By day 8, cultures contained 20–30% CD11c+ B220+ pDCs and 70–80% CD11c+ and CD11b+ cDCs (data not shown). GM-CSF–derived DCs and FLT3-ligand–derived DCs were plated into 96-well plates at 1 × 105 cells per well and infected with MCMV or MHV68 at the indicated multiplicities of infection (MOIs) and treated with phosphorothioate-linked CpG oligodeoxynucleotide (5′-TCCATGACGTTCCTGACGTT-3′) at 1 μM 2 h post infection.
Flow cytometry
GM-CSF DCs and FLT3-ligand DCs were plated into 24-well plates at 5 × 105 or 3 × 105 cells per well, respectively. DCs were infected with MHV68 at the indicated MOIs or MCMVat MOI 1 for 2 h and treated with TLR ligands (1μg/ml LPS, 1 μM phosphorothioate CpG oligodeoxynucleotide 5′-TCCATGACGTTCCTGACGTT-3′; 2 μg/ml R848) 2 h postinfection. Intracellular cytokine staining was performed using the BD Cytofix/Cytoperm Fixation/Permeabilization Solution Kit with BD GolgiPlug according to the manufacturer's instructions. Abs were obtained from BD Biosciences [CD11c clone HL3, CD11b clone M1/70, IL-12(p40/p70) clone C15.6] and eBioscience (TNF-α clone Mab11, B220 clone RA3-6B2). Data were collected on an LSR II flow cytometer (BD Bio-sciences) and analyzed using FlowJo software (Tree Star).
ELISA and IFN-I bioassay
IL-12p40 levels in sera and culture supernatants were measured by sandwich ELISA using anti–IL-12p40 Abs from eBioscience. IFN-I was measured in a bioassay using IFN-stimulated response element (ISRE) cells (44).
Briefly, ISRE cells were plated in 96-well plates at 5 × 104 cells per well. Mouse sera diluted in 1% BSA/PBS or culture supernatants were incubated with ISRE cells the following day for 6 to 8 h. Cells were lysed in 1× Passive Lysis Buffer (Promega) and luciferase activity measured using an LMAX II384 luminometer (Molecular Devices).
TLR9 reporter assay
HEK293 cells stably expressing TLR9N4C and an NF-κB responsive luciferase reporter construct were plated into 96-well plates at 4 × 104 cells per well. Cells were incubated with unmethylated or methylated viral genomic DNA or phosphodiester CpG oligodeoxynucleotides (5′-TCCNNRRCGYYNNTGATGCT-3′; N, any nucleotide; R, purine; Y, py-rimidine) at the indicated concentrations for 12 h. Viral genomic DNA was denatured for 15 min at 100°C and then immediately stored on ice prior to addition to TLR9N4C-HEK293 cells. Cells were lysed in 1× Passive Lysis Buffer (Promega) and luciferase activity measured using an LMAX II384 luminometer (Molecular Devices).
Isolation and methylation of viral genomic DNA
To obtain genomic DNA from viral particles, virus was concentrated and purified over a sucrose cushion as described earlier. The viral pellet was resuspended in PBS. An equal volume of 2× proteinase K buffer (20 mM Tris·Cl pH 8, 10 mM EDTA, 1% SDS, 100 μg/ml proteinase K) was added, and the mixture was incubated overnight at 50–60°C. Viral genomic DNA was extracted twice with phenol–chloroform and once with chloroform only. The DNA was precipitated with an equal volume of isopropanol, washed once with 70% ethanol, and resuspended in 10 mM Tris·Cl pH 8. The DNA was treated with RNase A at 25°C for 30 min and then again extracted with phenol–chloroform, precipitated with isopropanol, washed with ethanol, and resuspended in 10 mM Tris·Cl pH 8. Purified viral DNA was used for stimulation of TLR9N4C-HEK293 cells. Viral DNA was methylated using the bacterial methyltransferase M.SssI (NEB) according to the manufacturer's instructions. Methylated DNA was purified by phenol–chloroform extraction and isopropanol precipitation. Viral DNA from latent virus was copurified with genomic DNA from S11e cells, which are persistently but not exclusively latently infected with MHV68 (45). S11e cells were lysed in proteinase K buffer and genomic DNA extracted as described above for DNA from viral particles.
Methylation analysis
Viral DNA isolated from viral particles or latently infected cells was subjected to bisulfite treatment using the EZ DNA Methylation Kit (Zymo Research) according to the manufacturer's instructions. The following oligonucleotides were used to amplify a 307-bp region of the MHV68 genome (40242–40548): forward 5′-GAATATTGTTATTTTGGTTTAA-TGGA-3′ and reverse 5′-AAACCTTAAATTAAATTTTTCCCTCTAC-3′. The resulting PCR product was cloned using the PCR cloning Kit (Qiagen). Clones were analyzed by sequencing.
Statistical analysis
An unpaired, two-tailed Student t test was used to determine statistically significant differences.
Results
MHV68 is a poor inducer of IFN-I and IL-12p40 in vivo
DCs are some of the most prolific producers of IFN-I and IL-12 during the innate immune response and thus play a critical role in controlling virus replication as well as in shaping the adaptive antiviral immune response. Given the lack of DC activation and cytokine secretion previously reported in vitro and in vivo (36, 39, 40), we wanted to determine systemic cytokine responses to MHV68. As a positive control we used MCMV, a betaherpesvirus that elicits a robust innate immune response (22). C57BL/6 mice were infected i.p. with 2 × 106 PFU of cell culture-derived MCMV or MHV68. Serum levels of IL-12 and IFN-I were measured prior to and 36 and 72 h postinfection. As shown in Fig. 1, both cytokines were readily detectable in the sera of MCMV-infected mice. By contrast, IL-12 levels in the sera of MHV68-infected mice only slightly increased throughout the course of infection (Fig. 1A). Similar levels of serum IL-12p40 were observed previously upon intranasal infection with MHV68 (46). IFN-I in the sera of MHV68-infected mice was below the limit of detection in our bioassay (Fig. 1B). To determine if cytokine secretion could be observed locally at the site of infection, we measured IL-12p40 and IFN-I in i.p. lavage fluid of mice injected with PBS or infected with either MCMV or MHV68. Similar to systemic cytokine levels, IL-12p40 and IFN-I were readily detected in mice infected with MCMV but not in mice infected with MHV68 (Fig. 1C, 1D). This is in accordance with Weslow-Schmidt et al. (40) who were unable to detect IFN-I locally in bronchoalveolar lavage fluid after intranasal infection with MHV68. Importantly, IFN-I might still be secreted locally, at low levels, as mice lacking the IFN-I receptor are highly susceptible to MHV68 (41, 42).
Figure 1.

MHV68 is a poor inducer of innate cytokine responses in vivo. A and B, C57BL/6 mice were infected i.p. with 2 × 106 PFU of either MCMV or MHV68 (n = 6). Sera were obtained prior to infection as well as 36 and 72 h postinfection. Serum levels of IL-12p40 (A) and IFN-I (B) were determined by ELISA or ISRE bioassay, respectively. C and D, C57BL/6 mice were infected i.p. with 2 × 106 PFU of either MCMVor MHV68 (n = 5) or mock-infected with PBS (n = 3). Seventy-two hours postinfection, i.p. lavage with PBS was performed, and levels of IL-12p40 (C) and IFN-I (D) in the lavage were determined by ELISA or ISRE bioassay, respectively. The detection limits are 160 pg/ml for the IL-12p40 ELISA and 0.16 units/ml in the ISRE bioassay. Solid bars represent geometric means.
cDCs lytically infected with MHV68 suppress TLR-induced cytokine production
This apparent lack of activation in vivo was surprising as a previous study suggested that in the presence of pDCs, MHV68 can activate DCs and thus potentially initiate innate cytokine responses (40). We therefore decided to re-examine DC infections in vitro. Bone marrow cells were cultured in the presence of GM-CSF for 7 d to drive differentiation into cDCs (CD11c+, CD11b+). Both MCMV and MHV68 have previously been shown to replicate in cDCs with generation times greater than 24 h (39, 47). We therefore chose to measure IL-12 and IFN-I production in cDCs at 18 to 20 h postinfection, before progeny virions were released. As expected, MHV68 did not induce IL-12, whereas it was readily secreted upon stimulation with CpG or infection with MCMV (Fig. 2A). Importantly, the lack of IL-12 production by MyD88−/− cells (and MyD88/Toll-IL-1R domain-containing adaptor-inducing IFN-β [TRIF] DKO cells, data not shown) confirms that IL-12 in cDCs is produced in a TLR-dependent manner, suggesting that MHV68 may be able to inhibit TLR responses and/or is not recognized efficiently. Similarly to Weslow-Schmidt et al. (40), we were able to detect low levels of IFN-I in MHV68-infected cultures, albeit lower by at least one order of magnitude compared with that in MCMV-infected cultures (Fig. 2B). IFN-I production was not dependent on MyD88 (or TRIF, data not shown), suggesting that in cDCs intracellular nucleic acid sensors recognize MHV68. Activation of cytosolic nucleic acid sensors has been demonstrated to lead to IFN-I production in cDCs, whereas IFN-I production in response to TLR stimulation is predominately restricted to pDCs (48).
Figure 2.

MHV68 suppresses TLR signaling in cDCs. A and B, GM-CSF–derived DCs from wild-type (white bars) and MyD88−/− (black bars) mice were treated with CpG ODN or infected with MCMV (MOI 1) or MHV68 (indicated MOIs). Levels of IL-12p40 (A) and IFN-I (B) secreted into the supernatant were determined 20 h postinfection by ELISA or ISRE bioassay, respectively. C and D, GM-CSF–derived DCs were infected or not with MHV68 gM–eGFP at MOI 1 for 2 h and subsequently treated with LPS or CpG or left untreated. Cytokine production in infected cells was assessed by IL-12p40 ELISA of supernatants (C) or by intracellular cytokine staining for IL-12 (D, data shown is gated on CD11c+, CD11b+ cells). Percentages of IL-12+ cells in either infected (GFP+) or uninfected and latently infected (GFP−) cells after LPS and CpG stimulation are presented graphically on the left. Average and SD of triplicate wells are shown (data are representative of multiple independent experiments).
Previous studies have yielded conflicting results with regard to whether MHV68-infected cDCs are functionally altered (36, 39, 40). To determine whether MHV68 inhibits cytokine production in infected cDCs when exogenously stimulated with TLR ligands, we used an MHV68 reporter virus in which the endogenous gM protein is tagged with enhanced GFP (eGFP) on its C terminus. This virus can be used to detect lytically infected cDCs, which are marked by high GFP expression (37). GM-CSF–derived cDCs were infected or not with MHV68 gM–eGFP for 2 h. Uninfected and infected cells were then treated or not with either LPS or CpG. Cytokine production in cDCs was assessed by ELISA for IL-12p40 and by intracellular cytokine staining for IL-12 18 h postinfection. We observed inhibition of IL-12 induction in supernatants derived from infected cells that were treated with TLR ligands. Fig. 2C shows that the more cells were infected, the less IL-12 was produced when infected cells were treated with TLR ligands. As in Fig. 2A, neither lytically (GFP+) nor latently or uninfected (GFP−) cDCs produced IL-12 (Fig. 2D, bottom left). However, when infected cells were treated with LPS or CpG, a higher percentage of GFP− (latently infected or uninfected) cells produced IL-12 compared with lytically infected (GFP+) cells (Fig. 2D), suggesting that lytically infected cells may inhibit DC activation. Notably, we did observe about a 3-fold drop in lytic infection when cells were treated with TLR ligands. We speculate that in those cells in which lytic infection was not yet fully established when TLR ligands were added, the resulting DC activation and maturation halted progression to lytic infection, which may then have entered latency or been aborted altogether. Additionally, because MHV68 infects immature but not mature DCs (36, 37), infections that have not yet occurred within the 2-h window may have been prevented by the process of DC activation and maturation initiated upon TLR ligand addition. Altogether, our data confirm that MHV68 does not activate cDCs upon infection. Additionally, we show that lytically infected cells are capable of inhibiting TLR stimulation and thereby presumably activation that lytic infection itself causes in cDCs.
MHV68 neither replicates nor suppresses TLR signaling in pDCs
We next sought to re-examine TLR activation by MHV68 in pDCs, a specialized subset of DCs characterized by their ability to secrete large amount of IFN-I in response to TLR engagement by viral nucleic acid. FLT3-ligand DCs were treated with CpG or infected with either MCMV (MOI 1) or MHV68 at the indicated MOIs. Supernatants were recovered 20 h postinfection, and IFN-I was measured by bioassay or ELISA (data not shown). As before, the production of IFN-I by MHV68 was drastically reduced compared with that by MCMV (one to two orders of magnitude) (Fig. 3A). However, unlike in GM-CSF–derived cDCs, IFN-I induction by both viruses was severely diminished in MyD88-deficient cells (20- to 100-fold) and MyD88/TRIF-deficient cells (data not shown), suggesting that in FLT3-ligand DCs, IFN-I is predominately produced by the pDC population. The residual IFN-I observed in MyD88−/− FLT3-ligand DCs is likely secreted by the cDC population in these cultures owing to activation of cytosolic nucleic acid sensors in these cells (see Fig. 2B). Our observations indicate that compared with MCMV, MHV68 is a poor inducer of TLR-dependent responses in pDCs. Notably, MCMV does not replicate in pDCs (49, 50). To determine whether this is also the case for MHV68, we infected FLT3-ligand DCs with MHV68 gM– eGFP for 24 h and assessed infection by flow cytometry. As expected, cDCs (CD11c+, CD11b+) in FLT3-ligand DC cultures were lytically infected, whereas pDCs (CD11c+, B220+) did not seem to support lytic infection with MHV68 (Fig. 3B). Although we did not test this, it is likely that pDCs can be latently infected. Flaño et al. (39) were able to detect genome-positive cells in pDC populations isolated from mice 14 d and 3 mo postinfection. Because we are unable to detect latently infected pDCs, all subsequent experiments analyze whole populations of infected pDCs or FLT3-ligand DCs and will be referred to as “MHV68-exposed” to indicate this fact. Next, we wanted to understand whether MHV68-exposed pDCs are able to inhibit TLR stimulation. FLT3-ligand DCs were exposed to MHV68 for 2 h and subsequently treated with R848, a TLR7 ligand, for 7 h followed by intracellular cytokine staining for TNF-α. We found that TNF-α production did not diminish in pDCs (CD11c+, B220+) that were exposed to MHV68 and subsequently treated with R848 (Fig. 3C). We observed similar results in FLT3-ligand DCs that were exposed to MHV68 and subsequently treated with either CpG or infected with MCMV for 18 h (Fig. 3D). Unlike the decrease in IL-12p40 observed in the supernatants from MHV68-infected and TLR ligand-treated cDCs (Fig. 2D), TLR-dependent IFN-I in similarly treated FLT3-ligand DC cultures did not diminish. On the contrary, when MHV68-exposed cells were treated with CpG, we observed an additive increase in IFN-I production (Fig. 3D). Similar results have been reported previously (39, 40). Furthermore, treatment of MHV68-exposed FLT3-ligand DCs with the viral DNA polymerase inhibitor phosphonoacetic acid did not diminish cytokine production (data not shown).
Figure 3.

Lack of immunomodulation by MHV68 in pDCs. A, FLT3-ligand DCs from wild-type (white bars) and MyD88−/− (black bars) mice were treated with CpG ODN or infected with MCMV (MOI 1) or MHV68 (indicated MOIs). Levels of IFN-I secreted into the supernatant were determined 20 h postinfection by ISRE bioassay. B, FLT3-ligand DCs were infected or not with MHV68 gM–eGFP at MOI 1 for 24 h. Infection was assessed by flow cytometry. Data shown are gated on CD11c+, CD11b+ (cDCs) or CD11c+, B220+ (pDCs) cells. C, FLT3-ligand DCs were infected or not with MHV68 for 2 h and subsequently left untreated or treated with R848. Nine hours postinfection, TNF-α production was assessed by flow cytometry. Data shown are gated on CD11c+, B220+ cells. D, FLT3-ligand DCs were infected or not with MHV68 for 2 h and subsequently left untreated, treated with CpG, or infected with MCMV (MOI 1). Levels of IFN-I secreted into the supernatant were determined 20 h postinfection by ISRE bioassay. Average and SD of triplicate wells are shown (data are representative of multiple independent experiments).
The lack of robust IFN-I secretion by pDCs, a cell type that does not allow replication of MHV68, as well as the absence of inhibition upon exogenous TLR stimulation indicate that MHV68 may not actively interfere with TLR signaling in pDCs. Altogether, our data suggest that MHV68 is not efficiently recognized by TLRs. We predict a lack of recognition by TLR9 in particular, as it is the main TLR involved in the recognition of DNA viruses (17, 51).
The MHV68 genome poorly activates TLR9
We next sought to test the possibility that direct recognition of the MHV68 genome by TLR9 is impaired. We purified genomic DNA from MHV68 and MCMV viral particles and tested their ability to activate TLR9 using a TLR9N4C-HEK293 reporter cell line (29). These cells express a chimeric version of TLR9 (the extracellular domain of TLR9 fused to the transmembrane and intracellular domain of TLR4), which is localized to the cell surface, and encode a NF-κB–responsive luciferase reporter. Without this chimeric receptor, responses to extracellular genomic DNA are very difficult to detect. TLR9N4C-HEK293 cells were incubated with various concentrations of MHV68 or MCMV DNA, and TLR activation was measured using a luciferase reporter assay. As shown in Fig. 4, MHV68 genomic DNA stimulated the reporter cells poorly compared with that by MCMV DNA. As expected, methylation of MHV68 and MCMV genomes on CpG motifs, prior to exposure to TLR9N4C-HEK293 cells, drastically reduced activation. Previous studies have shown that gammaherpesviruses such as KSHV and EBV are methylated during latency but not during lytic replication (52–55). Therefore, DNA derived from viral particles is unlikely to be methylated. However, methylation of MHV68 DNA could explain the lack of efficient TLR9 activation. To test this hypothesis, we evaluated the methylation status of MHV68 genomic DNA isolated from viral particles. MHV68 DNA was treated with bisulfite, a reagent that converts unmethylated cytosines to uracil. Conversion of cytosine to uracil can then be detected by PCR and sequencing (cytosine to thymine). All cytosines present in the DNA sequence analyzed were converted to uracil, indicating that none of the original cytosines had been methylated (Table I). A similar experiment using MHV68 genomic DNA isolated from a cell line latently infected with MHV68 (45) showed that cytosines within CpG motifs were not converted upon bisulfite treatment and had thus been methylated (Table I). By contrast, cytosines outside CpG motifs were never methylated (data not shown). Altogether, these results indicate that, in contrast to MHV68 DNA extracted from latently infected cells, MHV68 DNA in viral particles is not methylated. Thus, the lack of TLR9 activation by MHV68 genomic DNA cannot be explained by the presence of methylation. This suggests that the primary sequence of the MHV68 genome itself is a poor TLR9 activator.
Figure 4.

MHV68 genomic DNA stimulates TLR9N4C-HEK293 cells poorly. TLR9N4C-HEK293 cells were incubated with increasing amounts of unmethylated or methylated viral genomic DNA from either MCMV or MHV68, and luciferase activity was measured 12 h later. Viral DNA was denatured prior to addition to the cells. Average and SD of triplicate wells are shown (data are representative of at least three independent experiments).
Table I. The MHV68 genome is methylated only during latency.
| Frequency of Methylation | ||
|---|---|---|
|
|
||
| CpG Motif (40242–40548)a | DNA from Lytic Virus (%) | DNA from Latently Infected Cells (%) |
| GACGCA | 0 | 75 |
| CCCGTG | 0 | 100 |
| GGCGTT | 0 | 75 |
| GGCGCT | 0 | 100 |
| AACGCT | 0 | 100 |
| ATCGCC | 0 | 25 |
A short sequence of the MHV68 genome corresponding to position 40242–40548 was subjected to bisulfite-based meth-ylation analysis. Percentages indicate how often a particular cytosine in a CpG dinucleotide was found to have been methylated in viral DNA isolated from either viral particles or latently infected S11e cells. Cytosines outside CpG dinucleotides were never methylated.
Boldface indicates the CpG dinucleotide that is invariant among all the motifs.
The MHV68 genome has few TLR9 stimulatory motifs
TLR9 is believed to recognize CpG dinucleotide motifs in ssDNA. The nature of the residues flanking CpG motifs has been shown to influence recognition by TLR9 (32, 56). However, mapping these residues has proved to be difficult mainly because natural oligodeoxynucleotides (ODN) are highly sensitive to DNases (which cleave phosphodiester bonds between adjacent nucleotides). Chemically modified phosphorothioate-linked ODN alleviate most of these problems. An optimal motif recognized by murine TLR9 (A/G-A/G-CpG-C/T-C/T) has been defined using these modified oligonucleotides (56). Consequently, the consensus sequence, defined as the best activator of TLR9 using chemically modified ODN, might not be the same for natural DNA. The precise sequence motifs or structural elements that are recognized by TLR9 in natural DNA remain poorly defined. We thus decided to establish a hierarchy of motifs based on their ability to stimulate the TLR9N4C-HEK293 reporter cells described above. Phosphodiester-linked ODN comprising all 16 possible variations of the A/G-A/G-CpG-C/T-C/T murine TLR9 stimulatory motifs were synthesized, and their ability to activate TLR9 was measured using the TLR9N4C-HEK293 reporter cells as before. As shown in Fig. 5A, these ODN exhibited a broad range of stimulatory capacity. The CpG ODN encoding the GACGTT context, which was previously described to be the optimal motif for murine TLR9 (57), is one of the most stimulatory motifs in our screen as well. Similar results were obtained with TLR9-HEK293 reporter cells (data not shown). However, a much higher oligonucleotide concentration was required, presumably due to inefficient uptake of the DNA.
Figure 5.

Suppression of stimulatory CpG motifs in the MHV68 and MCMV genomes. A, TLR9N4C-HEK293 cells were incubated with 16 distinct CpG ODN (1 μM) and luciferase activity measured 12 h later. Average and SD of triplicate wells are shown (data are representative of at least three independent experiments). B, Number of specific CpG motifs in either the MHV68 (black) or MCMV (gray) genome. Motifs are ordered as in A with most stimulatory motif on the left and least stimulatory motif on the right. C, Data as in A shown on primary y-axis (white bars) and frequency of specific CpG motifs in either the MHV68 (left) or MCMV (right) genome shown on secondary y-axis (black bars).
We next determined the number and frequency of each of the 16 motifs in the MHV68 and MCMV genomes. Fig. 5B and 5C display the stimulatory capacity of the 16 phosphodiester ODN from highest to lowest (white bars) on the primary y-axis and contrasts this with the either the number or frequency of each motif within either the MHV68 or MCMV genome (Fig. 5B, 5C). The genome sizes of MHV68 and MCMV are 120 kb and 230 kb, respectively. Strikingly, the total number of all 16 tested motifs as well as the 2 most stimulatory motifs (AACGTT and GACGTT) is 8 times lower (372 and 23) in the MHV68 genome compared with that of MCMV (2898 and 188) (Fig. 5B), thus correlating with the differences in the ability of these two viral genomes to stimulate TLR9. Importantly, in both viral genomes, the frequency of stimulatory motifs was reduced compared with that of the non-stimulatory motifs (Fig. 5C), suggesting that both viruses have suppressed the frequency of stimulatory motifs and possibly restricted their ability to stimulate TLR9.
Selective loss of stimulatory sequences in the MHV68 genome by cytosine to thymine conversion
The low overall CpG content within the genomes of gamma-herpesviruses is well documented and is believed to be largely the result of deamination, the spontaneous conversion of a 5-methyl cytosine to thymine (58). As a result, the dinucleotide TpG is overrepresented in comparison with the CpG dinucleotide in these genomes (58). Spontaneous deamination of methylated cytosines would occur at random throughout the genome and be subject to DNA repair. However, if 5-methyl cytosine to thymine conversions are not repaired, they have the potential to be manifested within the genome if the mutant virus was positively selected for. Deamination of methylated cytosines within TLR9 stimulatory sequences might present such a selective advantage. We calculated the TG/CG motif ratio for each of the 16 CpG motifs within MHV68 and MCMV genomes. In the MCMV genome, TG/CG ratios were essentially the same among all the 16 motifs, with a value close to one (Fig. 6A). However, comparing TG/CG ratios between stimulatory and nonstimulatory sequences in the MHV68 genome, we found that cytosine to thymine conversions were strikingly overrepresented among the stimulatory motifs (Fig. 6B), suggesting that positive selection of mutated TLR9-immuno-stimulatory motifs may have occurred in MHV68.
Figure 6.
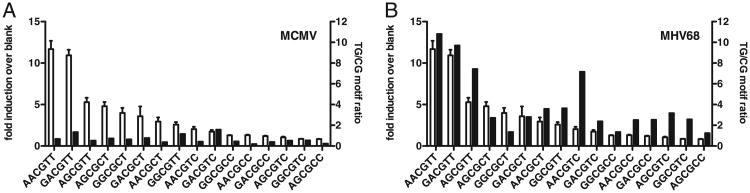
Deamination of stimulatory CpG motifs in MHV68 but not MCMV. A, MCMV. B, MHV68. Data from Fig. 5A (white bars) shown on the primary y-axis. Data on the secondary y-axis (black bars) shows the ratio of TpG motif frequency (CpG motif in which CG is replaced by TG) over corresponding CpG motif frequency.
Discussion
In this study, we show that TLR9 stimulatory motifs are underrepresented in the MHV68 genome in part due to deamination. The consequence of such suppression is magnified in MHV68 as it already has a low CpG content. As a result, DCs are poorly activated by MHV68, and the secretion of TLR-dependent cytokines is impaired. Furthermore, MHV68 uses active mechanisms to suppress innate cytokine responses in lytically infected DCs.
Together with previous studies, our data furthers our understanding of MHV68–DC interactions and innate immune responses to this gammaherpesvirus. Previous research has shown that MHV68 infects immature but not mature cDCs and that the majority of cDCs are latently infected (36, 37). Infection of cDCs with MHV68 does not lead to activation and maturation (36, 39, 40). However, whether this is due to an active immune evasion mechanism remains controversial. To address this question, we infected cDCs with MHV68 gM–eGFP, in which high GFP expression is restricted to lytically infected cells (37), and showed that only lytically infected cells were able to suppress TLR-mediated cytokine responses (Fig. 2C). We were not able to distinguish latently infected from uninfected cells. However, based on Smith et al. (37), the majority of cDCs in our cultures should be latently infected. The frequency of IL-12 producing, GFP− cells, which comprise uninfected as well as latently infected cells, treated with TLR ligands did not significantly diminish, suggesting that latently infected cells do not use an immune evasion strategy. This may be expected given the restricted gene expression profile during latency. Notably, MHV68-infected cDCs secreted IFN-I, likely due to intracellular nucleic acid sensor recognition of the virus, as this response was entirely MyD88/TRIF-independent (Fig. 2B). This is in agreement with a previous study showing that the 100-bp repeat region of MHV68 induces IFN-I when delivered into the cytosol (59). Furthermore, we observed a decrease in the amount of IFN-I secreted as the infectious dose of MHV68 increased (Fig. 2B), suggesting an active mechanism to suppress IFN-I responses could be involved. Although we are not aware of any proteins encoded by herpes-viruses that directly prevent TLR activation, some proteins interfere with the IFN-I response. This is the case for three MHV68 gene products, M2, ORF36, and possibly ORF45 (59–61). To what extent these MHV68 gene products contribute to IFN-I suppression during viral infection in cDCs is unknown and in some cases difficult to assess because a number of these genes are also essential for viral replication.
pDCs are activated after MHV68 infection and are necessary for activation of MHV68-infected cDCs (40). We also observed TLR-dependent IFN-I production in FLT3-ligand DCs, which comprise cDCs and pDCs, upon MHV68 infection (Fig. 3A). As IFN-I secretion by pDCs but not cDCs is largely TLR dependent, we speculate that the IFN-I observed in FLT3-ligand DCs is produced by activated pDC. However, the amount of IFN-I secreted in response to MHV68 is moderate at best compared with that in response to MCMV (50-fold higher), a very potent inducer of IFN-I (19, 21, 22, 24). Unlike cDCs, pDCs do not seem to support lytic infection with MHV68 (Fig. 2B). However, latent infection is likely, as it has previously been observed in vivo (39). We found that similar to uninfected and latently infected cDCs, pDCs do not inhibit TLR-mediated signaling when MHV68-exposed cells are exogenously stimulated with R848 or CpG (Fig. 3C, 3D). Treatment of MHV68-exposed pDCs with PAA, which inhibits herpesviral replication (62), did not affect IFN-I secretion (data not shown), further supporting the idea that MHV68 may not replicate in pDCs or actively interfere with its recognition.
IFN-I is a crucial cytokine in the defense against many viral infections including MHV68. Although the level of IFN-I in the serum of MHV68-infected mice is below detection level (Fig. 1B), IFN-I clearly plays a role as evidenced by the fact that mice deficient for the IFN-I receptor succumb rapidly to MHV68 infection (41, 42). The cell type mediating this IFN-I response in vivo is still unknown. Our observations show that both cDCs and pDCs are capable of producing IFN-I in response to MHV68 infection in vitro, albeit at very low levels.
Our in vitro data indicate that MHV68 uses active mechanisms to inhibit TLR signaling in lytically infected cells. However, these mechanisms appeared to be absent in latently infected cells, raising the question of why DC activation and cytokine secretion cannot be observed in latently infected cells. We suspected that in addition to immune evasion mechanisms, MHV68 might not be well recognized. Of all currently identified receptors, the PRR that is most likely playing a role in the recognition of MHV68 is TLR9. Does TLR9 participate at all in the control of MHV68 infection in vivo? We clearly showed that MHV68 genomic DNA is a weak activator of TLR9 (Fig. 4). Nevertheless, pDCs produce low levels of IFN-I in response to MHV68, and this production is dependent on MyD88, suggesting that TLR9 is capable of detecting MHV68 genomic DNA in these cells. Mice deficient for TLR9 or MyD88 do not show increased viral replication during acute replication after intranasal infection (34, 35), suggesting that TLR9 is not important under these conditions. However, viral loads were increased 10-fold in TLR9-deficient mice compared with that in wild-type mice when MHV68 was injected i.p. (34). Strikingly, in similar experiments performed with MCMV, viral titers increased three orders of magnitude (21). This is consistent with our observation that MCMV genomic DNA was three times as stimulatory as MHV68 genomic DNA when incubated with TLR9N4C-HEK293 cells. The question remains why differences between intranasal and i.p. infection with MHV68 in wild-type and TLR9−/− mice were observed. Intranasal infection is characteristic for herpesviral transmission (63) and is believed to be the natural route of infection. In addition, previous studies have shown that lung DCs express much lower levels of TLR9 compared with those of splenic DCs (64), the latter of which likely interact with MHV68 during i.p. infection. Thus, suppression of TLR9 immunostimulatory motifs in MHV68 masks infection and makes this virus uniquely adapted to the intranasal route of infection. Additionally, splenic DCs respond well to TLR9 stimulation, and because MHV68 is not as well adapted to the i.p. route of infection, the degree of suppression in TLR9 immunostimulatory motifs may simply not be sufficient to evade recognition by the innate immune system. The discrepancies between i.p. infection in TLR9−/− and MyD88−/− mice, the latter of which show no increase in latently infected cells and a dramatic reduction in reactivation from latency, remain unexplained and require further analysis.
Our studies did not address the involvement of other TLRs that have been implicated in the recognition of large dsDNA viruses. TLR 3 and 7, which sense dsRNA and ssRNA, respectively, contribute to the innate response to MCMV infection in addition to TLR9 (21, 24). However, TLR3 is dispensable during MHV68 infection (35). TLR2, which was initially found to recognize microbial lipopeptides, has since been shown to play a role in various viral infections including those with herpes simplex viruses types 1 and 2 (65, 66), varicella-zoster virus (67, 68), human CMV (69, 70), MCMV (71), vaccinia virus (72, 73), measles virus (74), hepatitis C virus (75), and lymphocytic choriomeningitis virus (76). Most recently, Michaud et al. (77) demonstrated that TLR2-deficient mice have elevated viral titers and reduced cytokine levels during acute infection with MHV68. Thus, TLRs other than TLR9 contribute to innate immune responses against MHV68.
Surprisingly, we observed that both MCMV and MHV68 have lower frequencies of highly stimulatory CpG motifs in their genomes, though only MHV68 DNA is able to evade TLR9 recognition efficiently. Although suppression of TLR9 stimulatory motifs might have been driven by TLR9 evasion in both viruses, it may have been sufficient for MCMV to eliminate only a few TLR9 stimulatory motifs to maintain a successful infection. Alternatively, other evolutionary forces may have contributed to the suppression of these stimulatory CpG motifs. Herpesviral genomes were shown to be methylated during some stages of the viral life cycle, in part to control viral gene expression (52–54). It is thus conceivable that viruses may have evolved to mutate certain CpG motifs within their genomes during evolution to allow for more targeted gene expression. Though currently unknown, cellular methyltransferases may be more specific for particular CpG contexts in order for the host to methylate preferentially TLR9 stimulatory CpG motifs in its own genome to avoid self-recognition by TLR9. Consequently, elimination of methylated sites in viral genomes through deamination would thus specifically remove TLR9 stimulatory motifs. Regardless of the means by which suppression of TLR9 stimulatory motifs was achieved, only gammaherpesviruses but not alphaherpesviruses or betaherpesviruses are generally CpG suppressed (58). As a result, the additional reduction of TLR9 stimulatory motifs renders MHV68 almost invisible to TLR9 recognition. One prediction from our observation is that TLR9 recognition of MHV68 may be improved by increasing the stimulatory CpG content in the viral genome. To this end, we introduced a 1.4-kb sequence containing 54 stimulatory CpG motifs into the MHV68 genome by BAC recombination, increasing the overall stimulatory CpG content of the virus 2-fold. However, it has to date been insufficient to increase innate immune recognition by this mutant virus (data not shown). Several reasons may account for these observations. First, it is possible that other as of yet uncharacterized stimulatory CpG motifs are present in both the MCMV and MHV68 genome, in which case a less than 2-fold increase in stimulatory CpG motifs would have been achieved. Thus, the level of CpG enrichment in our mutant CpG-MHV68 virus may simply not be sufficient to increase recognition by TLR9. Furthermore, the stimulatory CpG motifs were introduced in one continuous sequence. Because of the limited understanding of TLR9–DNA interaction, it is unknown whether this is would lead to TLR9 stimulation in the context of a large viral genome or in the context of a viral infection. It may be preferable to distribute stimulatory CpG motifs throughout the MHV68 genome. However, the genome size and the limitations of BAC mutagenesis make this approach not feasible at this time. During the revision of the manuscript for this article, a report described the loss of CpG motifs in U-rich contexts from certain influenza strains. The authors speculate that loss of these motifs leads to reduced TLR7-dependent IFN-α production. Notably, CpG-enriched influenza induced higher levels of IFN-α in pDCs compared with their CpG-low counterpart (78).
Differences in viral life cycles could play an important role in avoiding TLR9 recognition. Notably, virtually all small eukaryotic viruses are CpG suppressed (58). Among those with the lowest CpG content are polyomaviruses and papillomaviruses whose life cycles are characterized by persistent infections (58). Viral persistence is also a hallmark of herpesviruses. How might persistence or latency select for viruses that can more effectively evade innate recognition? It is unlikely that selection occurs during acute viral infection simply due to the vast amount of virus produced. Of note, it has previously been shown that establishment and maintenance of latency are independent of infectious dose and route of infection for MHV68 (79). Furthermore, several deletions in MHV68 genes important for acute replication did not affect establishment of latency (80–82). However, reactivation events during persistent infection are rare, rendering reactivating cells an easier target for the immune system. For successful dissemination, reactivation of persistent viruses needs to go unnoticed. Viruses that have evolved to reduce stimulation of innate PRRs like TLR9 may be able to spread more successfully and could thus be positively selected. Further studies are necessary to link definitively viral persistence with TLR9 evasion. Notably, evasion of adaptive immune responses during reactivation has previously been suggested to be important for virus spreading (83, 84).
There are several known examples of pathogens avoiding TLR recognition by modifying TLR ligands. Several human pathogens, such as Salmonella typhimurium and Yersinia pestis, produce modified lipid A moieties (the major signaling component of LPS) that are poorly recognized by TLR4 (85). To our knowledge, this strategy has not yet been described for viruses. Of note, modification of the herpesvirus genome as a mechanism of immune evasion has recently been documented. During latency, EBV expresses the EBNA-1 protein. Tellam et al. (86) showed that the coding sequence of EBNA-1 is enriched in purine residues in the third base wobble position. As a consequence, EBNA-1 is translated with a low efficiency, thus reducing the number of EBNA-1– derived peptides that can be displayed at the surface of an infected cell for recognition by CTL (86). Together with the work of Tellam et al., our study highlights the fact that during evolution, herpesviruses have modified their genomes to minimize immune recognition.
Acknowledgments
We acknowledge the Center for Host-Pathogen Studies Mouse/Virus core for providing mouse strains and viral stocks. We thank Laura Lau, Nicholas Arpaia, and Roman Barbalat for technical assistance and members of the Coscoy and Barton laboratories and Nadine Jarrousse for helpful discussions. MHV68 gM–eGFP was a generous gift from P.G. Stevenson.
This work was supported by National Institutes of Health grants (to L.C.).
Abbreviations used in this article
- cDC
conventional DC
- DC
dendritic cell
- eGFP
enhanced GFP
- IFN-I
type I IFN
- ISRE
IFN-stimulated response element
- MCMV
murine CMV
- MHV68
murine gammaherpesvirus 68
- MOI
multiplicity of infection
- ODN
oligodeoxynucleotide
- pDC
plasmacytoid DC
- PRR
pattern recognition receptor
- RIG-I
retinoic acid inducible gene I
- TRIF
Toll-IL-1R domain-containing adaptor-inducing IFN-β
Footnotes
Disclosures: The authors have no financial conflicts of interest.
References
- 1.O'Neill LA, Bowie AG. Sensing and signaling in antiviral innate immunity. Curr Biol. 2010;20:R328–R333. doi: 10.1016/j.cub.2010.01.044. [DOI] [PubMed] [Google Scholar]
- 2.Carpenter S, O'Neill LAJ. How important are Toll-like receptors for antimicrobial responses? Cell Microbiol. 2007;9:1891–1901. doi: 10.1111/j.1462-5822.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 3.Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2:947–950. doi: 10.1038/ni712. [DOI] [PubMed] [Google Scholar]
- 4.Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Microbes Infect. 2004;6:1382–1387. doi: 10.1016/j.micinf.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 5.Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- 6.Kawai T, Akira S. Innate immune recognition of viral infection. Nat Immunol. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- 7.Barton GM. Viral recognition by Toll-like receptors. Semin Immunol. 2007;19:33–40. doi: 10.1016/j.smim.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Koyama S, Ishii KJ, Coban C, Akira S. Innate immune response to viral infection. Cytokine. 2008;43:336–341. doi: 10.1016/j.cyto.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Takaoka A, Wang Z, Choi M, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 10.Wang Z, Choi MK, Ban T, Yanai H, Negishi H, Lu Y, Tamura T, Takaoka A, Nishikura K, Taniguchi T. Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc Natl Acad Sci USA. 2008;105:5477–5482. doi: 10.1073/pnas.0801295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat Immunol. 2009;10:1065–1072. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiu YH, Macmillan JB, Chen ZJ. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, Tsujimura T, Takeda K, Fujita T, Takeuchi O, Akira S. Cell typespecific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 15.Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–381. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Yokota S, Okabayashi T, Fujii N. The battle between virus and host: modulation of Toll-like receptor signaling pathways by virus infection. Mediators Inflamm. 20102010:184328. doi: 10.1155/2010/184328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumagai Y, Takeuchi O, Akira S. TLR9 as a key receptor for the recognition of DNA. Adv Drug Deliv Rev. 2008;60:795–804. doi: 10.1016/j.addr.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 18.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198:513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 20.Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004;103:1433–1437. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- 21.Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. 2005;175:6723–6732. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- 23.Lund JM, Linehan MM, Iijima N, Iwasaki A. Cutting Edge: Plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J Immunol. 2006;177:7510–7514. doi: 10.4049/jimmunol.177.11.7510. [DOI] [PubMed] [Google Scholar]
- 24.Zucchini N, Bessou G, Traub S, Robbins SH, Uematsu S, Akira S, Alexopoulou L, Dalod M. Cutting edge: Overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J Immunol. 2008;180:5799–5803. doi: 10.4049/jimmunol.180.9.5799. [DOI] [PubMed] [Google Scholar]
- 25.Fiola S, Gosselin D, Takada K, Gosselin J. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J Immunol. 2010;185:3620–3631. doi: 10.4049/jimmunol.0903736. [DOI] [PubMed] [Google Scholar]
- 26.Samuelsson C, Hausmann J, Lauterbach H, Schmidt M, Akira S, Wagner H, Chaplin P, Suter M, O'Keeffe M, Hochrein H. Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. J Clin Invest. 2008;118:1776–1784. doi: 10.1172/JCI33940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu J, Huang X, Yang Y. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J Virol. 2007;81:3170–3180. doi: 10.1128/JVI.02192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rocchi J, Ricci V, Albani M, Lanini L, Andreoli E, Macera L, Pistello M, Ceccherini-Nelli L, Bendinelli M, Maggi F. Torquetenovirus DNA drives proinflammatory cytokines production and secretion by immune cells via toll-like receptor 9. Virology. 2009;394:235–242. doi: 10.1016/j.virol.2009.08.036. [DOI] [PubMed] [Google Scholar]
- 29.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nat Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 30.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol. 2008;9:1407–1414. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 33.Hochrein H, Schlatter B, O'Keeffe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Suter M, Wagner H. Herpes simplex virus type-1 induces IFN-alpha production via Toll-like receptor 9-dependent and -independent pathways. Proc Natl Acad Sci USA. 2004;101:11416–11421. doi: 10.1073/pnas.0403555101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guggemoos S, Hangel D, Hamm S, Heit A, Bauer S, Adler H. TLR9 contributes to antiviral immunity during gammaherpesvirus infection. J Immunol. 2008;180:438–443. doi: 10.4049/jimmunol.180.1.438. [DOI] [PubMed] [Google Scholar]
- 35.Gargano LM, Moser JM, Speck SH. Role for MyD88 signaling in murine gammaherpesvirus 68 latency. J Virol. 2008;82:3853–3863. doi: 10.1128/JVI.02577-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hochreiter R, Ptaschinski C, Kunkel SL, Rochford R. Murine gammaherpesvirus-68 productively infects immature dendritic cells and blocks maturation. J Gen Virol. 2007;88:1896–1905. doi: 10.1099/vir.0.82931-0. [DOI] [PubMed] [Google Scholar]
- 37.Smith CM, Gill MB, May JS, Stevenson PG. Murine gammaherpesvirus-68 inhibits antigen presentation by dendritic cells. PLoS ONE. 2007;2:e1048. doi: 10.1371/journal.pone.0001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brawand P, Fitzpatrick DR, Greenfield BW, Brasel K, Maliszewski CR, De Smedt T. Murine plasmacytoid pre-dendritic cells generated from Flt3 ligand-supplemented bone marrow cultures are immature APCs. J Immunol. 2002;169:6711–6719. doi: 10.4049/jimmunol.169.12.6711. [DOI] [PubMed] [Google Scholar]
- 39.Flaño E, Kayhan B, Woodland DL, Blackman MA. Infection of dendritic cells by a gamma2-herpesvirus induces functional modulation. J Immunol. 2005;175:3225–3234. doi: 10.4049/jimmunol.175.5.3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weslow-Schmidt JL, Jewell NA, Mertz SE, Simas JP, Durbin JE, Flaño E. Type I interferon inhibition and dendritic cell activation during gammaherpesvirus respiratory infection. J Virol. 2007;81:9778–9789. doi: 10.1128/JVI.00360-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dutia BM, Allen DJ, Dyson H, Nash AA. Type I interferons and IRF-1 play a critical role in the control of a gammaherpesvirus infection. Virology. 1999;261:173–179. doi: 10.1006/viro.1999.9834. [DOI] [PubMed] [Google Scholar]
- 42.Barton ES, Lutzke ML, Rochford R, Virgin HW., IV Alpha/beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. J Virol. 2005;79:14149–14160. doi: 10.1128/JVI.79.22.14149-14160.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wagner M, Jonjic S, Koszinowski UH, Messerle M. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol. 1999;73:7056–7060. doi: 10.1128/jvi.73.8.7056-7060.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, et al. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 45.Usherwood EJ, Stewart JP, Nash AA. Characterization of tumor cell lines derived from murine gammaherpesvirus-68-infected mice. J Virol. 1996;70:6516–6518. doi: 10.1128/jvi.70.9.6516-6518.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elsawa SF, Bost KL. Murine gamma-herpesvirus-68-induced IL-12 contributes to the control of latent viral burden, but also contributes to viral-mediated leukocytosis. J Immunol. 2004;172:516–524. doi: 10.4049/jimmunol.172.1.516. [DOI] [PubMed] [Google Scholar]
- 47.Andrews DM, Andoniou CE, Granucci F, Ricciardi-Castagnoli P, Degli-Esposti MA. Infection of dendritic cells by murine cytomegalo-virus induces functional paralysis. Nat Immunol. 2001;2:1077–1084. doi: 10.1038/ni724. [DOI] [PubMed] [Google Scholar]
- 48.Takeuchi O, Akira S. Recognition of viruses by innate immunity. Immunol Rev. 2007;220:214–224. doi: 10.1111/j.1600-065X.2007.00562.x. [DOI] [PubMed] [Google Scholar]
- 49.Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med. 2003;197:885–898. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bozza S, Bistoni F, Gaziano R, Pitzurra L, Zelante T, Bonifazi P, Perruccio K, Bellocchio S, Neri M, Iorio AM, et al. Pentraxin 3 protects from MCMV infection and reactivation through TLR sensing pathways leading to IRF3 activation. Blood. 2006;108:3387–3396. doi: 10.1182/blood-2006-03-009266. [DOI] [PubMed] [Google Scholar]
- 51.Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem. 2007;141:137–145. doi: 10.1093/jb/mvm032. [DOI] [PubMed] [Google Scholar]
- 52.Chen J, Ueda K, Sakakibara S, Okuno T, Parravicini C, Corbellino M, Yamanishi K. Activation of latent Kaposi's sarcoma-associated herpes-virus by demethylation of the promoter of the lytic transactivator. Proc Natl Acad Sci USA. 2001;98:4119–4124. doi: 10.1073/pnas.051004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tao Q, Robertson KD. Stealth technology: how Epstein-Barr virus utilizes DNA methylation to cloak itself from immune detection. Clin Immunol. 2003;109:53–63. doi: 10.1016/s1521-6616(03)00198-0. [DOI] [PubMed] [Google Scholar]
- 54.Minarovits J. Epigenotypes of latent herpesvirus genomes. Curr Top Microbiol Immunol. 2006;310:61–80. doi: 10.1007/3-540-31181-5_5. [DOI] [PubMed] [Google Scholar]
- 55.Honess RW, Gompels UA, Barrell BG, Craxton M, Cameron KR, Staden R, Chang YN, Hayward GS. Deviations from expected frequencies of CpG dinucleotides in herpesvirus DNAs may be diagnostic of differences in the states of their latent genomes. J Gen Virol. 1989;70:837–855. doi: 10.1099/0022-1317-70-4-837. [DOI] [PubMed] [Google Scholar]
- 56.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 57.Bauer S, Kirschning CJ, Ha¨cker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc Natl Acad Sci USA. 2001;98:9237–9242. doi: 10.1073/pnas.161293498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karlin S, Doerfler W, Cardon LR. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J Virol. 1994;68:2889–2897. doi: 10.1128/jvi.68.5.2889-2897.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sanchez DJ, Miranda D, Jr, Arumugaswami V, Hwang S, Singer AE, Senaati A, Shahangian A, Song MJ, Sun R, Cheng G. A repetitive region of gammaherpesvirus genomic DNA is a ligand for induction of type I interferon. J Virol. 2008;82:2208–2217. doi: 10.1128/JVI.01718-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liang X, Shin YC, Means RE, Jung JU. Inhibition of interferon-mediated antiviral activity by murine gammaherpesvirus 68 latency-associated M2 protein. J Virol. 2004;78:12416–12427. doi: 10.1128/JVI.78.22.12416-12427.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hwang S, Kim KS, Flano E, Wu TT, Tong LM, Park AN, Song MJ, Sanchez DJ, O'Connell RM, Cheng G, Sun R. Conserved her-pesviral kinase promotes viral persistence by inhibiting the IRF-3-mediated type I interferon response. Cell Host Microbe. 2009;5:166–178. doi: 10.1016/j.chom.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang ES, Huang CH, Huong SM, Selgrade M. Preferential inhibition of herpes-group viruses by phosphonoacetic acid: effect on virus DNA synthesis and virus-induced DNA polymerase activity. Yale J Biol Med. 1976;49:93–99. [PMC free article] [PubMed] [Google Scholar]
- 63.Stevenson PG, Efstathiou S. Immune mechanisms in murine gammaherpesvirus-68 infection. Viral Immunol. 2005;18:445–456. doi: 10.1089/vim.2005.18.445. [DOI] [PubMed] [Google Scholar]
- 64.Chen L, Arora M, Yarlagadda M, Oriss TB, Krishnamoorthy N, Ray A, Ray P. Distinct responses of lung and spleen dendritic cells to the TLR9 agonist CpG oligodeoxynucleotide. J Immunol. 2006;177:2373–2383. doi: 10.4049/jimmunol.177.4.2373. [DOI] [PubMed] [Google Scholar]
- 65.Kurt-Jones EA, Chan M, Zhou S, Wang J, Reed G, Bronson R, Arnold MM, Knipe DM, Finberg RW. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci USA. 2004;101:1315–1320. doi: 10.1073/pnas.0308057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sato A, Linehan MM, Iwasaki A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci USA. 2006;103:17343–17348. doi: 10.1073/pnas.0605102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang JP, Kurt-Jones EA, Shin OS, Manchak MD, Levin MJ, Finberg RW. Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J Virol. 2005;79:12658–12666. doi: 10.1128/JVI.79.20.12658-12666.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Finberg RW, Wang JP, Kurt-Jones EA. Toll like receptors and viruses. Rev Med Virol. 2007;17:35–43. doi: 10.1002/rmv.525. [DOI] [PubMed] [Google Scholar]
- 69.Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, Finberg RW. Human cytomegalovirus activates inflammatory cyto-kine responses via CD14 and Toll-like receptor 2. J Virol. 2003;77:4588–4596. doi: 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boehme KW, Guerrero M, Compton T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol. 2006;177:7094–7102. doi: 10.4049/jimmunol.177.10.7094. [DOI] [PubMed] [Google Scholar]
- 71.Szomolanyi-Tsuda E, Liang X, Welsh RM, Kurt-Jones EA, Finberg RW. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J Virol. 2006;80:4286–4291. doi: 10.1128/JVI.80.9.4286-4291.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu J, Martinez J, Huang X, Yang Y. Innate immunity against vaccinia virus is mediated by TLR2 and requires TLR-independent production of IFN-beta. Blood. 2007;109:619–625. doi: 10.1182/blood-2006-06-027136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral butnot bacterial ligands. Nat Immunol. 2009;10:1200–1207. doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, Wagner H, Kirschning CJ, Ter Meulen V, Schneider-Schaulies S. Hemagglutinin protein of wild-type measles virus activates tolllike receptor 2 signaling. J Virol. 2002;76:8729–8736. doi: 10.1128/JVI.76.17.8729-8736.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang S, Dolganiuc A, Szabo G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J Leukoc Biol. 2007;82:479–487. doi: 10.1189/jlb.0207128. [DOI] [PubMed] [Google Scholar]
- 76.Zhou S, Kurt-Jones EA, Mandell L, Cerny A, Chan M, Golenbock DT, Finberg RW. MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur J Immunol. 2005;35:822–830. doi: 10.1002/eji.200425730. [DOI] [PubMed] [Google Scholar]
- 77.Michaud F, Coulombe F, Gaudreault E, Kriz J, Gosselin J. Involvement of TLR2 in recognition of acute gammaherpesvirus-68 infection. PLoS ONE. 2010;5:e13742. doi: 10.1371/journal.pone.0013742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jimenez-Baranda S, Greenbaum B, Manches O, Handler J, Rabadan R, Levine A, Bhardwaj N. Oligonucleotide motifs that disappear during the evolution of influenza virus in humans increase alpha interferon secretion by plasmacytoid dendritic cells. J Virol. 2011;85:3893–3904. doi: 10.1128/JVI.01908-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tibbetts SA, Loh J, Van Berkel V, McClellan JS, Jacoby MA, Kapadia SB, Speck SH, Virgin HW., IV Establishment and maintenance of gammaherpesvirus latency are independent of infective dose and route of infection. J Virol. 2003;77:7696–7701. doi: 10.1128/JVI.77.13.7696-7701.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moser JM, Farrell ML, Krug LT, Upton JW, Speck SH. A gammaherpesvirus 68 gene 50 null mutant establishes long-term latency in the lung but fails to vaccinate against a wild-type virus challenge. J Virol. 2006;80:1592–1598. doi: 10.1128/JVI.80.3.1592-1598.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kayhan B, Yager EJ, Lanzer K, Cookenham T, Jia Q, Wu TT, Woodland DL, Sun R, Blackman MA. A replication-deficient murine gamma-herpesvirus blocked in late viral gene expression can establish latency and elicit protective cellular immunity. J Immunol. 2007;179:8392–8402. doi: 10.4049/jimmunol.179.12.8392. [DOI] [PubMed] [Google Scholar]
- 82.Li H, Ikuta K, Sixbey JW, Tibbetts SA. A replication-defective gammaherpesvirus efficiently establishes long-term latency in macrophages but not in B cells in vivo. J Virol. 2008;82:8500–8508. doi: 10.1128/JVI.00186-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stevenson PG, May JS, Smith XG, Marques S, Adler H, Koszinowski UH, Simas JP, Efstathiou S. K3-mediated evasion of CD8(+) T cells aids amplification of a latent gamma-herpesvirus. Nat Immunol. 2002;3:733–740. doi: 10.1038/ni818. [DOI] [PubMed] [Google Scholar]
- 84.May JS, Bennett NJ, Stevenson PG. An in vitro system for studying murid herpesvirus-4 latency and reactivation. PLoS ONE. 2010;5:e11080. doi: 10.1371/journal.pone.0011080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 86.Tellam J, Smith C, Rist M, Webb N, Cooper L, Vuocolo T, Connolly G, Tscharke DC, Devoy MP, Khanna R. Regulation of protein translation through mRNA structure influences MHC class I loading and T cell recognition. Proc Natl Acad Sci USA. 2008;105:9319–9324. doi: 10.1073/pnas.0801968105. [DOI] [PMC free article] [PubMed] [Google Scholar]