

Abstract
A subset of enzymes that belong to the radical S-adenosylmethionine (SAM) superfamily are able to catalyze methylation reactions. Substrates of these enzymes are distinct from the nucleophilic substrates that undergo methylation by a polar mechanism. Recently, activities of several radical SAM methylating enzymes have been reconstituted in vitro and their mechanisms of catalysis investigated. The RNA modifying enzymes RlmN and Cfr catalyze methylation via a methyl synthase mechanism. These enzymes use SAM in two distinct roles: as a source of a methyl group transferred to a conserved cysteine and as a source of 5′-deoxyadenosyl radical (5′-dA•). Hydrogen atom abstraction by this species generates a thiomethylene radical which adds into the RNA substrate, forming an enzyme-substrate covalent adduct. In another recent study, methylation of the indole moiety of tryptophan by the radical SAM and cobalamin-binding domain enzyme TsrM has been reconstituted. Methylcobalamin serves as an intermediate methyl donor in TsrM, and is proposed to transfer the methyl group as a methyl radical. Interestingly, despite the presence of the radical SAM motif, no reductive cleavage of SAM has been observed in this methylation. These important reconstitutions set the stage for further studies on mechanisms of radical methylation.
Introduction
The radical SAM enzymes comprise a large and functionally diverse superfamily of proteins that use radical chemistry to effect substrate modifications [1]. The unifying feature of these enzymes is the use of 5′-dA• as a key catalytic intermediate [2]. This potent oxidant is formed via reductive cleavage of SAM, coordinated to an iron in the [4Fe-4S] cluster in these proteins [3]. A subset of radical SAM enzymes has been demonstrated, or is predicted, to catalyze methylation on a diverse set of biomolecules, some with electronic properties distinct from conventional nucleophilic methylation substrates. Akin to the profound biological roles of methyltransferses that use polar mechanisms [4], radical SAM methylating enzymes influence important cellular functions, such as regulation of translation, susceptibility to antibiotics, and maintenance of bacterial intracytoplasmic membranes [5–9]. Recently, significant progress has been made in the reconstitution and mechanistic analysis of radical SAM methylating enzymes. This work has expanded the repertoire of known biological methylation catalysts, as well as the scope of substrates that can undergo enzymatic methylation. This review summarizes these advances, and offers a perspective on outstanding questions regarding radical SAM-mediated methylation.
Classes of Radical SAM Methylating Enzymes
Radical SAM methylating enzymes are grouped into three classes based on their primary sequence [10]. The class A sub-family is comprised of RlmN and Cfr, enzymes that methylate an adenosine in RNA (Figure 1). These enzymes contain a radical SAM domain as their only domain. In addition to three cysteine residues required for the ligation of the [4Fe-4S] cluster, these enzymes contain two additional conserved cysteines required for catalysis [11–13]. Class B enzymes contain the radical SAM motif and a cobalamin-binding domain and modify a broad range of substrates, including heteroaromatic compounds, unactivated aliphatic carbons, and phosphinates [14–29]. For example, TsrM methylates the C2 indole carbon of tryptophan which is ultimately incorporated into the quinaldic acid moiety of thiostrepton [19,30] (Figure 1). In other examples highlighting class B substrate diversity, Fom3 is postulated to methylate an alcohol carbon in fosfomycin biosynthesis [14], while PoyB and PoyC are postulated to methylate a range of unactivated aliphatic carbons in polytheonamide biosynthesis [17] (Figure 1). Class C enzymes methylate heteroaromatic substrates [31–35]. One such example is NosN, an enzyme that methylates the indolic acid moiety in the biosynthesis of thiopeptide antibiotic nosiheptide [31,36] (Figure 1). These enzymes contain both the radical SAM motif and a coproporphyrinogen III oxidase HemN-like motif [37]. Given the differences in domain organization, it is likely that each of these three classes of enzymes use a distinct mechanism to catalyze methylation. Indeed, in recent years, different mechanisms have been shown to account for methylation by class A and class B enzymes. To date, no in vitro activity has been reconstituted for any of class C enzymes.
Figure 1.

Selected molecules that contain methyl groups or their derivatives introduced by radical SAM methylating enzymes. Groups introduced by these enzymes are shown in red. Polytheonamides A and B differ only in the absolute configuration of the sulfoxide [58].
RNA Methylation by RlmN and Cfr
RlmN and Cfr are related bacterial enzymes that catalyze methylation of the electrophilic amidine carbons of an adenosine nucleotide, A2503, in 23S ribosomal RNA (rRNA) [38–40]. The substrate adenosine is located at the entrance to the nascent peptide exit tunnel of the bacterial ribosome. RlmN modifies the C2 adenosine position, and the resulting modification has been shown to modulate interactions between the ribosome and the nascent peptide, contributing to regulation and fidelity of translation [5,6]. More recently, it was demonstrated that certain transfer RNAs (tRNAs) also serve as substrates for RlmN [6]. Cfr modifies the C8 amidine carbon of the same adenosine in 23S rRNA in pathogenic microorganisms [40]. The resulting modification leads to resistance to five major classes of antibiotics that target the peptidyltransferase center within the ribosome [7,8].
The activities of both RlmN and Cfr have been reconstituted in vitro. Both enzymes contain a [4Fe-4S] cluster, require the presence of a one electron reductant for activity, and generate both S-adenosyl-L-homocysteine (SAH) and 5′-deoxyadenosine (5′-dA) as byproducts, consistent with the formation of 5′-dA• and its role in hydrogen atom abstraction [41]. The mechanism of these enzymes was further investigated by the use of isotopically labeled substrates [42] (Figure 2). In these experiments, deuterium incorporation into the methylated adenosine product and 5′-dA was analyzed following incubation of the enzyme with RNA (containing either hydrogen or deuterium at the substrate carbon) in the presence of (methyl-d3)-SAM or unlabeled SAM (Figure 2A, B). Surprisingly, these experiments demonstrated that a methylene fragment, and not an intact methyl group, is incorporated into the methylated adenosine products. Furthermore, incorporation of deuterium into 5′-dA was only observed when (methyl-d3)-SAM was used in the reaction (Figure 2A), and not when the RNA substrate was deuterated (Figure 2B). This indicates that there is no direct activation of the RNA substrate by 5′-dA•. These experiments demonstrate that, rather than acting as methyltransferases, RlmN and Cfr are methyl synthases [42]. In a parallel effort, an RNA fragment containing seven nucleotides was used as a methylation substrate under single turnover conditions (Figure 2C, D) [12]. Incubation of this short fragment of RNA with (methyl-d3)-SAM showed no deuterium incorporated into the methyl group of the product (Figure 2C), leading to a proposal that the enzyme used in the reaction was pre-methylated under growth conditions. Expression of the enzyme in an E. coli methionine auxotroph grown in the presence of (methyl-d3)-methionine further supported pre-existing protein methylation. When this protein was incubated with RNA under single turnover conditions in the presence of unlabeled SAM, the product methyl group contained two deuterium atoms (Figure 2D), while the third deuterium was found in the 5′-dA (Figure 2E). Importantly, both sets of labeling experiments indicate that these enzymes transfer methylene fragments into their products, as opposed to intact methyl groups [12,42].
Figure 2.

Summary of labeling experiments used to elucidate the mechanism of methylation by radical SAM methyl synthases RlmN [12,42]. Experiments in panels A and B were performed under multiple turnover conditions, whereas experiments in panels C–E were performed under single turnover conditions. SDT = sodium dithionite. Flv = flavodoxin. Flx = flavodoxin reductase. R1 = nucleotides 2447–2778, 23S rRNA. R2 = nucleotides 2500–2506, 23S rRNA. R3 = nucleotides 2018–2788, 23S rRNA. The RlmN substrate adenosine is in position 2503 in all pictured RNA constructs.
Protein methylation was mapped to a conserved Cys in the C-terminal portion of RlmN (Cys 355). This was further supported by the crystallographic observation of the methylated Cys 355 in RlmN. Its location in a flexible loop presumably facilitates alkylation and subsequent 5’-dA•-mediated activation [43]. The observation that RlmN lacking the [4Fe-4S] cluster also lacks methylation indicates that the cluster is required for both SAM-dependent reactions in this enzyme [44].
Together, this data suggests an unprecedented mechanistic model for methylation by the RlmN/Cfr family (Figure 3). A key feature of these enzymes is their ability to cleave SAM both heterolytically and homolytically. The first molecule of SAM methylates a conserved cysteine (Cys 355 in RlmN), forming a protein-derived methyl thioether moiety (Figure 3). The second SAM is homolytically cleaved to generate the 5′-dA•. Rather than carrying out direct hydrogen atom abstraction from the RNA substrate or abstracting a hydrogen atom from the methyl group of SAM, the generated 5′-dA• abstracts a hydrogen atom from the cysteine-bound methyl group. Thermodynamically, this is the most favorable pathway [45]. Addition of the resultant thiomethylene radical into the amidine carbon of the substrate is followed by one electron oxidation and deprotonation, yielding a covalent RNA-protein adduct. The adduct is displaced by a second conserved cysteine (Cys 118 in RlmN), releasing an exocyclic olefin intermediate that undergoes aromatization and protonation, to form the methyl group. Reduction of the resulting disulfide species regenerates the enzyme for the next cycle of catalysis. Evidence for the proposed mechanism was obtained by the use of a RlmN Cys118Ala mutant enzyme, which allowed for the trapping of the covalent intermediate (inset, Figure 3) [13].
Figure 3.

Proposed mechanism of RNA methylation by RlmN [12]. The covalent intermediate, trapped by mutagenesis of Cys118 to Ala [13], is shown in the inset. SAM = S-adenosyl-L-methionine. SAH = S-adenosyl-L-homocysteine. 5′-dA• = 5′-deoxyadenosyl radical.
In summary, there are two key structural and mechanistic features that enable catalysis by the Class A enzymes: the presence of two conserved Cys residues involved in catalysis and the use of SAM both as a methyl donor and a source of the activating radical. The dual use of SAM in RlmN and Cfr catalysis resembles its dual function in the radical SAM methylthiotransferases MiaB and RimO [46–49]. Likewise, radical SAM enzyme NifB, responsible for carbide insertion into the nitrogenase M cluster, utilizes a similar mechanistic logic that exploits SAM as a methyl donor in an SN2 reaction and as a source of 5’-dA• used for subsequent hydrogen atom removals from the methyl group [50].
Tryptophan Methylation in Thiostrepton Biosynthesis
Thiostrepton A is a ribosomally synthesized and extensively post-translationally modified thiopeptide antibiotic that contains a tryptophan-derived quinaldic acid moiety [51]. It is proposed that this heterocycle is formed by an initial C2′ methylation of tryptophan which has been shown to proceed with net retention of configuration at the methyl group and with the incorporation of all three methyl hydrogens into the product [51,52]. The observed retention of configuration implies two consecutive inversion steps, suggesting an intermediate methyl carrier to which the methyl group is transferred from SAM prior to substrate methylation. This role is fulfilled by methylcobalamin [30].
Recently, the radical SAM methyltransferase TsrM was shown to catalyze the aforementioned transformation [30]. This enzyme contains both a cobalamin-binding domain and a radical SAM domain, and experiments have confirmed the requirement of methylcobalamin for the reaction. Substrate methylation by this enzyme is coupled to the production of SAH. Interestingly, in contrast to canonical radical SAM enzymes, product formation is not accompanied by formation of 5′-dA nor is one electron reductant required for the reaction. These observations strongly imply that SAM is utilized only as a methyl donor and does not undergo homolytic cleavage. Furthermore, when (methyl-d3)-SAM was used in the reaction, the product contained three deuterium atoms, even in the presence of a large excess of unlabeled methylcobalamin, leading to the hypothesis that the enzyme binds free cobalamin which is methylated by SAM in the active site. Similar efficiencies in the presence of either catalytic or stoichiometric amounts of methylcobalamin support its role as a cofactor, with SAM serving as a stoichiometric methyl source.
Several experiments were carried out to elucidate the functions of the different metal centers in catalysis [30]. UV-Vis spectroscopic monitoring of the reaction shows features characteristic of the direct conversion of methylcob(III)alamin to cob(II)alamin. In contrast to other cobalamin-dependent methyltransferases [53], no formation of cob(I)alamin was observed, suggesting that a polar mechanism of electrophilic aromatic substitution at the indole ring is unlikely. Rather, it is postulated that homolytic bond cleavage of the weak cobalt-carbon bond forms a methyl radical that adds into the substrate. The Co(II) formed in this reaction is proposed to be converted to Co(I) by the reduced [4Fe-4S] cluster, which is likely rapidly re-methylated, akin to the re-activation mechanism of the corrinoid iron-sulfur protein [54]. Other than its postulated role in the regeneration of the methylcobalamin cofactor, the precise role of the [4Fe-4S] cluster is yet to be fully understood. While the cluster is required for the formation of methylated Trp, its presence is not required for the formation of SAH. It is proposed that the cluster coordinates the substrate Trp, similar to coordination of this amino acid by the [4Fe-4S] cluster in NocL [55]. The proposed mechanism may be represented with the model shown in Figure 4.
Figure 4.
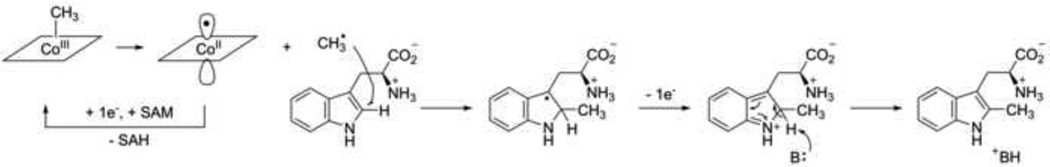
Proposed mechanism of the C2 methylation of the indole moiety of tryptophan by TsrM [30]. The Trp substrate is putatively coordinated by the iron-sulfur cluster. Reduction of Cob(II)alamin by the reduced [4Fe-4S] cluster may result in transient formation of the nucleophilic Co(I), which may react with SAM to regenerate methylcobalamin cofactor. Following addition of the methyl radical, oxidation of the Trp radical might be achieved by the oxidized cluster. SAM = S-adenosyl-L-methionine. SAH = S-adenosyl-L-homocysteine.
Conclusions and Future Directions
The recently accomplished reconstitution of the methylation activity of the radical SAM enzymes RlmN, Cfr and TsrM have uncovered novel mechanisms that nature uses to achieve methylation. These transformations complement the reactivity of conventional methyltransferases and expand the scope of methylation substrates. Importantly, these discoveries have set the stage for further work on reconstitution and detailed mechanistic studies of radical SAM methylating enzymes. In particular, further investigation of the roles of the two metal centers in catalysis by the Class B methyltransferases is needed. Another interesting aspect that awaits further exploration is the observed overall retention of configuration of the methyl group transferred from SAM. Model studies of methylcobalamin mediated methylation suggest that the methylation results from an SH2 type reaction, initiated by the attack of a substrate radical onto the methylcobalamin [56], thereby forming the product and Cob(II)alamin. Inversion of the methyl group configuration in this step, combined with an initial inversion required for methyl group transfer from SAM to cobalamin, can account for the observed overall retention. In the case of TsrM-mediated methylation, this model would require generation of a substrate-based radical, which then adds into methylcobalamin, as proposed previously [10]. Given that 5′-dA is not generated in this reaction, it remains unclear how this Trp radical may be generated under anaerobic conditions.
An additional intriguing question is whether all Class B enzymes use the same mechanism, or if multiple strategies might have evolved. The latter point is particularly important given that the aliphatic carbon atoms (for example, the β-carbons of Val and Ile in polytheonamides and the alcohol carbon in the proposed substrate of Fom3) are unlikely substrates for the addition of a methyl radical. A recent in vitro reconstitution of phosphinate methyltransferase PhpK from Kitasatospora phosalacinea, the first Class B enzyme to be reconstituted in vitro, shows several differences in reactivity between this enzyme and TsrM [22]. Unlike TsrM, PhpK uses exogenously supplied methylcobalamin as a source of the methyl group. Additionally, the presence of a strong one electron reductant, likely necessary for the conversion of the [4Fe-4S] cluster from +2 to +1 oxidation state, is required for methylation by PhpK, while dispensable for the activity of TsrM. Continued mechanistic work on this and other enzymes is expected to shed further light on the mechanism(s) that Class B enzymes utilize. Given the number of uncharacterized radical SAM methylating enzymes and recent identifications of new gene clusters that encode these proteins through metagenomic efforts [17,57], it is likely that more fascinating radical methylation chemistry is to be discovered.
Highlights.
A subset of radical SAM enzymes catalyzes methylation reactions
Radical SAM methylating enzymes expand the scope of methylation substrates
RlmN and Cfr add a methylene fragment, originating from SAM, into the substrate
Trp methylation by TsrM requires both methylcobalamin and a [4Fe-4S] cluster
Acknowledgements
I thank the members of my lab for their useful comments on this review and NSF (CAREER 1056143) and NIH (R01 AI095393) for providing funding for our research on radical SAM-mediated methylation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Radical SAM, a novel protein superfamily linking unresolved steps in familiar biosynthetic pathways with radical mechanisms: functional characterization using new analysis and information visualization methods. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frey PA, Magnusson OT. S-Adenosylmethionine: a wolf in sheep's clothing, or a rich man's adenosylcobalamin? Chem Rev. 2003;103:2129–2148. doi: 10.1021/cr020422m. [DOI] [PubMed] [Google Scholar]
- 3.Vey JL, Drennan CL. Structural insights into radical generation by the radical SAM superfamily. Chem Rev. 2011;111:2487–2506. doi: 10.1021/cr9002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le DD, Fujimori DG. Protein and nucleic acid methylating enzymes: mechanisms and regulation. Curr Opin Chem Biol. 2012;16:507–515. doi: 10.1016/j.cbpa.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vazquez-Laslop N, Ramu H, Klepacki D, Kannan K, Mankin AS. The key function of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. EMBO J. 2010;29:3108–3117. doi: 10.1038/emboj.2010.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benitez-Paez A, Villarroya M, Armengod ME. The Escherichia coli RlmN methyltransferase is a dual-specificity enzyme that modifies both rRNA and tRNA and controls translational accuracy. RNA. 2012;18:1783–1795. doi: 10.1261/rna.033266.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Long KS, Poehlsgaard J, Kehrenberg C, Schwarz S, Vester B. The Cfr rRNA methyltransferase confers resistance to Phenicols, Lincosamides, Oxazolidinones, Pleuromutilins, and Streptogramin A antibiotics. Antimicrob Agents Chemother. 2006;50:2500–2505. doi: 10.1128/AAC.00131-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toh SM, Xiong L, Arias CA, Villegas MV, Lolans K, Quinn J, Mankin AS. Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol Microbiol. 2007;64:1506–1514. doi: 10.1111/j.1365-2958.2007.05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welander PV, Summons RE. Discovery, taxonomic distribution, and phenotypic characterization of a gene required for 3-methylhopanoid production. Proc Natl Acad Sci U S A. 2012;109:12905–12910. doi: 10.1073/pnas.1208255109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Q, van der Donk WA, Liu W. Radical-mediated enzymatic methylation: a tale of two SAMS. Acc Chem Res. 2012;45:555–564. doi: 10.1021/ar200202c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaminska KH, Purta E, Hansen LH, Bujnicki JM, Vester B, Long KS. Insights into the structure, function and evolution of the radical-SAM 23S rRNA methyltransferase Cfr that confers antibiotic resistance in bacteria. Nucleic Acids Res. 2010;38:1652–1663. doi: 10.1093/nar/gkp1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ. A radically different mechanism for S-adenosylmethionine-dependent methyltransferases. Science. 2011;332:604–607. doi: 10.1126/science.1200877. Using single-turnover labeling experiments the authors demonstrate that a conserved C-terminal cysteine of RlmN is methylated, and that it serves as a precursor to a thiomethylene radical. Addition of this radical to an amidine carbon in the RNA substrates initiates the methylation.
- 13. McCusker KP, Medzihradszky KF, Shiver AL, Nichols RJ, Yan F, Maltby DA, Gross CA, Galonic Fujimori D. Covalent intermediate in the catalytic mechanism of the radical S-adenosyl-L-methionine methyl synthase RlmN trapped by mutagenesis. J Am Chem Soc. 2012;134:18074–18081. doi: 10.1021/ja307855d. By mutagenesis of a key catalytic cysteine, the authors have trapped a covalent intermediate between the RlmN and its RNA substrate. The identity of this intermediate was elucidated by mass spectrometric fragmentation.
- 14.Woodyer RD, Li G, Zhao H, van der Donk WA. New insight into the mechanism of methyl transfer during the biosynthesis of fosfomycin. Chem Commun (Camb) 2007:359–361. doi: 10.1039/b614678c. [DOI] [PubMed] [Google Scholar]
- 15.Kuzuyama T, Seki T, Dairi T, Hidaka T, Seto H. Nucleotide sequence of fortimicin KL1 methyltransferase gene isolated from Micromonospora olivasterospora, and comparison of its deduced amino acid sequence with those of methyltransferases involved in the biosynthesis of bialaphos and fosfomycin. J Antibiot (Tokyo) 1995;48:1191–1193. doi: 10.7164/antibiotics.48.1191. [DOI] [PubMed] [Google Scholar]
- 16.Welander PV, Coleman ML, Sessions AL, Summons RE, Newman DK. Identification of a methylase required for 2-methylhopanoid production and implications for the interpretation of sedimentary hopanes. Proc Natl Acad Sci U S A. 2010;107:8537–8542. doi: 10.1073/pnas.0912949107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Freeman MF, Gurgui C, Helf MJ, Morinaka BI, Uria AR, Oldham NJ, Sahl HG, Matsunaga S, Piel J. Metagenome mining reveals polytheonamides as posttranslationally modified ribosomal peptides. Science. 2012;338:387–390. doi: 10.1126/science.1226121. Using a metagenomic approach, authors identify a gene cluster responsible for modification of polytheonamide precursor peptide, uncovering several genes that encode unusual radical SAM enzymes, including putative class B methyltransferases.
- 18.Nunez LE, Mendez C, Brana AF, Blanco G, Salas JA. The biosynthetic gene cluster for the beta-lactam carbapenem thienamycin in Streptomyces cattleya. Chem Biol. 2003;10:301–311. doi: 10.1016/s1074-5521(03)00069-3. [DOI] [PubMed] [Google Scholar]
- 19.Kelly WL, Pan L, Li C. Thiostrepton biosynthesis: prototype for a new family of bacteriocins. J Am Chem Soc. 2009;131:4327–4334. doi: 10.1021/ja807890a. [DOI] [PubMed] [Google Scholar]
- 20.Westrich L, Heide L, Li SM. CloN6, a novel methyltransferase catalysing the methylation of the pyrrole-2-carboxyl moiety of clorobiocin. Chembiochem. 2003;4:768–773. doi: 10.1002/cbic.200300609. [DOI] [PubMed] [Google Scholar]
- 21.Kamigiri K, Hidaka T, Imai S, Murakami T, Seto H. Studies on the biosynthesis of bialaphos (SF-1293) 12. C-P bond formation mechanism of bialaphos: discovery of a P-methylation enzyme. J Antibiot (Tokyo) 1992;45:781–787. doi: 10.7164/antibiotics.45.781. [DOI] [PubMed] [Google Scholar]
- 22. Werner WJ, Allen KD, Hu K, Helms GL, Chen BS, Wang SC. In vitro phosphinate methylation by PhpK from Kitasatospora phosalacinea. Biochemistry. 2011;50:8986–8988. doi: 10.1021/bi201220r. This study describes first in vitro reconstitution of an enzyme belonging to class B radical SAM methyltransferases. The enzyme, PhpK, forms a carbon-phosphorus bond.
- 23.Blodgett JA, Zhang JK, Metcalf WW. Molecular cloning, sequence analysis, and heterologous expression of the phosphinothricin tripeptide biosynthetic gene cluster from Streptomyces viridochromogenes DSM 40736. Antimicrob Agents Chemother. 2005;49:230–240. doi: 10.1128/AAC.49.1.230-240.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unwin J, Standage S, Alexander D, Hosted T, Jr, Horan AC, Wellington EM. Gene cluster in Micromonospora echinospora ATCC15835 for the biosynthesis of the gentamicin C complex. J Antibiot (Tokyo) 2004;57:436–445. doi: 10.7164/antibiotics.57.436. [DOI] [PubMed] [Google Scholar]
- 25.Kudo F, Kasama Y, Hirayama T, Eguchi T. Cloning of the pactamycin biosynthetic gene cluster and characterization of a crucial glycosyltransferase prior to a unique cyclopentane ring formation. J Antibiot (Tokyo) 2007;60:492–503. doi: 10.1038/ja.2007.63. [DOI] [PubMed] [Google Scholar]
- 26.Rachid S, Scharfe M, Blocker H, Weissman KJ, Muller R. Unusual chemistry in the biosynthesis of the antibiotic chondrochlorens. Chem Biol. 2009;16:70–81. doi: 10.1016/j.chembiol.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Watanabe K, Hotta K, Nakaya M, Praseuth AP, Wang CC, Inada D, Takahashi K, Fukushi E, Oguri H, Oikawa H. Escherichia coli allows efficient modular incorporation of newly isolated quinomycin biosynthetic enzyme into echinomycin biosynthetic pathway for rational design and synthesis of potent antibiotic unnatural natural product. J Am Chem Soc. 2009;131:9347–9353. doi: 10.1021/ja902261a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funabashi M, Yang Z, Nonaka K, Hosobuchi M, Fujita Y, Shibata T, Chi X, Van Lanen SG. An ATP-independent strategy for amide bond formation in antibiotic biosynthesis. Nat Chem Biol. 2010;6:581–586. doi: 10.1038/nchembio.393. [DOI] [PubMed] [Google Scholar]
- 29.Wang ZX, Li SM, Heide L. Identification of the coumermycin A(1) biosynthetic gene cluster of Streptomyces rishiriensis DSM 40489. Antimicrob Agents Chemother. 2000;44:3040–3048. doi: 10.1128/aac.44.11.3040-3048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pierre S, Guillot A, Benjdia A, Sandstrom C, Langella P, Berteau O. Thiostrepton tryptophan methyltransferase expands the chemistry of radical SAM enzymes. Nat Chem Biol. 2012;8:957–959. doi: 10.1038/nchembio.1091. This manuscript describes reconstitution and mechanistic evaluation of TsrM, a methylcobalamin binding and radical SAM domain containing protein that methylates the indole moiety of tryptophan. The presented evidence suggests the addition of a cobalamin-derived methyl radical into the substrate.
- 31.Yu Y, Duan L, Zhang Q, Liao R, Ding Y, Pan H, Wendt-Pienkowski E, Tang G, Shen B, Liu W. Nosiheptide biosynthesis featuring a unique indole side ring formation on the characteristic thiopeptide framework. ACS Chem Biol. 2009;4:855–864. doi: 10.1021/cb900133x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris RP, Leeds JA, Naegeli HU, Oberer L, Memmert K, Weber E, LaMarche MJ, Parker CN, Burrer N, Esterow S, et al. : Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J Am Chem Soc. 2009;131:5946–5955. doi: 10.1021/ja900488a. [DOI] [PubMed] [Google Scholar]
- 33.Du L, Sanchez C, Chen M, Edwards DJ, Shen B. The biosynthetic gene cluster for the antitumor drug bleomycin from Streptomyces verticillus ATCC15003 supporting functional interactions between nonribosomal peptide synthetases and a polyketide synthase. Chem Biol. 2000;7:623–642. doi: 10.1016/s1074-5521(00)00011-9. [DOI] [PubMed] [Google Scholar]
- 34.Tao M, Wang L, Wendt-Pienkowski E, George NP, Galm U, Zhang G, Coughlin JM, Shen B. The tallysomycin biosynthetic gene cluster from Streptoalloteichus hindustanus E465-94 ATCC 31158 unveiling new insights into the biosynthesis of the bleomycin family of antitumor antibiotics. Mol Biosyst. 2007;3:60–74. doi: 10.1039/b615284h. [DOI] [PubMed] [Google Scholar]
- 35.Galm U, Wendt-Pienkowski E, Wang L, George NP, Oh TJ, Yi F, Tao M, Coughlin JM, Shen B. The biosynthetic gene cluster of zorbamycin, a member of the bleomycin family of antitumor antibiotics, from Streptomyces flavoviridis ATCC 21892. Mol Biosyst. 2009;5:77–90. doi: 10.1039/b814075h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mocek U, Knaggs AR, Tsuchiya R, Nguyen T, Beale JM, Floss HG. Biosynthesis of teh modified peptide antibiotic nosiheptide in Streptomyces actuosus. J. Am. Chem. Soc. 1993;115:7557–7568. [Google Scholar]
- 37.Layer G, Moser J, Heinz DW, Jahn D, Schubert WD. Crystal structure of coproporphyrinogen III oxidase reveals cofactor geometry of Radical SAM enzymes. EMBO J. 2003;22:6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kehrenberg C, Schwarz S, Jacobsen L, Hansen LH, Vester B. A new mechanism for chloramphenicol, florfenicol and clindamycin resistance: methylation of 23S ribosomal RNA at A2503. Mol Microbiol. 2005;57:1064–1073. doi: 10.1111/j.1365-2958.2005.04754.x. [DOI] [PubMed] [Google Scholar]
- 39.Toh SM, Xiong L, Bae T, Mankin AS. The methyltransferase YfgB/RlmN is responsible for modification of adenosine 2503 in 23S rRNA. RNA. 2008;14:98–106. doi: 10.1261/rna.814408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giessing AM, Jensen SS, Rasmussen A, Hansen LH, Gondela A, Long K, Vester B, Kirpekar F. Identification of 8-methyladenosine as the modification catalyzed by the radical SAM methyltransferase Cfr that confers antibiotic resistance in bacteria. RNA. 2009;15:327–336. doi: 10.1261/rna.1371409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan F, LaMarre JM, Rohrich R, Wiesner J, Jomaa H, Mankin AS, Fujimori DG. RlmN and Cfr are radical SAM enzymes involved in methylation of ribosomal RNA. J Am Chem Soc. 2010;132:3953–3964. doi: 10.1021/ja910850y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yan F, Fujimori DG. RNA methylation by radical SAM enzymes RlmN and Cfr proceeds via methylene transfer and hydride shift. Proc Natl Acad Sci U S A. 2011;108:3930–3934. doi: 10.1073/pnas.1017781108. The study describes use of isotope labeling experiments to elucidate origin of methyl group introduced into RNA by RlmN and Cfr. These experiments led to the conclusion that these enzymes are methyl synthases.
- 43. Boal AK, Grove TL, McLaughlin MI, Yennawar NH, Booker SJ, Rosenzweig AC. Structural basis for methyl transfer by a radical SAM enzyme. Science. 2011;332:1089–1092. doi: 10.1126/science.1205358. The crystal structure of RlmN provides evidence for the methylation of a conserved Cys in this enzyme and the presence of a single binding site for SAM.
- 44.Grove TL, Radle MI, Krebs C, Booker SJ. Cfr and RlmN contain a single [4Fe-4S] cluster, which directs two distinct reactivities for S-adenosylmethionine: methyl transfer by SN2 displacement and radical generation. J Am Chem Soc. 2011;133:19586–19589. doi: 10.1021/ja207327v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hioe J, Zipse H. Hydrogen transfer in SAM-mediated enzymatic radical reactions. Chemistry. 2012;18:16463–16472. doi: 10.1002/chem.201202869. [DOI] [PubMed] [Google Scholar]
- 46.Pierrel F, Douki T, Fontecave M, Atta M. MiaB protein is a bifunctional radical-S-adenosylmethionine enzyme involved in thiolation and methylation of tRNA. J Biol Chem. 2004;279:47555–47563. doi: 10.1074/jbc.M408562200. [DOI] [PubMed] [Google Scholar]
- 47.Lee KH, Saleh L, Anton BP, Madinger CL, Benner JS, Iwig DF, Roberts RJ, Krebs C, Booker SJ. Characterization of RimO, a new member of the methylthiotransferase subclass of the radical SAM superfamily. Biochemistry. 2009;48:10162–10174. doi: 10.1021/bi900939w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anton BP, Saleh L, Benner JS, Raleigh EA, Kasif S, Roberts RJ. RimO, a MiaB-like enzyme, methylthiolates the universally conserved Asp88 residue of ribosomal protein S12 in Escherichia coli. Proc Natl Acad Sci U S A. 2008;105:1826–1831. doi: 10.1073/pnas.0708608105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arragain S, Garcia-Serres R, Blondin G, Douki T, Clemancey M, Latour JM, Forouhar F, Neely H, Montelione GT, Hunt JF, et al. : Post-translational modification of ribosomal proteins: structural and functional characterization of RimO from Thermotoga maritima, a radical S-adenosylmethionine methylthiotransferase. J Biol Chem. 2010;285:5792–5801. doi: 10.1074/jbc.M109.065516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wiig JA, Hu Y, Lee CC, Ribbe MW. Radical SAM-dependent carbon insertion into the nitrogenase M-cluster. Science. 2012;337:1672–1675. doi: 10.1126/science.1224603. In this study, the authors demonstrate that, analogous to class A radical SAM methylating enzymes, NifB uses SAM both as a source of a methyl group and as a source of the 5'-deoxyadenosyl radical.
- 51.Zhou P, O'Hagan D, Mocek U, Zeng Z, Yuen L-D, Frenzel T, Unkefer CJ, Beale JM, Floss HG. Biosynthesis of the Antibiotic Thiostrepton. Methylation of Tryptophan in the Formation of the Quinaldic Acid Moiety by Transfer of the Methionine Methyl Group with Net Retention of Configuration. J. Am. Chem. Soc. 1989;111:7274–7276. [Google Scholar]
- 52.Frenzel T, Zhou P, Floss HG. Formation of 2-methyltryptophan in the biosynthesis of thiostrepton: isolation of S-adenosylmethionine:tryptophan 2-methyltransferase. Arch Biochem Biophys. 1990;278:35–40. doi: 10.1016/0003-9861(90)90227-p. [DOI] [PubMed] [Google Scholar]
- 53.Matthews RG, Koutmos M, Datta S. Cobalamin-dependent and cobamide-dependent methyltransferases. Curr Opin Struct Biol. 2008;18:658–666. doi: 10.1016/j.sbi.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Menon S, Ragsdale SW. Role of the [4Fe-4S] cluster in reductive activation of the cobalt center of the corrinoid iron-sulfur protein from Clostridium thermoaceticum during acetate biosynthesis. Biochemistry. 1998;37:5689–5698. doi: 10.1021/bi9727996. [DOI] [PubMed] [Google Scholar]
- 55.Zhang Q, Chen D, Lin J, Liao R, Tong W, Xu Z, Liu W. Characterization of NocL involved in thiopeptide nocathiacin I biosynthesis: a [4Fe-4S] cluster and the catalysis of a radical S-adenosylmethionine enzyme. J Biol Chem. 2011;286:21287–21294. doi: 10.1074/jbc.M111.224832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mosimann H, Krautler B. Methylcorrinoids Methylate Radicals-Their Second Biological Mode of Action? Angew Chem Int Ed Engl. 2000;39:393–395. doi: 10.1002/(sici)1521-3773(20000117)39:2<393::aid-anie393>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 57.Murphy K, O'Sullivan O, Rea MC, Cotter PD, Ross RP, Hill C. Genome mining for radical SAM protein determinants reveals multiple sactibiotic-like gene clusters. PLoS One. 2011;6:e20852. doi: 10.1371/journal.pone.0020852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hamada T, Matsunaga S, Yano G, Fusetani N. Polytheonamides A and B, highly cytotoxic, linear polypeptides with unprecedented structural features, from the marine sponge, Theonella swinhoei. J Am Chem Soc. 2005;127:110–118. doi: 10.1021/ja045749e. [DOI] [PubMed] [Google Scholar]