

Abstract
The structure activity relationships of N-(3-acyloxy-2-benzylpropyl)-N′-4-[(methylsulfonylamino)benzyl] thioureas, which represent simplified RTX-based vanilloids, were investigated by varying the distances between the four principal pharmacophores and assessing binding and antagonistic activity on rTRPV1. The analysis indicated that a 3-pivaloyloxy-2-benzylpropyl C-region conferred the best potency in binding affinity and antagonism. The molecular modeling of this best template with the tetrameric homology model of rTRPV1 was performed to identify its binding interactions with the receptor.
Keywords: Vanilloid Receptor 1, TRPV1, antagonist, resiniferatoxin, molecular modeling, capsaicin
Introduction
The transient receptor potential V1 (TRPV1) receptor1 is a molecular integrator of nociceptive stimuli, including protons,2 heat,3 inflammatory mediators such as anandamide4 and lipoxygenase products,5 and vanilloids such as capsaicin (CAP)6 and resiniferatoxin (RTX)7. The receptor functions as a non-selective cation channel with high Ca2+ permeability and its activation leads to an increase in intracellular Ca2+ that results in excitation of primary sensory neurons and ultimately the central perception of pain.
TRPV1 antagonists are promising drug candidates. Therapeutic applications include inhibiting the transmission of nociceptive signaling from the periphery to the CNS as well as blocking other pathological states associated with this receptor. TRPV1 antagonists have thus emerged as novel and promising analgesic and antiinflammatory agents, particularly for chronic pain and inflammatory hyperalgesia.8 The number of antagonists reported continues to increase and their clincal development has been extensively reviewed.9-13
Previously, we have reported that a series of N-(3-acyloxy-2-benzylpropyl)-N′-4-[(methylsulfonylamino)benzyl] thiourea analogues (e.g. 1, 2) were effective antagonists of the action of capsaicin on rat TRPV1 (Figure 1).14-16 We further found that 3-fluoro substitution on the 4-(methylsulfonylamino)phenyl group in the A-region enhanced the extent of antagonism.14 Thus, the compounds 3 and 4 showed high binding affinities and potent antagonism in the rTRPV1/CHO system.
Figure 1. Pharmacophoric regions of RTX and sRTX.

The principal pharmacophores in the template, denoted as P1-P4 in Figure 2, were derived from our RTX-derived pharmacophore model in which 4-hydroxy-3-methoxyphenyl (A-region), C20-ester (B-region), orthophenyl (C1-region) and C3-keto (C2-region) groups in RTX correspond to P1-P4, respectively.17
Figure 2. The four principal pharmacophores of sRTX.

In order to find the optimal template and its active conformation for an sRTX (a simplified RTX having the four principal pharmacophores of RTX), we have investigated the structure-activity relationships of the template with regard to the spatial arrangement of key pharmacophores by lengthening or shortening the distances between the pharmacophores. We have already established from previous analysis that the distance between P1 and P2 was optimal when the linker is one-carbon (X =1).15
In a continuation of these efforts, we have now optimized the distances between P2-P4 in the template (corresponding to Y and Z in Figure 2) with a (3-fluoro-4-methylsulfonylamino)benzyl A-region to obtain the preferred active spatial arrangement of four pharmacophores. We describe here the syntheses and functional TRPV1 activities of the designed ligands and our docking study of the resulting active template with our homology model.
Chemistry
General syntheses of the final target thioureas were achieved by the coupling between the corresponding isothiocyanates (or thiocarbamate) and amines.
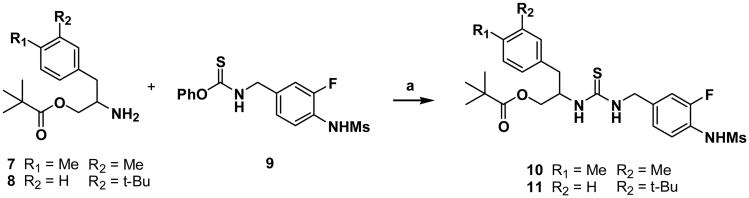
For the template having Y=0 and Z=1, compounds 10 and 11 were synthesized by the coupling of the C-region amines 7 and 8 previously reported15 with the phenylthiocarbamate of A-region 9 (Scheme 1).
Scheme 1. Reactions and conditions: (a) NEt3, CH2Cl2.
For the synthesis of a template having Y=0 and Z=2, the amines of the C-region 12 and 13 were prepared starting from N-(diphenylmethylene)glycine ethyl ester by the procedure reported previously15 and then were converted to the corresponding isothiocyanates, 14 and 15. The isothiocyanates reacted with the amines of the A-region 16 and 1714 to afford thioureas 18-21, respectively (Scheme 2).
Scheme 2. Reactions and conditions: (a) 1,1′-Thiocarbonyl diimidazole, NEt3, DMF; (b) NEt3, CH2Cl2.

For the synthesis of a template having Y=0 and Z=0, the isothiocyanate of the C-region 25 was prepared starting from vinylbenzene 22 in four steps (Scheme 3). The dihydroxyation of 22 followed by selective pivaloylation provided 23 whose hydroxyl group was converted to the corresponding azide 24. The azide 24 was then converted to the isothiocyanate 25 efficiently with carbon disulfide and triphenylphosphine;18 the isothiocyanate 25 was coupled with the amines of the A-region 16 and 17 to afford the final compounds 26 and 27.
Scheme 3. Reactions and conditions: (a) OsO4, NMO, acetone-H2O (1:1); (b) pivaloyl chloride, NEt3, DMAP, CH2Cl2; (c) DPPA, DBU, toluene; (d) CS2, PPh3, THF; (e) 16 or 17, NEt3, CH2Cl2.

For the synthesis of a template having Y=1 and Z=0, the isothiocyanate of the C-region 31 was prepared starting from 2-phenylacetate 28 in five steps (Scheme 4). The C-acylation of 28, the reduction of the diester followed by selective pivaloylation provided alcohol 29. By following the route represented in Scheme 3, compound 29 was converted to the final thioureas 32 and 33.
Scheme 4. Reactions and conditions: (a) NaOEt, diethyl carbonate, toluene; (b) LiAlH4, ether; (c) pivaloyl chloride, N(C2H5)3, DMAP, CH2Cl2; (d) MsCl, NEt3, CH2Cl2; (e) NaN3, DMF; (f) CS2, PPh3, THF; (e) 16 or 17, N(C2H5)3, CH2Cl2.

Biological Activity
The binding affinities and potencies as agonists/antagonists of the synthesized TRPV1 ligands were assessed in vitro by a binding competition assay with [3H]RTX and by a functional 45Ca2+ uptake assay using rat TRPV1 heterologously expressed in Chinese hamster ovary (CHO) cells, as previously described.19 The results are summarized in Table 1, together with the potencies of the previously reported thiourea antagonists 1-6.14-16
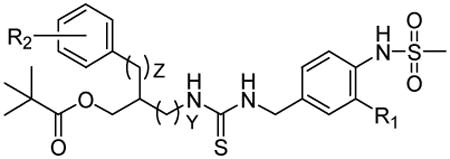
Table 1. Potencies of vanilloid ligands for binding to rat TRPV1 and for inducing calcium influx in CHO/TRPV1 cells.
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Y | Z | R1 | R2 | Ki (nM)a Binding Affinity | Agonism b (%) | Ki (nM)c Antagonism (%) | |
| 1 | 1 | 1 | H | 3,4-Me2 | 29.3 ±7.6 | (15%) | 67 ±25 |
| 2 | 1 | 1 | H | 4-t-Bu | 64 ±21 | (40%) | 86 ±17 |
| 3 | 1 | 1 | F | 3,4-Me2 | 54 ±28 | NE | 7.8±3.0 |
| 4 | 1 | 1 | F | 4-t-Bu | 22.6 ±2.7 | (5%) | 52 ±17 |
| 5 | 0 | 1 | H | 3,4-Me2 | 580 (±130 | (19%) | (48%) |
| 6 | 0 | 1 | H | 4-t-Bu | 416 ±44 | (8%) | (31%) |
| 10 | 0 | 1 | F | 3,4-Me2 | 233 ±18 | NE | 650 ±210 |
| 11 | 0 | 1 | F | 4-t-Bu | 115 ±2.7 | NE | 179 ±20 |
| 32 | 1 | 0 | H | 4-t-Bu | 143 ±43 | NE | 91 ±25 |
| 33 | 1 | 0 | F | 4-t-Bu | 69 ±18 | NE | 116 ±40 |
| 18 | 0 | 2 | H | 3,4-Me2 | 390 ±100 | (5%) | 33.7 ±2 |
| 19 | 0 | 2 | H | 4-t-Bu | 366 ±8.1 | NE | 286 ±48 |
| 20 | 0 | 2 | F | 3,4-Me2 | 120 ±4.6 | NE | 111 ±43 |
| 21 | 0 | 2 | F | 4-t-Bu | 190 ±38 | NE | 152 ±46 |
| 26 | 0 | 0 | H | 4-t-Bu | 197 ±56 | (46%) | 940±420(46% |
| 27 | 0 | 0 | F | 4-t-Bu | 244 ±46 | (35%) | 213±18 (72%) |
NE: not effective, WE: weakly effective at 30 μM
mean ± SEM of at least three experiments
if any agonism was detected, value in parentheses indicates fractional calcium uptake compared to that by 30 μM capsaicin
mean ± SEM of at least three experiments for measuring Ki. Value in parenthesis indicates measured extent of partial antagonism if observed
Previously, the analysis of the SAR for the spacing between P1 ((4-methylsulfonylamino)phenyl) and P2 (thiourea) (the A and B-regions, respectively) indicated that either one-carbon elongation or one carbon deletion between them caused a dramatic loss of binding affinity regardless of the distances between P2-P4. Thereby, the SAR in the present study was conducted based on the template of the N-[(4-methylsulfonylamino)benzyl]thiourea template corresponding to X=1 in Figure 2.
In the variation of the Y part of the template, one-carbon shortening of 1 and 2 (Y=1, Z=1) provided N-(2-pivaloyl-1-benzylethyl) analogues 5 and 6 (Y=0, Z=1), which were found to be partial antagonists with weaker binding affinities by 20- and 6.5-fold, respectively. Modification of the A-region through incorporation of a fluoro group into 5 and 6 at the 3-position in the A-region led not only to stronger binding affinities (2.5-fold for 10, 3.6-fold for 11) but also to enhanced antagonism, as we would predict from previous studies.14
In the variation of the Z part of the template, one-carbon shortening of 2 and 4 (Y=1, Z=1) provided N-(3-pivaloyloxy-2-phenylpropyl) analogues 32 and 33 (Y=1, Z=0), which were found to be full antagonists despite a modest loss in binding affinities of 2.2- and 3-fold, respectively.
The combination of one-carbon shortening of the Y part and one-carbon elongation of the Z part of analogues 1-4 (Y=1, Z=1) provided N-[2-pivaloyl-1-(phenylethyl)ethyl] analogues 18-21 (Y=0, Z=2), which exhibited reduced potencies in binding affinity and variable effects on antagonistic potencies. The extremes were a 2-fold enhancement in antagonistic potency for 18 compared to 1 and a 14-fold reduction in potency for 20 compared to 3. On the other hand, one-carbon shortening of both the Y and Z parts from 2 and 4 (Y=1, Z=1) afforded N-(2-pivaloyl-1-phenylethyl) analogues 26 and 27 (Y=0, Z=0), which also showed weaker binding affinities but became partial agonists/antagonists in contrast to the above template.
Overall, the SAR analysis of Y and Z parts in the C-region provided the order of potencies in their binding affinities as follows: (Y, Z) = (1, 1) > (1, 0) > (0, 0 ≈ 0, 1≈ 0, 2). We conclude that the C-region having X=1, Y=1 together with Z=1 for the A-region provided the optimal sRTX for interacting with the rTRPV1.
Molecular Modeling
We previously constructed the tetramer homology model of rat TRPV1 (rTRPV1) and reported the docking results of representative TRPV1 ligands, including capsaicin, resiniferatoxin (RTX) and sRTX.20 Using this rTRPV1 model, we performed a flexible docking study with compound 4, which possesses the highest binding affinity in this series.21 As shown in Figure 3, this molecule fitted well into the binding site located at the interface of the two monomers. The 4-(methylsulfonylamino)phenyl group (A-region) occupied the deep bottom hole at the binding site, and the two oxygen atoms and NH of the sulfonamide group participated in hydrogen bonding with Ser512 and Gly558, respectively. The sulfur of the thiourea group (B-region) made a hydrogen bond with Tyr511 and also contributed to the appropriate positioning of the C-region for the hydrophobic interactions. The bulky and hydrophobic 4-t-butylbenzyl and pivaloyloxymethyl groups in the C-region extended toward Met547 in the upper hydrophobic area and the adjacent monomer's hydrophobic region, composed of Phe587 and Phe591, and formed hydrophobic interactions.
Figure 3.

Predicted binding mode of compound 4 in the rTRPV1 model with surface representations.
(A) Binding mode of compound 4. The key residues are marked and displayed as capped-stick with carbon atoms in white. The helices are colored in gray and the helices of the adjacent monomer are displayed in line ribbon. Compound 4 is depicted as ball-and-stick with carbon atoms in magenta. The van der Waals surface of the ligand is presented with the lipophilic potential property. Hydrogen bonds are shown in black dashed lines, and non-polar hydrogens are undisplayed for clarity. (B) Surface representations of the docked compound 4 and rTRPV1. The Fast Connolly surface of rTRPV1 was generated by MOLCAD and colored by the lipophilic potential property. The surface of rTRPV1 is Z-clipped, and that of the ligand is in its carbon color for clarity. (C) Van der Waals surface of compound 4 is colored by the lipophilic potential property.
In summary, we have investigated the structure activity relationship of a series of N-(3-acyloxy-2-benzylpropyl)-N′-4-[(methylsulfonylamino)benzyl] thioureas as TRPV1 antagonists in which we varied the distances between the four principal pharmacophores (P1-P4). The analysis indicated that the C-region template having X=1, Y=1, and Z=1 in figure 2 displayed the best potency in binding affinity and antagonism for rTRPV1, reflecting that its pharmacophores provided the optimal spacing for rTRPV1 interaction. The docking study of compound 4 with the tetrameric homology model established by us provided useful information on the binding interactions with the receptor, including the hydrophilic interactions with Tyr 511 and Ser512 and the hydrophobic ones with the two separate pockets composed of Met547 and Phe587/591, respectively.
Acknowledgments
This research was supported by Grants R11-2007-107-02001-0 from the NRF, Grants 2011-0028885 from the NLRL program and Frontier Research Program 2011-K000289 funded by MEST and NRF, and was supported in part by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, National Cancer Institute (Project Z1A BC 005270).
References and notes
- 1.Szallasi A, Blumberg PM. Pharmacol Rev. 1999;51:159. [PubMed] [Google Scholar]
- 2.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. Neuron. 1998;21:531. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 3.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. Nature. 1997;389:816. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 4.Zygmunt PM, Petersson J, Andersson DA, Chuang HH, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Nature. 1999;400:452. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]
- 5.Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U. Proc Natl Acad Sci USA. 2000;97:6155. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walpole CSJ, Wrigglesworth R. Capsaicin in the Study of Pain. Vol. 63 Academic Press; 1993. [Google Scholar]
- 7.Appendino G, Szallasi A. Life Sci. 1997;60:681. doi: 10.1016/s0024-3205(96)00567-x. [DOI] [PubMed] [Google Scholar]
- 8.Szallasi A, Cruz F, Geppetti P. Trends in Mol Med. 2006;12:545. doi: 10.1016/j.molmed.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Voight EA, Kort ME. Exp Opin Ther Pat. 2010;20:1. doi: 10.1517/13543776.2010.497756. [DOI] [PubMed] [Google Scholar]
- 10.Lazar J, Gharat L, Khairathkar-Joshi N, Blumberg PM, Szallasi A. Exp Opin Drug Discov. 2009;4:159. doi: 10.1517/17460440802681300. [DOI] [PubMed] [Google Scholar]
- 11.Gunthorpe MJ, Chizh BA. Drug Disc Today. 2009;14:56. doi: 10.1016/j.drudis.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 12.Wong GY, Gavva NR. Brain Res Rev. 2009;60:267. doi: 10.1016/j.brainresrev.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Kym PR, Kort ME, Hutchins CW. Biochem Pharmacol. 2009;78:211. doi: 10.1016/j.bcp.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 14.Lee J, Lee J, Kang M, Shin MY, Kim JM, Kang SU, Lim JO, Choi HK, Suh YG, Park HG, Oh U, Kim HD, Park YH, Ha HJ, Kim YH, Toth A, Wang Y, Tran R, Pearce LV, Lundberg DJ, Blumberg PM. J Med Chem. 2003;46:3116. doi: 10.1021/jm030089u. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, Kim SY, Lee Ji, Kang M, Kil MJ, Choi HK, Jin MK, Wang Y, Toth A, Pearce LV, Lundberg DJ, Tran R, Blumberg PM. Bioorg Med Chem. 2004;12:3411. doi: 10.1016/j.bmc.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 16.Ryu H, Jin MK, Kang SU, Kim SY, Kang DW, Lee J, Pearce LV, Pavlyukovets VA, Morgan MA, Tran R, Toth A, Lundberg DJ, Blumberg PM. J Med Chem. 2008;51:57. doi: 10.1021/jm701049p. [DOI] [PubMed] [Google Scholar]
- 17.Lee J, Lee J, Kim J, Kim SY, Chun MW, Cho H, Hwang SW, Oh U, Park YH, Marquez VE, Beheshti M, Szabo T, Blumberg PM. Bioorg Med Chem. 2001;9:19. doi: 10.1016/s0968-0896(00)00216-9. [DOI] [PubMed] [Google Scholar]
- 18.Isoda T, Hayashi K, Tamai S, Kumagai T, Nagao Y. Chem Pharm Bull. 2006;54:1616. doi: 10.1248/cpb.54.1616. [DOI] [PubMed] [Google Scholar]
- 19.Min KH, Suh YG, Park MK, Park HG, Park YH, Kim HD, Oh U, Blumberg PM, Lee J. Mol Pharm. 2002;62:947. published erratum appears in Mol Pharmacol 2003, 63, 958. [Google Scholar]
- 20.Lee JH, Lee Y, Ryu H, Kang DW, Lee J, Lazar J, Pearce LV, Pavlyukovets VA, Blumberg PM, Choi S. J Comput Aided Mol Des. 2011;25:317. doi: 10.1007/s10822-011-9421-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The docking study was conducted with the active (R)-isomer of compound 4. The syntheses and biological characterization of chiral isomers will be published elsewhere. The 3D structure of the ligand was generated with Concord and energy minimized using the MMFF94s force field and MMFF94 charge until the rms of the Powell gradient was 0.05 kcal mol-1A-1 in SYBYL-X 1.2 (Tripos Int., St. Louis, MO, USA). The flexible docking study on the rTRPV1 model20 was performed by GOLD v.5.0.1 (Cambridge Crystallographic Data Centre, Cambridge, UK), which uses a genetic algorithm (GA) and allows for full ligand flexibility and partial protein flexibility. The binding site was defined as 8 Å around the capsaicin docked in the rTRPV1 model. The side chains of the seven residues which are important for ligand binding (i.e., Tyr511, Ser512, Leu515, Phe543, Leu547, Thr550, and Asn551) were set to be flexible with ‘crystal mode’ in GOLD. The ligand was docked using the GoldScore scoring function with 30 GA runs. Other parameters were set as default. All computation calculations were undertaken on an Intel® Xeon™ Quad-core 2.5 GHz workstation with Linux Cent OS release 5.5.